

Abstract
Metformin lowers blood glucose levels in type 2 diabetic patients. To evaluate the insulin sensitizing action of metformin on skeletal muscle cells, we have used C2C12 skeletal muscle cells differentiated in chronic presence or absence of insulin.
Metformin was added during the last 24 h of differentiation of the C2C12 myotubes. Insulin-stimulated tyrosine phosphorylation of insulin receptor (IR) and insulin receptor substrate-1 (IRS-1) was determined.
Chronic insulin treatment resulted in 60 and 40% reduction in insulin-stimulated tyrosine phosphorylation of IR and IRS-1, respectively. Treatment with metformin was able to increase the tyrosine phosphorylation of IR and IRS-1 by 100 and 90% respectively.
Chronic insulin treatment drastically reduced (45%) insulin-stimulated phosphatidyl inositol 3-kinase (PI 3-kinase) activity. Metformin treatment restored PI 3-kinase activity in insulin-resistant myotubes.
Insulin-stimulated glucose uptake was impaired in chronically insulin-treated myotubes. Metformin increased basal glucose uptake to significant levels (P<0.05), but metformin did not increase insulin-stimulated glucose transport.
All the three mitogen-activated protein kinases (MAPK) were activated by insulin in sensitive myotubes. The activation of p38 MAPK was impaired in resistant myotubes, while ERK and JNK were unaffected. Treatment with metformin enhanced the basal activation levels of p38 in both sensitive and resistant myotubes, but insulin did not further stimulate p38 activation in metformin treated cells.
Treatment of cells with p38 inhibitor, SB203580, blocked insulin- and metformin-stimulated glucose uptake as well as p38 activation.
Since the effect of metformin on glucose uptake corresponded to p38 MAPK activation, this suggests the potential role p38 in glucose uptake.
These data demonstrate the direct insulin sensitizing action of metformin on skeletal muscle cells.
Keywords: Insulin resistance, skeletal muscle, insulin receptor, insulin receptor substrate-1, phosphatidyl inositol 3-kinase, glucose uptake, metformin, p38 MAPK
Introduction
Metformin, a biguanide, reduces hyperglycaemia by a number of mechanisms, including inhibition of hepatic output and stimulation of glucose uptake in a variety of tissues, such as skeletal muscle (Wiernsperger & Bailey, 1999). The precise mechanism of action of metformin is controversial and whether it can act directly on skeletal muscle is still unclear. Given the fact that skeletal muscle accounts for more than 80% of insulin-stimulated glucose uptake, the impaired glucose uptake in skeletal muscle due to insulin resistance is responsible for the development of type 2 diabetes.
The mechanism by which metformin stimulate glucose uptake is poorly understood. At the cellular level, it has been shown in L6 skeletal muscle cells that metformin action involves the translocation of glucose transporters from an intracellular pool to the plasma membrane (Hundal et al., 1992). Hyperinsulinaemia in cell culture models has been shown to develop insulin resistance (Pryor et al., 2000; Ricort et al., 1995; Thomson et al., 1997). Insulin-stimulated tyrosine phosphorylation of insulin receptor (IR) and insulin receptor substrate-1 (IRS-1), activation of phosphatidyl inositol 3-kinase (PI 3-kinase) and glucose uptake is impaired by chronic treatment with insulin (Pryor et al., 2000; Ricort et al., 1995; Thomson et al., 1997). It has been reported that metformin can alleviate the defects in insulin signalling caused by chronic insulin treatment in rat adipocytes (Pryor et al., 2000).
PI 3-kinase pathway (Farsee, 2001) and more recently p38 activation has been implicated in glucose uptake (Somwar et al., 2000; 2001). The present study was undertaken to determine whether metformin could directly act on insulin resistant skeletal muscle to improve insulin-signalling pathway. The effect of metformin was also studied on all three mitogen-activated protein kinases (MAP kinases).
Methods
Cell culture and treatment
C2C12 cells were cultured in DMEM supplemented with 15% foetal calf serum and antibiotics (Penicillin 100 IU ml−1, Streptomycin 100 μg ml−1) in 5% CO2 at 37°C. The cells were differentiated in an equal mixture of two serum free media (MCDB 201 and Ham's F-12 medium) along with 0.05% BSA in the absence (MF) or chronic presence of 100 nm insulin (MFI) for 3 days. The media was changed after every 12 h. Metformin was added during last 24 h of differentiation where indicated. Characteristics of the differentiated myotubes were checked by testing the expression of various muscle markers, like myosin, myogenin, myo D, creatine kinase enzyme activity and were found to be correct.
Preparation of extracts of C2C12 muscle cells for immunoblotting and immunoprecipitation
Media in the differentiated cells was changed 1 h before the start of experiment. The cells were washed twice with the Krebs-Ringer phosphate buffer (KRP) (mM): phosphate (pH 7.2) 10, NaCl 136, KCl 4.7, CaCl2 1.25, MgSO4 1.25, containing 5 mM glucose and 0.05% BSA. The cells were further incubated twice with KRP buffer at 37°C for 30 min. After that the cells were stimulated with 100 nm of insulin for 5 min at 37°C or left unstimulated. The cells were washed with ice-cold phosphate buffered saline (PBS) and lysed in lysis buffer (mM: HEPES 50, pH 7.4, NaCl 150, MgCl2 1.5, EGTA 1, sodium pyrophosphate 10, sodium fluoride 50, β-glycerophosphate 50, Na3VO4 1, 1% Triton X-100, phenylmethylsulphonyl fluoride 2, 10 μg ml−1 each of leupeptin, aprotinin and soyabean trypsin inhibitor. Lysis was carried out at 4°C for 30 min. Cell lysate were clarified at 16,000×g at 4°C for 15 min. 500 μg of protein was immunoprecipitated with antibody against either anti-IR-β or anti-IRS-1 with the addition of Protein A-agarose. The cell lysates or immunoprecipitates were boiled with Laemmli sample buffer (Laemmli, 1970) for 5 min, resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS–PAGE) under reducing conditions, transferred to PVDF membranes. The membranes were blocked and incubated with the indicated antibodies, followed by incubation with HRP-conjugated secondary antibodies. The bands were visualized with enhanced chemiluminescence. Blots were stripped in stripping buffer (62.5 mM Tris-HCl, pH 6.7, 2% SDS and 100 mM β-mercaptoethanol) at 50°C for 30 min and reprobed with either antibodies against IR-β or IRS-1.
PI 3-kinase assay
To study PI 3-kinase activity associated with IRS-1, 1 mg of cell lysate was used. Cell lysates were prepared after 10 min of 100 nM insulin stimulation. Immunoprecipitates were washed twice with lysis buffer, and twice with (mM): Tris-HCl, pH 7.5 10, NaCl 100, EDTA 1 and 100 μM Na3VO4. Immunoprecipitates were resuspended in 50 μl of 20 mM HEPES, pH 7.5, 180 mM NaCl followed by addition of 25 μl of assay buffer (mM) HEPES 28, pH 7.5, NaCl 50, 0.15% Nonidet P40, MgCl2 12.5, EGTA, 0.8 mg ml−1 L-α-phosphatidylinositol, [γ-32P]-ATP 0.4 (10 μCi per assay). Reactions were terminated after 15 min at 30°C by the addition of 50 μl of 2 M HCl followed by 160 μl of chloroform, vortexed and centrifuged briefly. 60 μl of lower phase was applied to TLC plates. TLC plates were developed in CHCl3 : CH3OH : NH4OH : H2O (60 : 47 : 11 : 5 by volume), dried, and visualized by autoradiography.
2-deoxyglucose (2-DOG) uptake
After washing with KRP buffer, cells were stimulated with 100 nm insulin in KRP buffer without glucose for 15 min. 2-DOG uptake (0.2 μCi ml−1 in 1 μM of unlabelled 2-deoxyglucose) was added and cells were incubated for 10 min. Cells were washed in ice-cold PBS three times and solubilized in 0.1 N NaOH. Protein concentration was measured in each sample by Bicinchoninic acid (BCA) method (Smith et al., 1985) followed by liquid scintillation counting. The uptake measurement was made in duplicates. The results were corrected for non-specific uptake in the presence of 10 μM of cytochalasin B. Non-specific uptake and absorption were always less than 10% of the total uptake.
Materials
Mouse skeletal muscle cell line, C2C12 was kindly provided by Dr H. Blau, Stanford University, School of Medicine, Stanford, U.S.A. and Dr J. Dhawan, CCMB, India. Nutrient Mixture F-12 Ham, MCDB 201 medium, bovine albumin (cell culture grade) and protein A-agarose were from Sigma. Dulbecco's modified Eagle's medium (DMEM) was purchased from GIBCO BRL, U.S.A. Foetal calf serum (FCS) was purchased from Biological Industries, Israel. Bovine insulin and SB203580 were purchased from Calbiochem, U.S.A. Anti-pp38, anti-pERK and anti-JNK antibodies were from New England Biolabs, U.S.A. Monoclonal anti-phosphotyrosine, anti-IR-β, anti-IRS-1, anti-pJNK, anti-p38, anti-ERK and horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit antibodies were purchased from Santa Cruz Biotechnology, U.S.A. [γ32P]-ATP was purchased from Bhaba Atomic Research Centre, India. 2-deoxy-D-glucose-1-3H and L-α-phosphatidylinositol was purchased from Sigma Chemical Company, U.S.A. TLC plates were from Merck, Germany (gift from Dr K.K. Bhutani, NIPER). Other reagents were obtained from Sigma Chemical Company, U.S.A., Roche Molecular Biochemicals, Germany and Bio-Rad, U.S.A.
Densitometric analysis
Densitometric analysis of the Western blots were done by using a GS-670 Imaging Densitometer (BioRad, U.S.A.) and Molecular Analyst software (version 1.3). The relative values of the samples were determined by giving an arbitrary value of 1.0 to the control samples.
Statistics
The data are expressed as mean±s.e.mean. For comparison of two groups, P-values were calculated by two-tailed unpaired student's t-test. In all cases, P<0.05 was considered to be statistically significant.
Results
Effect of metformin on tyrosine phosphorylation of IR-β
Eight hundred μM concentration of metformin has been shown to enhance glucose uptake in L6 cells (Hundal et al., 1992) and 1 mM concentration has been reported to reverse the defects in insulin signaling due to chronic presence of insulin in rat adipocytes (Pryor et al., 2000). The primary mechanism of action of metformin seems to inhibit the production of glucose from liver, but whether it improves insulin sensitivity in skeletal muscle is still debatable (Moller, 2001). To see whether metformin can enhance the insulin signalling in C2C12 skeletal muscle cells, MF and MFI cells were treated with various concentrations of metformin (100–800 μM) during the last day of differentiation. Immunoprecipitated IR-β samples were immunoblotted against phosphotyrosine. Insulin-stimulated tyrosine phosphorylation of chronically insulin treated cells (MFI) was impaired as compared to control cells (MF) (Figure 1A) and 60% (Figure 1C) reduction in the tyrosine phosphorylation of IR-β was observed in insulin stimulated MFI cells as compared to insulin stimulated MF (control) samples (P<0.001). 400 μM of metformin stimulated tyrosine phosphorylation of IR-β maximally (data not shown). Therefore, 400 μM concentration was used for further experiments. Treatment with 400 μM of metformin resulted in 100% increase in insulin-stimulated tyrosine phosphorylation of IR under the resistant conditions (MFI) as compared to untreated MFI cells stimulated with insulin (P<0.05). Fifty-five per cent increase was observed by insulin stimulation in the tyrosine phosphorylation of IR in MF cells (Figure 1C) when treated with 400 μM of metformin as compared to untreated MF cells stimulated with insulin (P<0.05). Chronic insulin treatment resulted in down-regulation of IR-β expression by 80%, however, metformin did not affect the expression of IR-β (Figure 1B). Inoue et al. (1996) reported that chronic insulin treatment leads to 90% reduction in IR-β expression.
Figure 1.
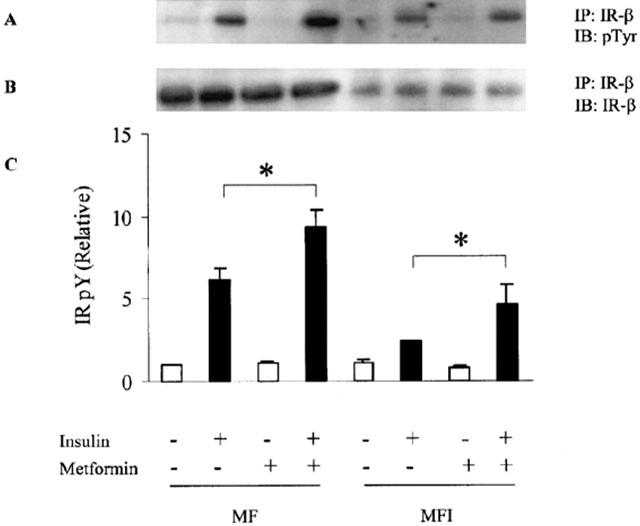
Effect of insulin stimulation and metformin on tyrosine phosphorylation of IR-β in C2C12 myotubes. Metformin (400 μM) was added during the last day of differentiation for 24 h to C2C12 cells differentiated in absence (MF) or in chronic presence of insulin (MFI). Then, the cells were stimulated with 100 nM of insulin for 5 min and lysed. Cell lysate were immunoprecipitated (IP) with antibodies against IR-β and Western immunoblotted (IB) with anti-phosphotyrosine antibody (A). The blots were stripped and reprobed with IR-β (B). Experiments were repeated thrice and representative blots are shown. Phosphorylation levels of IR (C) were quantified by densitometry and expressed relative to MF (control) samples. Error bars represent s.e.m. of three independent experiments (*P<0.05).
Effect of metformin on tyrosine phosphorylation of IRS-1
MF and MFI cells were treated with 400 μM of metformin and then lysed and immunoprecipitated with IRS-1 followed by immunoblotting with phosphotyrosine. Insulin-stimulated IRS-1 phosphorylation was impaired in MFI cells as compared to MF cells (Figure 2A). Chronic treatment of insulin resulted in 40% reduction in insulin stimulated tyrosine phosphorylation of IRS-1 as compared to insulin stimulated MF cells (Figure 2C) (P<0.001). Treatment with metformin restored insulin-stimulated IRS-1 tyrosine phosphorylation and it was increased by 90% in MFI cells (Figure 2C) as compared to untreated MFI cells stimulated with insulin (P<0.05). Metformin treatment resulted in 45% increase in insulin-stimulated tyrosine phosphorylation of IRS-1 in MF cells (Figure 2C) as compared to untreated MF cells stimulated with insulin (P<0.05). Expression of IRS-1 was unaffected with treatment by either chronic presence of insulin or metformin (Figure 2B).
Figure 2.
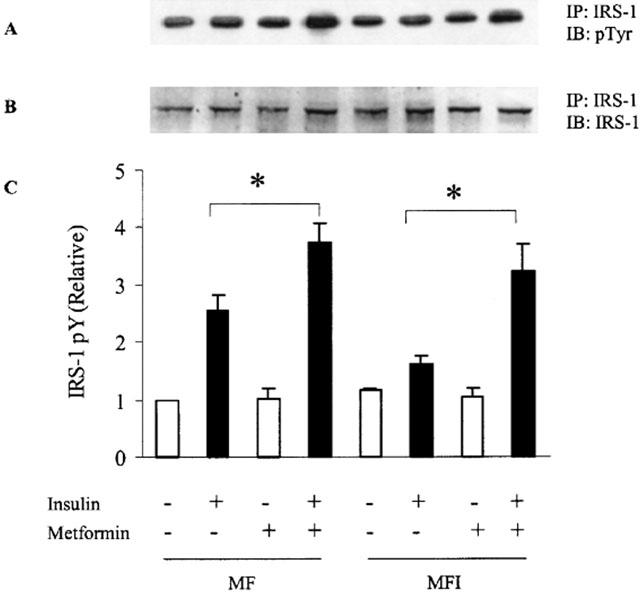
Effect of insulin stimulation and metformin on tyrosine phosphorylation of IRS-1 in C2C12 myotubes. Samples were treated as described in Figure 1. Cell lysate were immunoprecipitated (IP) with antibodies against IRS-1 and Western immunoblotted (IB) with anti-phosphotyrosine antibody (A). The blots were stripped and reprobed with IRS-1 (B). Experiments were repeated thrice and representative blots are shown. Phosphorylation levels of IRS-1 (C) were quantified by densitometry and expressed relative to MF (control) samples. Error bars represent s.e.m. of three independent experiments (*P<0.05).
Effect of metformin on PI 3-kinase activity
PI 3-kinase plays a key role in insulin-stimulated glucose uptake in insulin-responsive tissues (Farsee, 2001) and is down stream to IRS-1. Therefore we determined whether metformin could affect IRS-1 associated PI 3-kinase activity. Insulin-stimulated PI 3-kinase activity was severely reduced in MFI cells as compared to MF cells (Figure 3A). Insulin stimulation resulted in only 30% increase in PI 3-kinase activity in MFI samples and 280% increase in MF samples against the non-insulin stimulated control samples. The treatment of insulin resistant cells with metformin was able to restore insulin-stimulated IRS-1 associated PI 3-kinase activity and 60% increase was observed in MFI cells (Figure 3B) as compared to untreated MFI cells stimulated with insulin.
Figure 3.
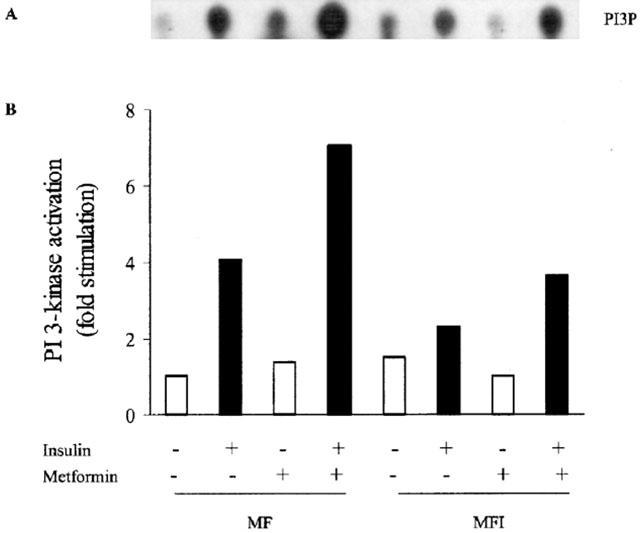
Effect of insulin stimulation and metformin on PI 3-kinase activity in C2C12 myotubes. C2C12 cells, differentiated in MF or MFI, were washed with KRP buffer as described in Methods, followed by stimulation with insulin (100 nM) for 10 min. PI 3-kinase activity in anti-IRS-1 immunoprecipitates was measured as described under Methods. Cells were treated with 400 μM metformin during last 24 h of differentiation. A representative autoradiogram for two independent experiments is shown (A). Relative density against control (MF) of PI3P spots (B) was quantified by densitometry and expressed as an average of two experiments. PI3P refers to PI 3-phosphate.
Effect of metformin on 2-deoxyglucose uptake
Insulin-stimulated glucose transport in skeletal muscle of NIDDM has been shown to be down regulated (Farsee, 2001). So, the effect of chronic presence of insulin on the basal and insulin-stimulated glucose uptake was tested in MF and MFI in the absence or presence of metformin. Insulin stimulated 20% increase in 2-DOG uptake in MF samples as compared MF samples not stimulated with insulin (P<0.05); however, we could not observe any increase in insulin stimulated 2-DOG uptake in the MFI cells (Figure 4). The basal level of glucose uptake in MFI samples were increased as compared to MF samples, but it could not reach statistically significant levels. The treatment with the metformin resulted in enhancement of basal 2-DOG uptake by 18% in both MF and MFI cells (P<0.05) as compared to the basal 2-DOG uptake in untreated MF and MFI cells, respectively. However, there was no significant difference in 2-DOG uptake by insulin stimulation in MF and MFI cells treated with metformin as compared to the basal glucose uptake in metformin treated MF and MFI cells, respectively.
Figure 4.
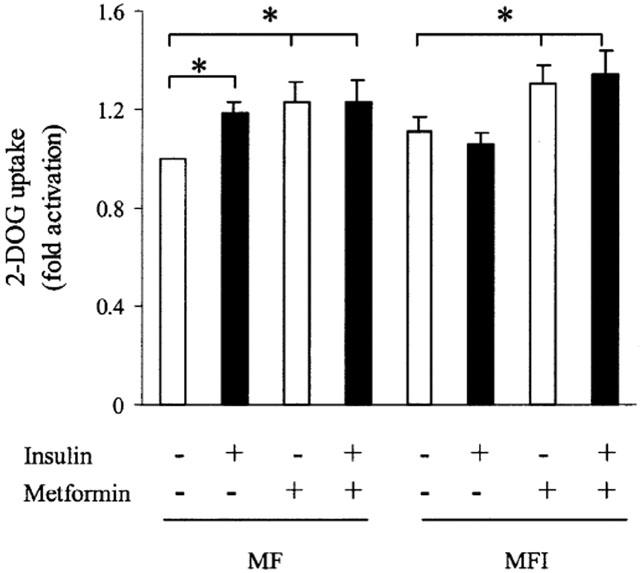
Effect of insulin stimulation and metformin on 2-DOG uptake in C2C12 myotubes. C2C12 cells, differentiated in MF or MFI, were washed with KRP buffer as described in Methods, followed by stimulation with insulin (100 nM) for 15 min. [3H]-2-DOG uptake was measured as described in Methods. Cells were treated with 400 μM of metformin during last 24 h of differentiation. Error bars represent s.e.m. of four independent experiments (*P<0.05).
Effect of metformin on MAPK phosphorylation in insulin-resistant skeletal muscle
Although metformin was able to increase PI 3-kinase activity associated with IRS-1 in MFI cells, we could not observe any increase in the insulin-stimulated glucose uptake in resistant cells on metformin treatment. Recent studies have proposed the potential role of p38 MAPK activation in glucose uptake and stimulation of p38 activity by insulin in skeletal muscle cells (Somwar et al., 2000; 2001). Therefore, we next studied the effect of metformin on p38 activation and related MAP kinases. The results (Figure 5A,C) showed that insulin-stimulation increased ERK activation in both MF and MFI cells. Metformin treatment did not enhance the basal activation of ERK (Figure 5A,C). Similarly, JNK was significantly activated by insulin stimulation in MF and MFI cells (Figure 5D,F). Metformin treatment resulted into increase in JNK activation; however, the increase was not statistically significant (Figure 5D,F). The insulin-stimulated activation of p38 was impaired in MFI cells (Figure 5G,I). Treatment with metformin resulted in increased basal activation of p38 in both MF and MFI cells. However, we could not observe any significant increase in the p38 activation by insulin stimulation in MF and MFI cells treated with metformin. The expression of all the three MAP kinases was unaffected by the chronic presence of insulin or with the treatment of metformin (Figure 5B,E,H).
Figure 5.
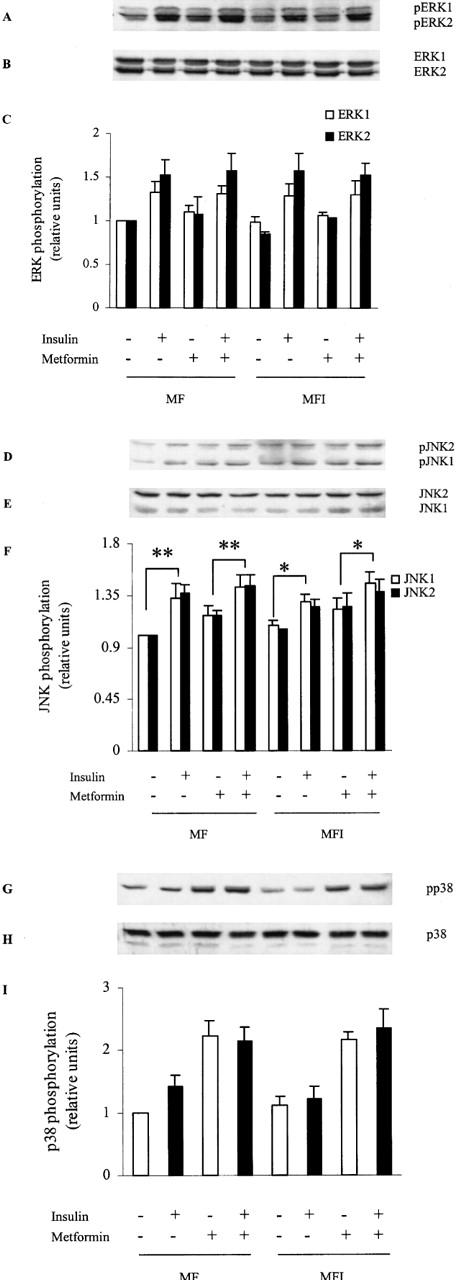
Effect of insulin stimulation and metformin on ERK, JNK and p38 activation: C2C12 cells, differentiated in MF or MFI, were washed with KRP buffer as described in Methods, followed by stimulation with insulin (100 nM) for 5 min. Lysates were prepared from cells treated with 400 μM of metformin during last 24 h of differentiation. Proteins were resolved by SDS–PAGE and immunoblotted either to phospho-specific ERK (A), phospho-specific JNK (D), phospho-specific p38 (G), or ERK (B), JNK (E), p38 (H) antibodies. Phospho-specific blots are representative of experiments performed three times. Error bars of quantified data for pERK (C), pJNK (F) and pp38 (I) represents s.e.m. of three independent experiments (*P<0.05, **P<0.01).
Effect of SB203580 on glucose uptake and p38 activation
SB203580, a specific inhibitor of p38 MAPK, has been shown to reduce insulin-stimulated glucose uptake in L6 myotubes and 3T3-L1 adipocytes in culture (Sweeney et al., 1999). To determine whether activation of p38 can be blocked, cells were treated with 10 μM of SB203580 for 30 min. Results in Figure 6A shows that SB203580 effectively blocked p38 activation by insulin in MF cells. However, during this SB203580 treatment, p38 activation was not blocked due to the metformin treatment in MF and MFI cells. To determine the role of p38 in glucose uptake under the same conditions, glucose transport was determined in the presence of SB203580. The results (Figure 6B) show that there was insignificant increase in insulin-stimulated 2-DOG uptake in MF cells as compared to significant increase (P<0.05) observed in MF cells in the absence of p38 inhibitor (Figure 4). However, basal 2-DOG uptake in metformin treated MF and MFI cells was significant as compared to respective basal glucose uptake in untreated MF or MFI cells even in the presence of p38 inhibitor. These results implicate the potential role of p38 in glucose uptake in skeletal muscle cells.
Figure 6.
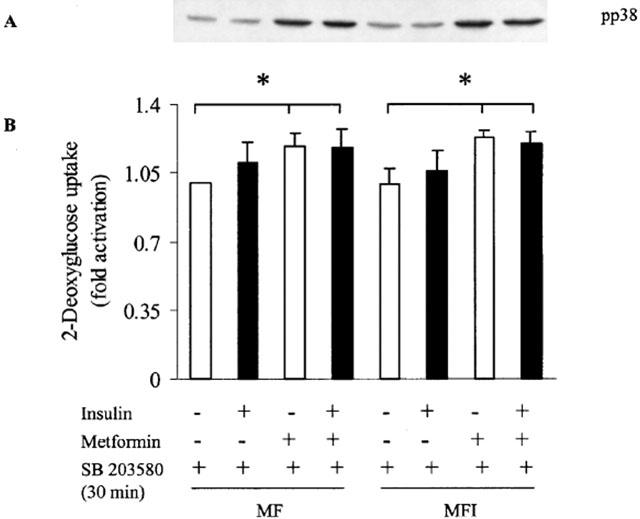
Effect of SB203580 on 2-DOG uptake and p38 activation. C2C12 cells, differentiated in MF or MFI, were incubated in KRP buffer for 30 min followed by another incubation of 30 min in presence of 10 μM of SB203580. Cells were stimulated with 100 nM of insulin in presence of SB203580, followed by (A) immunoblot analysis of p38 activation or (B) 2-DOG uptake for 10 min. Cells were treated with 400 μM of metformin during last 24 h of differentiation. Error bars represent s.e.m. of three independent experiments. A representative blot of pp38 is shown.
To eliminate the possibility that 24 h of metformin pretreatment might have already activated p38, and SB203580, being a competitive inhibitor, could not possibly block the activation of p38, cells were simultaneously treated with metformin and 10 μM of SB203580 during the last 24 h of differentiation. Data shows that the treatment with SB203580 blocked the activation of p38 by metformin in MF as well as MFI cells (Figure 7A). Enhanced basal glucose uptake by metformin treatment observed in MF and MFI cells in absence of SB203580, was also blocked by treatment of SB203580 for 24 h during the addition of drug (Figure 7B). These results strongly suggest the involvement of p38 in glucose uptake.
Figure 7.
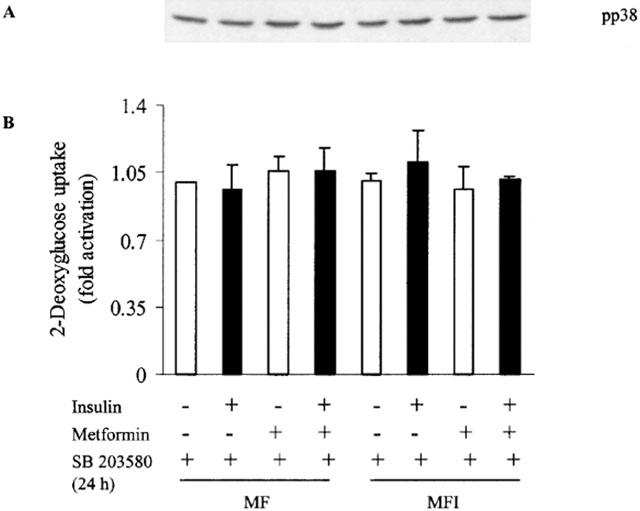
Effect of chronic presence of SB203580 on stimulation of 2-DOG uptake and p38 activation by metformin. C2C12 cells, differentiated in MF or MFI, were incubated in KRP buffer twice for 30 min each in presence of 10 μM of SB203580. Cells were stimulated with 100 nM of insulin in presence of SB203580, followed by (A) immunoblot analysis of p38 activation or (B) 2-DOG uptake for 10 min. Cells were treated with 10 μM of SB203580 with or without 400 μM of metformin during last 24 h of differentiation. Error bars represent s.e.m. of three independent experiments. A representative blot of pp38 is shown from three independent experiments.
Discussion
Metformin is antidiabetic drug widely used to improve glycemic control, and its major mechanism of action is the inhibition of glucose production from the liver (Moller, 2001). Evidence points to the potential role of metformin in glucose uptake in skeletal muscle (Hundal et al., 1992; Klip et al., 1992; Borst & Snellen, 2001; Galuska et al., 1994; Inzucchi et al., 1998). This study was undertaken to examine whether metformin can improve insulin signalling in insulin resistant skeletal muscle cells.
We examined the effect of metformin on the tyrosine phosphorylation of IR and IRS-1. It has been previously reported that hyperinsuliniaemic conditions leads to the development of insulin resistance in cells in culture (Pryor et al., 2000; Ricort et al., 1995; Thomson et al., 1997). In this study, insulin resistance was developed in serum free medium in the absence or chronic presence of insulin in C2C12 cells, although, L6 skeletal muscle cells has been shown to express relatively more GLUT-4 (Sarabia et al., 1992). However, as reported earlier (Lawson & Purslow, 2000; Pinset et al., 1982) L6 cells could not be differentiated in serum free medium, but C2C12 cells can be differentiated in serum free medium (Conejo et al., 2001; Goto et al., 1999). So, it was pertinent to choose C2C12 cell line to study the effect of hyperinsuliniaemic conditions in serum free medium. Recently, it has been shown that metformin can reverse the chronic insulin induced defects in insulin signalling (Pryor et al., 2000). However, it has been reported that glucose transport and insulin-stimulated Akt serine phosphorylation was unaltered from pretreatment in isolated adipocytes from metformin treated subjects (Ciaraldi et al., 2002). The role of metformin in skeletal muscle of treated patients is controversial with Inzucchi et al. (1998) suggesting that metformin increased glucose disposal in skeletal muscle. However, Kahn and co-workers recently reported that metformin treatment had no effect on PI 3-kinase and Akt activity in skeletal muscle of type 2 diabetic subjects (Kim et al., 2002). We show in this study that metformin can enhance the insulin-stimulated tyrosine phosphorylation of IR and IRS-1, PI 3-kinase activity associated with IRS-1 at 400 μM concentration in C2C12 myotubes. Metformin is required at high concentrations for in vitro responses as compared to required therapeutic doses used for type 2 diabetic patients (Galuska et al., 1994). Based on the data of circulating levels of metformin measured 2 h after an oral dose (1.0 g day−1), the therapeutic level of metformin has been estimated to be in the order of 10 μM (Tucker et al., 1981). Considering the facts that (i) clinically used metformin concentration being up to 3 g day−1, (ii) metformin demonstrated its effect on isolated human skeletal muscle strips at 100 μM concentration (Galuska et al., 1994), (iii) in vitro concentrations of metformin on other cell systems have been used is up to 1.0 mM (Sarabia et al., 1992; Klip et al., 1992; Pryor et al., 2000), (iv) while determining the dose response of metformin we have also observed the stimulatory effect at 100 μM concentration of metformin (however, the maximum effect was observed at 400 μM) (data not shown), our in vitro model seems promising as it is closer to the concentration being effective in isolated human skeletal muscle strips. Moreover, it has been postulated that hypoglycemic effect of metformin in vivo at lower concentration may be due to an accumulation of drug in the extracellular space of skeletal muscle (Galuska et al., 1994). Therefore, a direct correlation between in vivo and in vitro conditions may not be straightforward.
It had been reported in rat adipocytes that a 2 h of metformin treatment had no effect on tyrosine phosphorylation of IR (Matthaei et al., 1991). In contrast, Pryor et al. (2000) had reported increase in IR tyrosine phosphorylation due to 24 h of metformin treatment in rat adipocytes. Another study by Matthaei et al. (1993) demonstrated that in metformin treated obese zucker rats, there was no enhancement of IR tyrosine phosphorylation in adipocytes. Contradictory evidence for in vivo enhancement of IR tyrosine kinase activity by metformin treatment exists in erythrocytes of type 2 diabetic human subjects (Santos et al., 1995). Similar observation of enhancement of IR tyrosine phosphorylation by metformin treatment was also reported in the muscles of streptozotocin diabetic rats (Rossetti et al., 1990). Our study suggests that metformin can sensitize insulin signalling in skeletal muscle cells by enhancing tyrosine phosphorylation of IR and IRS-1.
PI 3-kinase pathway has been implicated in the regulation of glucose transport by insulin (Farsee, 2001). Protein kinase B and atypical protein kinase C isoforms, ζ and λ, has been proposed to serve as downstream effectors for PI 3-kinase in regulation of glucose transport (Farsee, 2001). Klip and co-workers has recently shown the role of p38 in glucose transport in muscle cells (Somwar et al., 2000; 2001). We report that PI 3-kinase activity associated with IRS-1 is restored by metformin treatment in insulin resistant myotubes. Although the insulin-stimulated glucose uptake was impaired in insulin resistant myotubes, treatment with metformin increased the basal glucose uptake to significant levels in metformin treated cells. But there was no significant difference in the insulin-stimulated glucose uptake in normal and insulin resistant myotubes treated with metformin. The retreating effect on insulin stimulated glucose uptake even in sensitive cells treated with metformin might be due to maximum ability of these cells to uptake glucose in under the conditions used.
Insulin-stimulated activation of ERK and JNK was unaffected by chronic presence of insulin and treatment with metformin. The effect observed by us on ERK was in line with the study by Kahn and co-workers (Cusi et al., 2000) where it was shown that ERK pathway is unaffected in skeletal muscle of type 2 diabetic subjects. White and co-workers has implicated the role of JNK in insulin resistance caused by TNF-α in Chinese hamster ovary cells overexpressing human insulin receptor (Aguirre et al., 2000) and in human muscle caused during hyperinsulinemic euglycemic clamp (Rui et al., 2001). Our study suggests that JNK activation is not involved in insulin resistance induced by chronic insulin presence in C2C12 skeletal muscle cells. It has been shown that TNF-α does not activate JNK in 3T3-L1 adipocytes (Engelman et al., 2000; Jain et al., 1998). Activation of p38 was impaired in insulin resistant myotubes. The treatment of cells with metformin enhanced the basal p38 activation but no significant increase in p38 activation by insulin in metformin treated cells was observed. This data is in accord with the data obtained on glucose uptake. The treatment of cells with SB203580, for a short period of time, blocked insulin stimulated p38 activation as well as glucose uptake in sensitive cells. Moreover, treatment of sensitive and resistant myotubes with SB203580 for 24 h prevented insulin and metformin stimulated effects on p38 activation and glucose uptake. Although, an increase in insulin-stimulated tyrosine phosphorylation of IR and IRS-1, PI 3-kinase activity in resistant myotubes treated with metformin, the corresponding increase in glucose uptake or insulin-stimulated p38 activation was not observed. Metformin enhanced the basal activation of p38 and resulted in higher basal glucose uptake in the C2C12 myotubes and based on the data obtained in these experiments strongly suggest a role of p38 in glucose uptake and the generation of insulin resistance in C2C12 myotubes.
In conclusion, our study shows that metformin can enhance insulin signalling in insulin resistant skeletal muscle by increasing tyrosine phosphorylation of IR and IRS-1 and PI 3-kinase activity associated with IRS-1. Metformin enhanced basal glucose uptake in insulin-resistant myotubes as well as basal p38 activation. These results indicate that action of metformin is insulin-dependent on tyrosine phosphorylation of IR, IRS-1 and activation of PI 3-kinase; however, it is insulin-dependent on glucose uptake and p38 activation in insulin resistant myotubes. In conclusion, p38 plays a major role in glucose uptake and it can be a potential therapeutic target for novel anti-diabetic drugs. However, further studies need to be carried out to decipher the status of p38 in insulin resistance in type 2 diabetic subjects.
Acknowledgments
We would like to thank Dr C.L. Kaul, Director, NIPER, for his constant encouragement in this work. A. Khurana, H.L. Goel, Jayanarayan K.G. and R. Singh are being acknowledged for their support. N. Kumar is a recipient of a senior research fellowship from Council of Scientific and Industrial Research, New Delhi.
Abbreviations
- DMEM
Dulbecco's modified Eagle's medium
- 2-DOG
2-deoxyglucose
- EDTA
ethlenediamine tetraacetic acid
- EGTA
ethylene glycol bis (2-aminoethyl ether)-N,N,N′N′tetraacetic acid
- FCS
foetal calf serum
- IR
insulin receptor
- IRS-1
insulin receptor substrate-1
- KRP
Kreb's Ringer phosphate
- MAP kinases
mitogen-activated protein kinases
- NIDDM
non-insulin-dependent diabetes mellitus
- PBS
phosphate buffered saline
- PI 3-kinase
phosphatidyl inositol 3-kinase
- PVDF
polyvinylidene difluoride
- SDS–PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
References
- AGUIRRE V., UCHIDA T., YENUSH L., DAVIS R., WHITE M.F. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J. Biol. Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- BORST S.E., SNELLEN H.G. Metformin, but not exercise training, increases insulin responsiveness in skeletal muscle of Sprague-Dawley rats. Life Sci. 2001;69:1497–1507. doi: 10.1016/s0024-3205(01)01225-5. [DOI] [PubMed] [Google Scholar]
- CIARALDI T.P., KONG A.P.S., CHU N.V., KIM D.D., BAXI S., LOVISCACH M., PLODKOWSKI R., REITZ R., CAULFIELD M., MUDALIAR S., HENRT R.R. Regulation of glucose transport and insulin signaling by troglitazone or metformin in adipose tissue of type 2 diabetic subjects. Diabetes. 2002;51:30–36. doi: 10.2337/diabetes.51.1.30. [DOI] [PubMed] [Google Scholar]
- CONEJO R., VALVERDE A.V., BENITO M., LORENZO M. Insulin produces myogenesis in C2C12 myoblasts by induction of NF-κB and downregulation of AP-1 activities. J. Cell. Physiol. 2001;186:82–94. doi: 10.1002/1097-4652(200101)186:1<82::AID-JCP1001>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- CUSI K., MAEZONO K., OSMAN A., PENDERGRASS M., PATTI M.E., PRATIPANAWATR T., DEFRONZO R.A., KAHN C.R., MANDARINO L.J. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J. Clin. Invest. 2000;105:311–320. doi: 10.1172/JCI7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENGELMAN J.A., BERG A.H., LEWIS R.Y., LISANTI M.P., SCHERER P.E. Tumor necrosis factor α-mediated insulin resistance, but not dedifferentiation, is abrogated by MEK1/2 inhibitors in 3T3-L1 adipocytes. Mol. Endocrinol. 2000;14:1557–1569. doi: 10.1210/mend.14.10.0542. [DOI] [PubMed] [Google Scholar]
- FARSEE R.V. Insulin-sensitive phospholipid signaling systems and glucose transport. Update II. Exp. Biol. Med. 2001;226:283–295. doi: 10.1177/153537020122600404. [DOI] [PubMed] [Google Scholar]
- GALUSKA D., NOLTE L.A., ZIERATH J.R.., WALBERG-HENRIKSSON H. Effect of metformin on insulin-stimulated glucose transport in isolated skeletal muscle obtained from patients with NIDDM. Diabetologia. 1994;37:826–832. doi: 10.1007/BF00404340. [DOI] [PubMed] [Google Scholar]
- GOTO S., MIYAZAKI K., FUNABIKI T., YASUMITSU H. Serum-free culture conditions for the analysis of secretory proteinases during myogenic differentiation of mouse C2C12 myoblasts. Anal. Biochem. 1999;272:135–142. doi: 10.1006/abio.1999.4163. [DOI] [PubMed] [Google Scholar]
- HUNDAL H.S., RAMLAL T., RETES R., LEITER L.A., KLIP A. Cellular mechanism of metformin action involves glucose transporter translocation from an intracellular pool to the plasma membrane in L6 muscle cells. Endocrinol. 1992;131:1165–1173. doi: 10.1210/endo.131.3.1505458. [DOI] [PubMed] [Google Scholar]
- INOUE G., CHEATHAM B., KAHN C.R. Different pathways of postreceptor desensitization following chronic insulin treatment and in cells overexpressing constitutively insulin receptors. J. Biol. Chem. 1996;271:28206–28211. doi: 10.1074/jbc.271.45.28206. [DOI] [PubMed] [Google Scholar]
- INZUCCHI S.E., MAGGS D.G., SPOLLETT G.R., PAGE S.L., RIFE F.S., WALTON V., SHULMAN G.I. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N. Engl. J. Med. 1998;338:867–872. doi: 10.1056/NEJM199803263381303. [DOI] [PubMed] [Google Scholar]
- JAIN R.G., MEREDITH M.J., PEKALA P.H. Tumor necrosis factor-α mediated activation of signal transduction cascades and transcription factors in 3T3-L1 adipocytes. Adv. Enzyme Regul. 1998;38:333–347. doi: 10.1016/s0065-2571(97)00007-1. [DOI] [PubMed] [Google Scholar]
- KIM Y.-B., CIARALDI T.P., KONG A., KIM D., CHU N., MOHIDEEN P., MUDALIAR S., HENRY R.R., KAHN B.B. Troglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases p110β protein levels in skeletal muscle of type 2 diabetic subjects. Diabetes. 2002;51:443–448. doi: 10.2337/diabetes.51.2.443. [DOI] [PubMed] [Google Scholar]
- KLIP A., GUMA A., TAMLAL T., BILAN P.J., LAM L., LEITER L.A. Stimulation of hexose transport by metformin in L6 muscle cells in culture. Endodrionology. 1992;130:2535–2544. doi: 10.1210/endo.130.5.1572281. [DOI] [PubMed] [Google Scholar]
- LAEMMLI U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- LAWSON M.A., PURSLOW P.P. Differentiation of myoblasts in serum-free media: effects of modified media are cell line-specific. Cells Tissues Organs. 2000;167:130–137. doi: 10.1159/000016776. [DOI] [PubMed] [Google Scholar]
- MATTHAEI S., HAMANN A., KLEIN H.H., BENECKE H., KREYMANN G., FLIER J.S., GRETEN H. Association of Metformin's effect to increase insulin-stimulated glucose transport with potentiation of insulin-induced translocation of glucose transporters from intracellular pool to plasma membrane in rat adipocytes. Diabetes. 1991;40:850–857. doi: 10.2337/diab.40.7.850. [DOI] [PubMed] [Google Scholar]
- MATTHAEI S., REIBOLD J.P., HAMANN A., BENECKE H., HARING H.U., GRETEN H., KLEIN H.H. In vivo metformin treatment ameliorates insulin resistance: evidence for potentiation of insulin-induced translocation and increased functional activity of glucose transporters in obese (fa/fa) Zucker rat adipocytes. Endocrinology. 1993;133:304–311. doi: 10.1210/endo.133.1.8391425. [DOI] [PubMed] [Google Scholar]
- MOLLER D.E. New drug targets for type 2 diabetes and the metabolic syndrome. Nature. 2001;414:821–827. doi: 10.1038/414821a. [DOI] [PubMed] [Google Scholar]
- PINSET C., GROS F., WHALEN R. Proliferation and differentiation of a myogenic line in synthetic media. C.R. Seances Acad. Sci. 1982;295:727–732. [PubMed] [Google Scholar]
- PRYOR P.R., LIU S.C.H., CLARK A.E., YAND J., HOLMAN G.D., TOSH D. Chronic insulin effects on insulin signaling and GLUT4 endocutosis are reversed by metformin. Biochem. J. 2000;348:83–91. [PMC free article] [PubMed] [Google Scholar]
- RICORT J.M., TANTI J.F., OBBERGHEN E.V., LE MARCHAND-BRUSTEL Y. Alterations in insulin signaling pathway induced by prolonged insulin treatment of 3T3-L1 adipocytes. Diabetologia. 1995;38:1148–1156. doi: 10.1007/BF00422363. [DOI] [PubMed] [Google Scholar]
- ROSSETTI L., DEFRONZO R.A., GHERZI R., STEIN P., ANDRAGHETTI G., FALZETTI G., SHULMAN G.I., KLEIN-ROBBENHAAR E., CORDERA R. Effect of metformin treatment on insulin action in diabetic rats: in vivo and in vitro correlations. Metab. Clin. Exp. 1990;39:425–435. doi: 10.1016/0026-0495(90)90259-f. [DOI] [PubMed] [Google Scholar]
- RUI L., AGUIRRE V., KIM J.K., SHULMAN G.I., LEE A., CORBOULD A., DUNAIF A., WHITE M.F. Insulin/IGF-1 and TNF-α stimulate tyrosine phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J. Clin. Invest. 2001;107:181–189. doi: 10.1172/JCI10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANTOS R.F., NOMIZO R., WAJHENBERG B.L., REAVEN G.M., AZHAR S. Changes in insulin receptor tyrosine kinase activity associated with metformin treatment of type 2 diabetes. Diabetes Metab. 1995;21:274–280. [PubMed] [Google Scholar]
- SARABIA V., LAM L., BURDETT E., LEITER L.A., KLIP A. Glucose transport in human skeletal muscle cells in culture: stimulation by insulin and metformin. J. Clin. Invest. 1992;90:1386–1395. doi: 10.1172/JCI116005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH P.K., KROHN R.I., HERMANSON G.T., MALLIA A.K., GARTNER F.H., PROVENZANO M.D., FUJIMOTO E.K., GOEKE N.M., OLSON B.J., KLENK D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- SOMWAR R., KIM D.Y., SWEENEY G., HUANG C., NIU W., LADOR C., RAMLAL T., KLIP A. GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: potential activation of GLUT4 via p38 mitogen-activated protein kinase. Biochem. J. 2001;359:639–649. doi: 10.1042/0264-6021:3590639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOMWAR R., PERREAULT M., KAPUR S., TAHA C., SWEENET G., RAMLAL T., KIM D.Y., KEEN J., COTE C.H., KLIP A., MARETTE A. Activation of p38 mitogen-activated protein kinase α and β by insulin and contraction in rat skeletal muscle. Diabetes. 2000;49:1794–1800. doi: 10.2337/diabetes.49.11.1794. [DOI] [PubMed] [Google Scholar]
- SWEENEY G., SOMWAR R., RAMLAL T., VOLCHUK A., UEYAMA A., KLIP A. An inhibitor of p38 mitogen-activated protein kinase prevents insulin-stimulated glucose transport but not glucose transporter translocation in 3T3-L1 adipocytes and L6 myotubes. J. Biol. Chem. 1999;274:10071–10078. doi: 10.1074/jbc.274.15.10071. [DOI] [PubMed] [Google Scholar]
- THOMSON M.J., WILLIAMS M.G., FROST S.C. Development of insulin resistance in 3T3-L1 adipocytes. J. Biol. Chem. 1997;272:7759–7761. doi: 10.1074/jbc.272.12.7759. [DOI] [PubMed] [Google Scholar]
- TUCKER G.T., CASEY C., PHILLIPS P.J., CONNOR H., WARD J.D., WOODS H.F. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br. J. Clin. Pharmacol. 1981;12:235–246. doi: 10.1111/j.1365-2125.1981.tb01206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIERNSPERGER N.F., BAILEY C.J. The antihyperglycaemic effect of metformin: therapeutic and cellular mechanisms. Drugs. 1999;58:31–39. doi: 10.2165/00003495-199958001-00009. [DOI] [PubMed] [Google Scholar]