

Abstract
Cyclic hexapeptides represent a class of compounds with important, diverse biological activities. We report herein that the antibody 16G3 catalyzes the cyclization of d-Trp-Gly-Pal-Pro-Gly-Phe⋅p-nitrophenyl ester (8a) to give c-(d-Trp-Gly-Pal-Pro-Gly-l-Phe) (11a). The antibody does not, however, catalyze either epimerization or hydrolysis. The resulting rate enhancement of the cyclization by 16G3 (22-fold) was sufficient to form the desired product in greater than 90% yield. In absolute rate terms, the turnover of 16G3 is estimated to be 2 min−1. The background rate of epimerization of 8a was reduced from 10 to 1% and hydrolysis from 50 to 4% in the presence of 16G3. As expected, the catalytic effects of 16G3 were blocked by the addition of an amount of the hapten equal to twice the antibody concentration. We also synthesized three diastereomers of 8a: the d-Trp1-d-Phe6 (8b), l-Trp1-l-Phe6 (8c), and l-Trp1-d-Phe6 (8d) hexapeptides as well as d-Trp′-l-Trp6 (12) and d-Phe′-l-Phe6 (13). As expected, the rate enhancement by 16G3 was greatest for 8a, because the stereochemistry of Trp1 and Phe6 matches that of the corresponding residues on the hapten used to induce the biosynthesis of 16G3. A model of the variable domain of 16G3 was generated from the primary sequence using the antibody structural database to guide the model construction. The resulting model provided support for some previously proposed interpretations of the kinetic data, while providing valuable new insights for others.
Several years ago, we undertook the generation of antibodies to catalyze peptide bond formation in part because man-made catalysts of peptide bond formation had not been described (1). We were also intrigued by the contrast between medicinal chemistry and antibody design. Generally, the former seeks by design or screening to discover small molecules that interact with macromolecules such as enzymes or receptors leading to enzyme inhibitors or to hormone/neurotransmitter agonists/antagonists. In contrast, catalytic antibody research involves the synthesis of haptens, designed to generate novel macromolecules (antibodies), which in turn are capable of catalyzing predetermined chemical reactions.
The Design of Hapten 1a: Novel Chemistry and the Generation of Antibody 16G3 for Bimolecular Peptide Bond Formation.
We reported the synthesis of hapten 1a (Fig. 1), designed to induce the formation of antibodies capable of catalyzing the formation of dipeptides of the general structure acetyl-XXX-d-Trp⋅NH2, wherein XXX represents hydrophobic l-amino acids typified by l-Phe (1). This endeavor generated antibody 16G3, which gave rate enhancements on the order of 2 × 104 over the background reaction with pleasingly high turnover rates (≈2 min−1). Our study of the kinetics implicated a sequential mechanism with no preferred order of substrate binding and no evidence for acylation of the antibody by the ester before peptide bond formation. Antibody 16G3 did not, however, catalyze either the hydrolysis or racemization of the active ester. The generation of antibodies that catalyze nonsolvolytic bimolecular bond formation is a greater challenge than the production of antibodies designed to catalyze solvolysis because an important requirement for the former is to prevent hydrolysis. The acylating agents employed in these coupling reactions were the p-nitrophenyl esters of acetyl-XXX. Although the hapten had been designed to generate antibodies that can catalyze the formation only of dipeptides, we subsequently found that antibody 16G3 also catalyzes the coupling of the p-nitrophenyl ester of N-acetyl-l-Phe with d-Trp-Gly⋅NH2 and the p-nitrophenyl ester of N-acetyl-Gly-l-Phe with d-Trp-Gly⋅NH2 to afford the corresponding tri- and tetrapeptides, respectively (2). Further, hapten 1a was designed so that 16G3 would catalyze the coupling of the activated ester only with the α-amino group of the nucleophile. Indeed, 16G3 catalyzed the coupling of the p-nitrophenyl ester of N-acetyl-l-Phe with the bisnucleophilic d-Trp-α,β-diaminopropionic acid amide only at the α-amino group, whereas the uncatalyzed reaction provided the α- and β-coupled products in equal amounts (2). This selectivity holds the promise that ɛ-amino groups of lysine will not have to be protected and lends credence to the rationale of the hapten design. As required by the design, the hapten inhibited catalysis at concentrations equal to twice that of the antibody. Initially, we interpreted the failure of 16G3 to catalyze the coupling of the p-nitrophenyl ester of N-acetyl-Phe with d-Trp-l-Lys⋅NH2 to mean that the hapten sculpted a pocket that could accommodate d-Trp-α,β-diaminopropionic acid amide but not the larger d-Trp-l-Lys⋅NH2. Recently, molecular modeling has suggested a different interpretation of these results (see below).
Figure 1.

Structures of phosphonyl derivatives 1a,b and 2a,b.
At the outset, we had sought unsuccessfully to prepare hapten 1b (Fig. 1). In the course of the unsuccessful attempts to prepare 1b and the successful synthesis of 1a, we discovered a novel class of phosphonylating agents typified by the triethylammonium salt 2b, which proved superior to the phosphonyl chloride 2a (Fig. 1) for the preparation of both phosphonates and phosphonamides (3). Nevertheless, 2b, like its phosphonyl chloride counterpart (2a), failed to generate 1b, presumably because of a combination of electronic and steric factors (3). In an important paper, Greenhalg (4) showed that the more positively charged the phosphorus atom of the phosphonylating agent, the more phosphonate ester formation is favored over phosphonamide formation. Subsequently, we showed (3) that steric factors are also important in this regard. Thus, steric crowding in the attempted coupling of 2b with d-Trp⋅NH2 prevents formation of the P—N bond of 1b whereas the stronger P—O bond of 1a provides the energy required to overcome the energy necessary for both the charge transfer and the steric crowding in the transition state. These considerations now lead us to question whether 1b can in fact exist and to suspect that 1b would collapse to generate a phosphinate (4).
During our synthesis of 1a, we also devised a versatile protocol for the construction of highly enantioenriched α-amino phosphonate diesters (5, 6). Addition of lithium diethyl phosphite to the chiral chelating imine derived from cyclohexanecarboxaldehyde and the chiral auxiliary (R)-(–)-1-amino-1-phenyl-2-methoxyethane (3) after hydrogenolysis generated the α-amino phosphonate diester (i.e., 7) required for the synthesis of 1a (Scheme S1). Thus, catalytic antibody research can also contribute to synthetic organic chemistry and enzymology.
Scheme 1.

Synthesis of highly enantioenriched α-amino phosphonate diesters.
Unexpectedly, 16G3 revealed no strong substrate preferences vis-à-vis the stereogenicity at the α centers.¶ These findings were rationalized (2) by the fact that the antibody was programmed to provide binding pockets for the nitroaryl group, the indole ring, and a variety of hydrophobic α-carbon side chains of the reactants. The sterically smaller C-terminal carboxamide, the N-terminal acetamide, and the α-protons are likely to be readily accommodated independent of their relative configurations, explaining why the catalysis is not substrate-selective (Fig. 2).
Figure 2.

Depiction of the sculpting of three binding pockets by the hapten (Left) and the fit of the N-terminal Phe, the C-terminal Trp, and the p-nitrophenyl ester activating group, irrespective of stereochemistry (Right).
The Synthesis of Enantiomerically Pure Hexapeptide Active Esters and Authentic Cyclic Specimens.
Given the general access of antibody–antigen complexes to external reagents, we reasoned that 16G3 might also catalyze an intramolecular reaction, if a Phe p-nitrophenyl ester electrophile and a Trp nucleophile were at the respective C- and N-terminal ends of a linear peptide. We envisioned that the chain connecting the donor/acceptor termini would loop outside the active site (Fig. 3).
Figure 3.

Cartoon depicting the proposed tetrahedral intermediate for the 16G3 catalyzed cyclization of a linear hexapeptide. Only the diastereomer containing l-Phe and d-Trp will bind pockets B and A optimally; stereochemical integrity is retained in all cases. The remaining four amino acids are presumed to reside largely outside the binding pocket.
We predicted that, if 16G3 can catalyze cyclizations, the yield of such cyclizations should be highest for that substrate in which the stereochemistry at the C and N termini corresponds to that of the hapten. Furthermore, these cyclizations might be more substrate selective because of the steric constraints imposed by having all binding epitopes in a single molecule. Herein, we report the testing of these surmises.
Our initial target for cyclization, d-Trp-Gly-Phe-Pro-Gly-Phe⋅p-nitrophenyl ester, proved too insoluble in the pH 7.0 Mops buffer (2). We therefore constructed the more soluble analog 8a in which Phe3 is replaced by 3-pyridylalanine (Pal) (Scheme S2). We also prepared the three isomers diastereomeric at the C and N termini (8b–d). Conversion of the diastereomerically pure N-Boc protected hexapeptide free acids to their p-nitrophenyl esters by treatment with p-nitrophenol in the presence of N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and 4-dimethylaminopyridine (DMAP) (the method that we had successfully employed in the preparation of enantiomerically pure Ac-Gly-l-Phe⋅p-nitrophenyl ester) was accompanied by epimerization at the C terminus.‖ However, the “backing-off” strategy, first described by Goodman (7), afforded the desired diastereomerically pure hexapeptide active esters via coupling of a mixed anhydride of the C-terminal glycine pentapeptide 9a** with the p-nitrophenyl esters of l- and d-Phe (10a and 10b, respectively) (Scheme S2).‡‡ Authentic samples of the diastereomerically pure c-hexapeptides c-(d-Trp-Gly-Pal-Pro-Gly-Phe) (11a) and the d-Phe diastereomer (11b) were prepared from the corresponding linear hexapeptide free acids by using our previously reported protocol [i.e., diphenyl phosphoryl azide (DPPA), NaHCO3, N,N-dimethylformamide (DMF) with vigorous exclusion of moisture] (8).
Scheme 2.

Synthesis of hexapeptide active esters 8a–d.
16G3 Catalyzed Cyclizations.
The results summarized in Table 1 demonstrate that, as expected, antibody 16G3 displayed a marked preference for diastereomer 8a, in which the C- and N-terminal amino acids have the l- and d-configurations, respectively. At an assay time of 6 min, essentially all of 8a was converted to the cyclic peptide 11a. In absolute rate terms, the turnover of 16G3 with 8a (assuming saturation of 16G3 at 10 μM) is estimated at 2 min−1,†† so that 2 μM of cyclic product is formed per 1 μM of antibody active sites per minute. This relatively high velocity is attributable to the intrinsic reactivity of the ester substrate. The marked improvement in the yield of the desired product is attributable to the fact that the catalytic antibody acts as a template to channel the activated linear peptide ester into formation of the desired cyclic product, without catalyzing background epimerization or hydrolysis. Thus, the modest increase in the rate of cyclization (≈22-fold) is sufficient to form the cyclic product in >90% yield. As expected, the catalytic effects of 16G3 are completely blocked by the hapten. Although we were able to predict that rate enhancement should be optimal for 8a, the effect of 16G3 on the rate enhancement of the remaining three diastereomers was more difficult to predict. Modeling studies (see below) showed that the antibody cannot optimally accommodate the side chains without generating trajectories for the termini, which are less than optimal. As shown in Table 1, the l-Trp, d-Phe isomer (8d) gave a rate enhancement of 5.7, whereas the effects on 8b and 8c were negligible, indicating that, for these latter compounds, there is no acceptable compromise between binding of the side chains and the trajectories of the amino groups and the active esters. A different binding mode must, however, also be considered wherein the Trp and Phe side chains occupy each others pockets. As shown below, this interchange does not occur. To this end, we synthesized two additional linear hexapeptides in which the optimal stereochemistry is retained at both the C and N termini, and in which either Trp or Phe is present at both ends (i.e., 12 and 13, respectively). As shown in Table 1, the d-Trp, l-Trp activated ester revealed a rate enhancement comparable to that of 8d whereas the d-Phe, l-Phe activated ester showed no rate enhancement in the presence of 16G3. The enantiomer 8d is likewise cyclized to the corresponding cyclic peptide (11d) at a rate of ≈5.7-fold over the spontaneous rate, which is sufficient to provide 11d as the major product (>90%). Again, the catalysis proceeds without epimerization. For the two additional diastereomeric hexapeptides d-Trp, d-Phe (8b) and l-Trp, l-Phe (8c), the rate of antibody-catalyzed reaction is only slightly above background (1.4- and 1.7-fold, respectively); these substrates are thus either weakly bound or are not bound in a reactive conformation for ring closure. We point out that the lack of selectivity in substrate recognition is not a shortcoming, because cyclization does not generate a new stereogenic center. Stereochemical integrity of the cyclic peptide product derives from the linear active ester and from the fact that the antibody does not catalyze epimerization; only cyclization occurs. Indeed, the lack of selectivity enhances the scope of the reaction. To be sure, exquisite selectivity is one of the characteristics of enzymes, although this is not always the case even for enzymes (10).
Table 1.
Rate enhancements for 16G3 catalyzed cyclizations

| Hexapeptide | Xaa | Yaa | Rate enhancement |
|---|---|---|---|
| 8a | d-Trp | l-Phe | 22 |
| 8d | l-Trp | d-Phe | 5.7 |
| 8c | l-Trp | l-Phe | 1.7 |
| 8b | d-Trp | d-Phe | 1.4 |
| 12 | d-Trp | l-Trp | 5.9 |
| 13 | d-Phe | l-Phe | 1.0 |
Molecular Modeling.
To investigate the structural basis for 16G3 catalysis in more detail, a model of the variable domain of 16G3 was constructed, using the antibody structural database (11) as a guide (see the supplemental data on the PNAS web site, www.pnas.org). Because of strong structural conservation, the antibody variable domain may be the most accurate to model of all proteins. Of the six loops [called complementarity determining regions (CDRs)] forming the antigen-binding site, five adopt specific conformations depending on their sequence (12). Although the third CDR of the heavy chain, CDR H3, shows substantial variation in length and structure among antibodies, the structure at the N- and C-terminal regions of CDR H3 is usually well defined (13, 14), making only the top of the loop difficult to predict. High-resolution antibody structures having high sequence identity to 16G3 were used as the structural templates to build the 16G3 variable domain. Only the top of the 16G3 CDR H3 loop lacked a template; this loop was constructed by computer graphics.
The hapten and substrate were docked into the 16G3 antibody model. A model of the hapten built with computer graphics was energy minimized to relax bond and valence angles, and was fit into the binding site of 16G3 while adjusting flexible torsion angles. The final complex (Fig. 4) was obtained after energy minimization. The p-nitrobenzyl group lies in a well defined pocket at the bottom of the antigen-binding site (Fig. 5), close to the position found for the p-nitrobenzyl group in the crystallographic structure of the catalytic antibody 28B4 complexed with hapten (15). This places the phosphonate group at the bottom of the antigen-binding pocket, in a position similar to that found for related haptens bound to esterolytic antibodies (16). The hexapeptide substrate was then built and docked in the antibody by simultaneous superposition of the d-Trp indole ring and p-nitrophenyl group onto the corresponding positions of the docked hapten. Energy minimization provided the final model of 16G3 with docked hexapeptide substrate (see Fig. 7).
Figure 4.
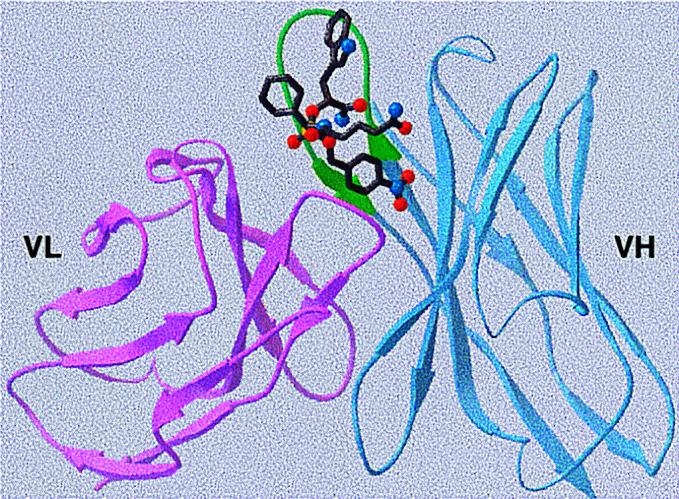
Model of the variable domain of 16G3 constructed from the antibody structural database with bound hapten.
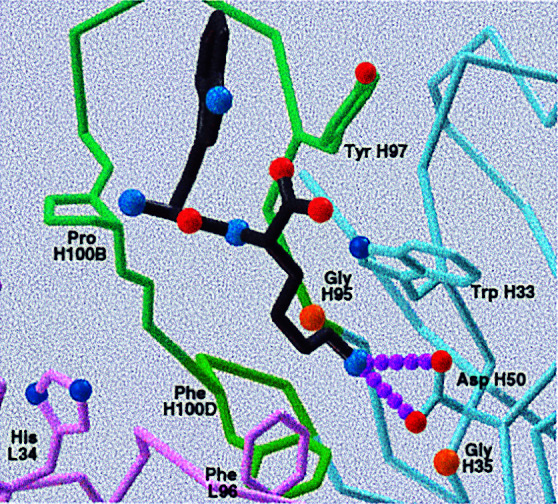
Figure 5.

Model of 16G3 with bound hapten showing the three sculpted binding pockets (Left) and key residues (Right). His L34 serves as a H-bond donor for the P O of the hapten and for the oxygen anion of the transition state. Gly H95 and Gly H35 allow easy access for the p-nitrophenyl ester group of the electrophile to its binding pocket. Pro H100B and a Tyr (not shown) sandwich the cyclohexyl group of the hapten. The binding site for the Trp of the nucleophile is not well defined but includes Tyr H97.
Figure 7.

Model of 16G3 with d-Trp-Gly-Phe-Pro-Gly-Phe·p-nitrophenyl ester (8a) showing binding pockets (Left) and key contacts (Right). The amino terminus (dark blue sphere) is about 3.5 Å from the electrophilic carbon of the ester (yellow sphere), generating an optimal trajectory for ring closure.
Molecular modeling (Fig. 5) reveals the p-nitrobenzyl group of the hapten to be deeply embedded in the antibody 16G3, in a pocket that serves as the anchor for binding of both the hapten and the electrophiles. Not unexpectedly, modeling also suggests (see Fig. 7) that the active esters of the acylating agents do not penetrate the pocket as deeply as the hapten. The modeling studies thus provide support for our surmise that the pleasingly favorable turnover numbers and low product inhibition observed in the 16G3-catalyzed formation of dipeptides may be the result of the fact that the p-nitrophenyl group is bound tightly by the pocket sculpted by the p-nitrobenzyl ester of the hapten but, because it lacks a methylene group, not so tightly to lead to extensive product inhibition.§§ Thus, as we suggested earlier (2), reasonable binding of the substrates is required for good catalysis; a perfect fit is likely to cause unwanted product inhibition. That we achieved a favorable balance is the result partly of design and partly of serendipity. The design of the hapten differed from the conventional protocol, which seeks to maximize the overlap of the hapten with the transition state of the reactants. As we pointed out (2), the cyclohexyl side chain in 1a does not match perfectly the side chains of any of the electrophiles employed by us so far. This tactic should further reduce product inhibition. In addition, we believe that we were fortunate in that the hapten was designed to generate a pocket to accommodate the p-nitrobenzyl esters of the electrophile (2). These esters proved to be insufficiently reactive to result in 16G3-induced catalysis. Switching from p-nitrobenzyl to p-nitrophenyl esters served to overcome the reactivity problem and, as discussed above, also reduced product inhibition.
In this context, the results reported at about the same time by Jacobsen and Schultz are of interest (18). They sought to catalyze dipeptide formation using antibody 9B5.1 and benzyl esters of amino acids as electrophiles. The Ab was sculpted by a hapten containing a benzyl ester. As in the case of our p-nitrobenzyl esters, no catalysis was observed. Jacobsen and Schultz therefore turned to the more reactive azides as the acylating agents, just as we turned to p-nitrophenyl esters. Not unexpectedly, the binding affinity of the azides for the pocket sculpted by the benzyl esters was poor (Km = 15 μM), leading to poor catalysis (efficiency of 102 rather than the reported 104) (18). Thus, we believe that haptens must be designed to generate binding pockets that strike a balance between very tight binding and weak binding of the transition state, because excessive binding will induce poor turnover numbers, and very weak binding leads to minimal catalysis.
The 16G3 model suggests a second reason why the product p-nitrophenolate anion does not inhibit catalysis. Asp H50 lines one side of the p-nitrophenyl binding pocket (Fig. 5). Electrostatic interactions between the Asp H50 carboxylate and p-nitrophenolate may help eject the product from the antibody. The presence of Asp H50 also provides a possible explanation for the failure of the coupling of the p-nitrophenyl ester of N-acetyl-Phe with d-Trp-l-Lys·NH2 (see above). With the Trp indole ring positioned as in the bound substrate, the Lys side chain of d-Trp-l-Lys·NH2, but not the amino containing side chain of d-Trp-α,β-diaminopropionic acid, is long enough to reach Asp H50. This results in the formation of a salt bridge that causes d-Trp-l-Lys⋅NH2 to fill partially the p-nitrobenzyl binding pocket, thereby preventing binding of the p-nitrophenyl ester of N-acetyl-Phe (Fig. 6). Thus, our initial interpretation (see above) that the larger, lysine-containing bisnucleophile is not tolerated by 16G3 is probably not valid.
Figure 6.
Model of 16G3 with d-Trp-l-Lys·NH2 showing the hypothetical salt bridge between the ε-amino group of the Lys side chain and Asp H50. The dislodged electrophile can no longer couple with the nucleophile.
It is noteworthy that 16G3 catalyzed the cyclization of 12 but failed to catalyze the cyclization of 13 (Table 1). The data suggest that l-Trp is accommodated by the pocket sculpted for the smaller l-Phe, but d-Phe is not bound by the pocket sculpted for the larger d-Trp. The model shows the d-Trp pocket to be very shallow (Fig. 7), and this would explain, therefore, why it does not bind the smaller d-Phe whereas the smaller pocket sculpted for l-Phe is sufficiently plastic to accommodate l-Trp. In other words, the d-Trp pocket is too large to allow tight binding of d-Phe. These results are consistent with our initial design because a binding pocket was sculpted specifically for the Trp side chain and another that could accommodate a variety of hydrophobic amino acid side chains. Fig. 7 provides an additional rationalization for this result. If Tyr H97 comprises the binding site for the d-Trp of 12, the d-Phe of 13 may actually be too small to make contact with Tyr H97. As mentioned above, exchange of pockets does not occur. This illustrates that the fields of medicinal chemistry and catalytic antibody research can complement each other.
Discussion.
The results described herein have shown that antibodies programmed to bind specific side chains can catalyze the synthesis of small cyclic peptides under mild conditions. This represents another interesting difference between enzymes and catalytic antibodies. For example, enzyme-derived subtiligases can effect cyclization only if the linear peptide precursor contains more than 12 amino acids (19), whereas 16G3 can cyclize hexapeptides. It is also interesting that proteases similarly do not cleave small cyclic peptides, such as c-hexapeptides (cf. MK-678) (20).
Based on our experience with bimolecular coupling catalysis, ɛ-amino side chain protection will not be required. We expected that the size and components of the linear peptide should not be a limitation for antibody catalysis because antibodies can be tailor-made to recognize those particular side chains that are involved in the ring closure, and, apparently, the remaining peptide chain lies off the antibody surface during cyclization. The scope of such cyclization reactions remains to be determined experimentally.
Finally, we never intended our catalytic antibodies to equal enzymes in specificity or extent of catalysis. Nature's enzymes evolved over a very long period of time. Thus, comparing the relative rate enhancements of enzymes and of catalytic antibodies is not a particularly fruitful endeavor. We believe, however, that studies such as those reported herein, in our earlier papers, and by others (for comprehensive reviews, see refs. 21–24) have contributed in a small way to defining the factors that influence the interaction of small molecules with proteins. In this sense, such research has relevance to medicinal chemistry. In addition, work that we described previously has generated novel phosphonate chemistry, a subject matter of considerable importance in its own right. That catalytic antibody research, first conceived by Jencks (25) and pioneered by Lerner and colleagues (26) and by Schultz and colleagues (27), has achieved some degree of success is, we believe, quite remarkable. That Benkovic could explain the experimental results on the basis of the principles of enzymology is highly significant. From these perspectives, it is less noteworthy that enzymes do it all much better.
Supplementary Material
Acknowledgments
We acknowledge with pleasure Chu-Young Kim for the preparation and purification of the Fab fragments used in these studies and the intellectual contributions of Paul A. Sprengeler and Joseph Barbosa, including the generation of Figs. 2 and 3. R.H. thanks Leo Benoiton for very helpful discussions. We are pleased to acknowledge financial support provided by the National Institutes of Health (Institute of General Medical Sciences through Grants GM-45611 and GM-48877) and the Merck and DuPont Research Laboratories. S.I. also wishes to thank the Japan Society for the Promotion of Science (JSPS) for financial support.
Abbreviation
- CDR
complementarity determining region
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on April 27, 1999
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040534397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040534397
The S-configuration at the phosphonate center of the hapten was assigned based on an x-ray crystal structure of the p-bromobenzoyl derivative.
We attribute the difference to the lower solubility of 8a and 8b compared with that of Ac-Gly-Phe. At lower concentrations, the p-nitrophenylation of the latter, too, afforded the racemized active ester, presumably because at lower concentrations the rate of racemization competes effectively with the slower rate of the coupling reaction.
It should be appreciated that the success of the Goodman procedure does not depend on the presence of the achiral Gly5 because methods exist that permit the coupling of peptides with little or no racemization. The p-nitrophenyl esters of N-acylated amino acids are not in this category.
The mixture of the labile diastereomeric active esters could not be separated, but the pure chemical entities 8a and 8b could be easily distinguished by NMR. The chemical shifts of the Pro α-protons are 4.42 and 4.35 ppm for the l-Phe and d-Phe active esters, respectively.
This is typical of more reactive catalytic antibodies. For comparison, the antibody catalyzing a cationic cyclization to a six-membered ring has a turnover number of 0.01 min−1 (9).
Tawfik et al. (17) subsequently reported p-nitrophenyl ester hydrolytic antibodies elicited by a p-nitrobenzyl phosphonate hapten.
References
- 1.Hirschmann R, Smith A B, III, Taylor C M, Benkovic P A, Taylor S D, Yager K M, Sprengeler P A, Benkovic S J. Science. 1994;265:234–237. doi: 10.1126/science.8023141. [DOI] [PubMed] [Google Scholar]
- 2.Smithrud D B, Benkovic P A, Benkovic S J, Taylor C M, Yager K M, Witherington J, Philips B W, Sprengeler P A, Smith A B, III, Hirschmann R. J Am Chem Soc. 1997;119:278–282. [Google Scholar]
- 3.Hirschmann R, Yager K M, Taylor C M, Witherington J, Sprengeler P A, Phillips B W, Moore W, Smith A B., III J Am Chem Soc. 1997;119:8177–8190. [Google Scholar]
- 4.Greenhalgh R, Heggie R M, Weinberger M A. Can J Chem. 1970;48:1351–1357. [Google Scholar]
- 5.Smith A B, III, Yager K M, Taylor C M. J Am Chem Soc. 1995;117:10879–10888. [Google Scholar]
- 6.Smith A B, III, Yager K M, Phillips B W, Taylor C M. Org Synth. 1997;75:19–30. [Google Scholar]
- 7.Goodman M, Stueben K C. J Am Chem Soc. 1959;81:3980–3983. [Google Scholar]
- 8.Hirschmann R, Yao W, Cascieri M A, Strader C D, Maechler L, Cichy-Knight M A, Hynes J, Jr, van Rijn R D, Sprengeler P A, Smith A B., III J Med Chem. 1996;39:2441–2448. doi: 10.1021/jm960281e. [DOI] [PubMed] [Google Scholar]
- 9.Li T, Janda K D, Lerner R A. Nature (London) 1996;379:326–327. doi: 10.1038/379326a0. [DOI] [PubMed] [Google Scholar]
- 10.Horecker B L, Tsolas O, Lai C V. In: The Enzymes. Boyer P, editor. Vol. 7. New York: Academic; 1972. , ch. 6. [Google Scholar]
- 11.Roberts V A, Stewart J, Benkovic S J, Getzoff E D. J Mol Biol. 1994;235:1098–1116. doi: 10.1006/jmbi.1994.1060. [DOI] [PubMed] [Google Scholar]
- 12.Chothia C, Lesk A M. J Mol Biol. 1987;196:901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 13.Shirai H, Kidera A, Nakamura H. FEBS Lett. 1996;399:1–8. doi: 10.1016/s0014-5793(96)01252-5. [DOI] [PubMed] [Google Scholar]
- 14.Morea V, Tramontano A, Rustici M, Chothia C, Lesk A M. J Mol Biol. 1998;275:269–294. doi: 10.1006/jmbi.1997.1442. [DOI] [PubMed] [Google Scholar]
- 15.Hsieh-Wilson L C, Schultz P G, Stevens R C. Proc Natl Acad Sci USA. 1996;93:5363–5367. doi: 10.1073/pnas.93.11.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thayer M M, Olender E H, Arvai A S, Koike C K, Canestrelli I L, Stewart J D, Benkovic S J, Getzoff E D, Roberts V A. J Mol Biol. 1999;291:329–345. doi: 10.1006/jmbi.1999.2960. [DOI] [PubMed] [Google Scholar]
- 17.Tawfik D S, Lindner A B, Chap R, Eshhar Z, Green B S. Eur J Biochem. 1997;244:619–626. doi: 10.1111/j.1432-1033.1997.00619.x. [DOI] [PubMed] [Google Scholar]
- 18.Jacobsen J R, Schultz P G. Proc Natl Acad Sci USA. 1994;91:5888–5892. doi: 10.1073/pnas.91.13.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson D Y, Burnier J P, Wells J A. J Am Chem Soc. 1995;117:819–820. [Google Scholar]
- 20.Veber D F, Saperstein R, Nutt R F, Freidinger R M, Brady S F, Curley P, Perlow D S, Paleveda W J, Colton C D, Zacchei A G, et al. Life Sci. 1984;34:1371–1378. doi: 10.1016/0024-3205(84)90009-2. [DOI] [PubMed] [Google Scholar]
- 21.Lerner R A, Benkovic S J, Schultz P G. Science. 1991;252:659–667. doi: 10.1126/science.2024118. [DOI] [PubMed] [Google Scholar]
- 22.Benkovic S J. Annu Rev Biochem. 1992;61:29–54. doi: 10.1146/annurev.bi.61.070192.000333. [DOI] [PubMed] [Google Scholar]
- 23.Schultz P G, Lerner R A. Acc Chem Res. 1993;26:391–395. [Google Scholar]
- 24.MacBeath G, Hilvert D. Chem Biol. 1996;3:433–445. doi: 10.1016/s1074-5521(96)90091-5. [DOI] [PubMed] [Google Scholar]
- 25.Jencks W P. Catalysis in Chemistry and Enzymology. New York: McGraw–Hill; 1969. [Google Scholar]
- 26.Tramontano A, Janda K D, Lerner R A. Science. 1986;234:1566–1570. doi: 10.1126/science.3787261. [DOI] [PubMed] [Google Scholar]
- 27.Pollack S J, Jacobs J W, Schultz P G. Science. 1986;234:1570–1573. doi: 10.1126/science.3787262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.