

Abstract
This study compares the effect of three chemically unrelated cystic fibrosis transmembrane conductance regulator (CFTR) activators on epithelial cell monolayers expressing the G551D-CFTR mutant.
We measured Cl− transport as the amplitude of short-circuit current in response to the membrane permeable cAMP analogue 8-(4-chlorophenylthio)adenosine-3′-5′-cyclic monophosphate (CPT-cAMP) alone or in combination with a CFTR opener. The correction of G551D-CFTR defect was quantified by comparison with maximal activity elicited in cells expressing wild type CFTR. To this end we used Fisher rat thyroid (FRT) cells transfected with wild type or G551D CFTR, and primary cultures of human nasal epithelial cells.
In both types of epithelia, cAMP caused activation of Cl− transport that was inhibited by glibenclamide and not by 4,4′-diisothiocyanato-stilbene-2,2′-disulfonic acid. After normalising for CFTR expression, the response of FRT-G551D epithelia was 1% that of wild type monolayers.
Addition of genistein (10–200 μM), but not of 8-cyclopentyl-1,3-dipropylxanthine (CPX, 1–100 μM) or of the benzo[c]quinolizinium MPB-07 (10–200 μM) to FRT-G551D epithelia pre-treated with cAMP, stimulated a sustained current that at maximal genistein concentration corresponded to 30% of the response of wild type epithelia.
The genistein dose-response curve was bell-shaped due to inhibitory activity at the highest concentrations. The dose-dependence in G551D cells was shifted with respect to wild type CFTR so that higher genistein concentrations were required to observe activation and inhibition, respectively.
On human nasal epithelia the correction of G551D-CFTR defective conductance obtained with genistein was 20% that of wild type. The impressive effect of genistein suggests that it might correct the Cl− transport defect on G551D patients.
Keywords: CFTR, CF-G551D, monolayers, nasal epithelia, CFTR openers, genistein, CPX, MPB-07
Introduction
Cystic fibrosis (CF) is the most common lethal genetic disorder in Caucasian populations, affecting one in 2000 births (Welsh et al., 1995). The disease is autosomic recessive and the corresponding gene encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein, a cAMP-activated Cl− channel present in the apical membrane of surface and glandular epithelia. In CF the ability of epithelia to secrete Cl− is greatly reduced, and the consequences are severe, in particular in the respiratory and gastrointestinal tracts. The most common CF associated mutation, which accounts for 70% of all disease causing CF alleles, is a deletion of phenylalanine 508 (F508del) and yields a protein that is unable to exit from the endoplasmic reticulum and to traffic to the plasma membrane. Among the other mutations, the glycine-to-aspartic acid change at codon 551 (G551D) is the third commonest mutation with a worldwide frequency of 3.1% among CF chromosomes (Hamosh et al., 1992) although populations of Celtic descent display frequencies as high as 8% (Cashman et al., 1995). The phenotype of patients carrying one F508del and one G551D allele is characterized by a clinical course as severe as that of homozygous for F508del but with a reduced risk of meconium ileus (Hamosh et al., 1992). The G551D is a class III mutation that produces a protein that is well synthesized, processed and correctly inserted in the plasma membrane but with a strongly reduced channel activity (Welsh & Smith, 1993). G551D is localized in the first nucleotide binding domain (NBD) of CFTR and interferes with ATP hydrolysis (Howell et al., 2000; Li et al., 1996; Logan et al., 1994).
There is an increasing interest in the pharmacological approach to CF as an alternative or complementary strategy to gene therapy. Some compounds belonging to the families of flavonoids, xanthines, and substituted benzo[c]quinoliziniums are able to increase wild type CFTR activity with unknown mechanisms (Schultz et al., 1999). Genistein is a soybean-derived isoflavone that can reach micromolar concentrations in the serum of individuals who eat Asian diets (Barns et al., 1996). Genistein is known as an inhibitor of ATP utilizing enzymes such as tyrosine kinases (Diener & Hug, 1996), topoisomerase II (Markovits et al., 1989), and histone kinase (Huang et al., 1992). Recent studies have shown that genistein activates both wild type and mutant CFTR (Diener & Hug, 1996; Gondor et al., 1998; Illek et al., 1995; 1999). This effect seems to involve a direct binding of the drug to CFTR (French et al., 1997; Weinreich et al., 1997) and the inhibition of the ATPase activity of the second nucleotide binding domain (Howell et al., 2000; Randak et al., 1999). The inhibition observed at higher concentrations of genistein suggests the existence of a second binding site that reduces the open rate of CFTR (Wang et al., 1998).
Several xanthines have also been found to activate CFTR (Bulteau et al., 2000; Haws et al., 1996). Among them, CPX has been proposed as a pharmacological lead agent for CF given the low concentrations required to activate Cl− efflux (Eidelman et al., 1992). CPX activates CFTR with a mechanism not involving phosphodiesterase or adenosine receptor inhibition but probably a direct interaction at the NBD1 (Arispe et al., 1998; Cohen et al., 1997). Also the recently synthesized substituted benzo[c]quinolizinium MPB-07 activates CFTR by a mechanism not involving modification of cAMP and ATP levels or protein phosphatases (PP1, PP2A, PP2C or alkaline) activity (Becq et al., 1999).
Recent data indicate that the isoflavone genistein is particularly effective on the G551D-CFTR Cl− channel (Illek et al., 1999) and we showed that also the benzo[c]quinolizinium MPB-91 does activate this mutation (Derand et al., 2001). It is not clear however, if the other CFTR openers are similarly effective on this mutation and, in particular, the amount of absolute correction that can be obtained with respect to the normal function. This information is important for the development of improved CFTR activators effective on G551D and similar mutations.
Here we determine, on polarized monolayers, the pharmacological properties of genistein, CPX and MPB-07, which are representative of three major classes of CFTR openers. Our results, obtained on a heterologous expression system, show that the three compounds are similarly effective on wild type CFTR but that only genistein is active on the G551D mutant. This result is confirmed on nasal monolayers obtained from a G551D CF patient, where the transport correction by genistein is as high as 20% that of wild type CFTR.
Methods
Plasmid construction
For expression of human CFTR gene in FRT cells we cloned the CFTR cDNA in the expression vector pTracer-CMV (Invitrogen). The CMV promoter controls CFTR gene expression while SV40 promoter controls synthesis of the GFP-Zeocin fusion protein. Site-directed mutagenesis was done using the GeneEditor kit (Promega).
Cell transfection
Fisher rat thyroid (FRT) epithelial cells were cultured on 60 mm Petri dishes with Coon's Modified F12 containing 5% serum, 2 mM L-glutamine, 50 U ml−1 penicillin and 50 μg ml−1 streptomycin. Cells were stably transfected using 10 μl Effectene (Qiagen) with 1 μg of the CFTR plasmid according to the manufacturer's instruction. Clones were initially selected in 800 μg ml−1 and then maintained in 600 μg ml−1 of Zeocin. For Ussing chamber experiments, cell were seeded at high density (5·105 cells cm−2) on Snapwell inserts (Costar, Corning) and maintained at 37°C in a 5% CO2/95% air atmosphere. Apical and basolateral media were replaced every 48 h. Transepithelial resistance was daily measured with an epithelial voltohmeter (Millipore-ERS, Millipore) using chopstick-like electrodes. After 4–7 days, FRT monolayers developed a transepithelial resistance in the range of 2–4 kΩ cm2. Experiments were done at days 6 to 8 after seeding.
Ussing chamber experiments
Filters were mounted into an Ussing-like Vertical Diffusion Chamber (Corning, Costar). To detect CFTR activity, the basolateral membrane of FRT epithelia was permeabilized with 250 μg ml−1 amphotericin B and a transepithelial Cl− gradient was applied as previously reported (Sheppard et al., 1993). Membrane permeabilization was monitored measuring the current response to a 10 mV stimulus. The resistance progressively decreased and reached a stable value after 30–40 min. We assume this is the condition for maximal permeabilization. The apical chamber was bathed with a low Cl− containing solution (in mzM): Na-gluconate 140, MgSO4 1, CaCl2 2, HCl 1, glucose 10 and NaHCO3 24 (pH 7.4). The basolateral chamber was instead bathed with (in mM): NaCl 126, NaHCO3 24, KH2PO4 0.38, K2HPO4 2.13, MgSO4 1, CaCl2 1 and glucose 10 (pH 7.4). Experiments were done at 37°C and solutions were bubbled with 5% CO2/95% air. The transepithelial potential difference (PD) was short-circuited at 0 mV with a voltage-clamp amplifier (Bioengineering, The University of Iowa) connected to the chambers through Ag-AgCl electrodes and agar bridges. All experiments were done in the presence of 10 μM amiloride on the apical chamber. For nasal epithelial cells, Ussing chamber experiments were similar to those performed for FRT cells except for the absence of permeabilization with amphotericin B and the use of symmetrical chloride solutions (in mM): NaCl 126, NaHCO3 24, KH2PO4 0.38, K2HPO4 2.13, MgSO4 1, CaCl2 1 and glucose 10 (pH 7.4).
CFTR expression
Total RNA (14 μg sample−1) extracted with trizol was used for Northern analysis. CFTR and G3PDH probes were labelled using the kit Megaprime Labelling System (Amersham, Buckinghamshire, U.K.) and hybridization was performed at 68°C overnight in Quikhyb solution (Stratagene, La Jolla, CA, U.S.A.). Image analysis of hybridization signals was done using appropriate software (NIH Image; Igor PRO, Wavemetrics). After background subtraction, CFTR and G3PDH signals were fitted with gaussian functions and the function integral was determined. It has been well established that G551D mutation yields an amount of correctly processed protein similar to that of wild type CFTR (Logan et al., 1994; Welsh & Smith, 1993) therefore normalisation by CFTR mRNA relative levels (CFTR/G3PDH ratio) was considered a good estimation of CFTR expression. Therefore, values of short-circuit current (Isc) reported throughout the manuscript have been obtained by dividing the raw Isc measured by the CFTR/G3PDH ratio of the respective clone. All figures show Isc values already corrected by CFTR expression. Two wild type (N8 and N10) and two G551D clones (6E and A1) were used in this study.
Immunoprecipitation/PKA Assay
Immunoprecipitation was done using the monoclonal antibody (mAb) 24-1 (R&D Systems), recognizing the C-terminus of CFTR, in agreement with the manufacturer's instructions. Briefly, cell lysates were prepared from two confluent stably-transfected dishes (60 mm diameter) and mixed with 0.4 μg mAb 24-1 and Pansorbin (Calbiochem). Immunoprecipitated proteins were phosphorylated in vitro with 5 units of the catalytic subunit of protein kinase A (Promega) and 10 μCi [γ-33P]-ATP (Amersham), and separated by 5% SDS–PAGE, dried and quantitated by radioanalytic scanning (Molecular Dynamics PhosphoImager).
CF patient clinical feature and genotype, and nasal polyp cell cultures
The CF patient is a 12-year-old female carrying the G551D mutation on one chromosome. The second mutation is presently unknown, but is not one of the 15 most frequent mutations found in the CF patients of Northeast Italy, namely F508del, I507del, R1162X, 2183AA>G, N1303K, 3849+10KbC>T, G542X, 1717-1G>A, R553X, Q552X, G85E, 711+5G>A, 3132delTG, 2789+5G>A, W1282X. At present, clinical features are characterized by respiratory disease with Staphylococcus aureus colonization and pancreatic insufficiency. At the ages of 4 and 5 years the patient was subjected to polypectomy in order to relieve recurrent nasal obstruction and polyps were conserved in liquid nitrogen. For nasal polyp cell culture, one polyp was removed from liquid nitrogen, washed in warm (37°C) physiological solution and incubated overnight at 4°C in Hank's balanced salt solution containing 0.25% type XIV protease. The polyp was removed from protease solution and epithelial cells were mechanically detached. Collected cells were plated on collagen-coated Petri dishes in LHC9-RPMI 1640 (1 : 1), 2 mM L-glutamine, 100 u ml−1 penicillin, 100 μg ml−1 streptomycin and 2.5 μg ml−1 amphotericin B. After two trypsinization passages, 54×106 cells were obtained and most of them were stored on liquid nitrogen. Cell culture of nasal polyps from four different non-CF subjects was done using the same protocol but with freshly excised polyps. For Ussing chamber experiments, cells were seeded at high density (5 105 cells cm−2) on Snapwell inserts (Costar, Corning). The medium was Dulbecco's Modified Eagle Medium and Ham's F12 (1 : 1), 2% serum, 2 mM L-glutamine, 100 u ml−1 penicillin, 100 μg ml1− streptomycin, 2.5 μg ml−1 insulin, 2.5 μg ml−1 transferrin, 0.02 μM hydrocortisone, 1 nM triiodothyronine, 3.3 nM retinoic acid, 0.25 μM ethanolamine, 0.25 μM phosphoethanolamine, and 0.25% (vol : vol) bovine pituitary extract. The apical and basolateral media were replaced every 24 h. Experiments were done 8 days after seeding.
Sample collection from human airways, done during surgical procedures, and the use of epithelial cells for experimental purposes, was approved by the Ethical Committee of Gaslini Institute.
Chemicals
Amphotericin B, 8-(4-chlorophenylthio)adenosine-3′-5′-cyclic monophosphate (CPT–cAMP), 8-cyclopentyl-1,3-dipropylxanthine (CPX), genistein, glibenclamide, insulin and transferrin were from Sigma Chemical (St.Louis, MO, U.S.A.). 4,4′-diisothiocyanato-stilbene-2,2′-disulphonic acid (DIDS) was from Molecular Probes (Eugene, OR, U.S.A.). LHC, triiodothyronine and retinoic acid were from Biofluids. Trizol was from GIBCO BRL. All other chemicals were from Sigma except DMEM, RPMI, Ham's F12 and serum that were from Euroclone (Paignton, Devon, U.K.).
Statistics
Results are presented as raw data or as mean plus standard error of the mean (SE). Significance was determined using two-tail Student t-test.
Results
Northern blot and immunoprecipitation analysis of CFTR expression
To assess the effect of different CFTR openers, we stably expressed wild type (wt) and G551D-CFTRs in Fisher rat thyroid (FRT) epithelial cells. These cells were chosen because of their ability to form polarized epithelia when grown on permeable supports and because they have no cAMP-dependent Cl− channels in their apical membrane. Therefore, CFTR activity can be studied using short-circuit current (Isc) experiments as already shown (Sheppard et al., 1993). We considered two clones with wild type CFTR, labelled as N8 and N10, and two clones with G551D, A1 and 6E. Northern blot analysis with a CFTR probe showed in the N8 clone (Figure 1, lane 1) a strong hybridization signal, which was absent in untransfected cells. Under the same conditions, the clone N10 appeared as negative. However, a longer exposition time revealed a band of the same size as in N8 cells (Figure 1, lane labelled N10*). The G551D clones A1 and 6E showed a CFTR expression lower than N8, but considerably stronger than N10. We compared CFTR mRNA expression among the different clones relative to the expression of the housekeeping gene G3PDH. The ratio of CFTR to G3PDH expression was 2.37, 0.12, 0.72 and 0.98 for wt-N8, wt-N10, G551D-6E and G551D-A1, respectively. Analysis of CFTR protein by immunoprecipitation confirmed that all clones produced a fully glycosylated (band C) form of the protein and that N10 express low levels of CFTR with respect to N8, A1 and 6E. The abundance of CFTR mRNA showed correlation with the amount of total CFTR protein determined by the mean of three immunoprecipitation assays.
Figure 1.
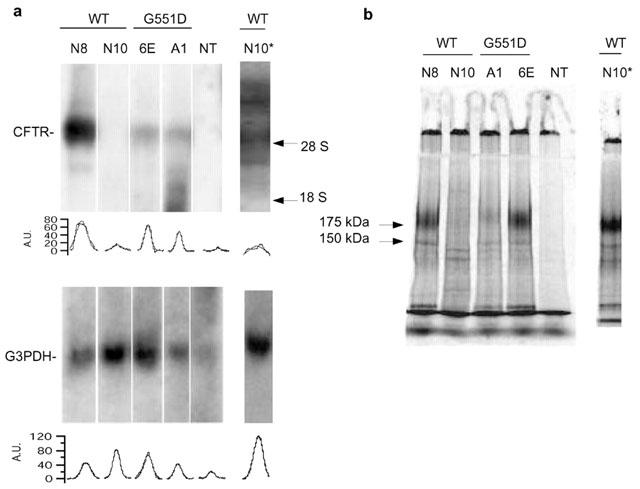
Northern blot analysis of CFTR transcripts and immunoprecipitation. (a) Northern analysis of RNA obtained from non transfected (NT) FRT cells and from cells stably transfected with G551D-CFTR (clones A1 and 6E) or with wt-CFTR (clones N8 and N10) CFTR. Size markers are indicated at the right. The exposure time, chosen to avoid saturation, does not allow to see a clear CFTR signal from the wild type N10 clone. Nevertheless, with longer exposure time the signal was evident (see lane wtN10*). A densitometric analysis for each band is shown below corresponding lanes (A.U. are arbitrary units). After background subtraction, CFTR and G3PDH signals were fitted with gaussian functions. (b) Immunoprecipitation of G551D- and wt-CFTR proteins was done using a monoclonal antibody that recognises the C-terminus of CFTR. Fully glycosylated (band C, 175 kDa) and core glycosylated (band B, 150 kDa) CFTR can be observed in wild type and G551D clones, but not in non-transfected FRT cells (lane NT). Molecular weight standards are indicated at left. Lane N10* was obtained by loading 10 fold more protein than in the other lanes to show that a clear C band could be obtained also from the low-expression clone.
Response of wt- and G551D-CFTR monolayers to cAMP elevating agents
Application of the cAMP analogue CPT–cAMP to intact FRT monolayers of cells transfected with wt- or G551D-CFTR in symmetrical Cl− was unable to stimulate a measurable Isc (Figure 2). A lack of anion secretion activation in intact FRT monolayers was previously reported by Sheppard et al. (1993) and is presumably due to the lack of active basolateral K+ channels. In contrast, after permeabilizing the basolateral membrane of wt-CFTR with amphotericin B and imposing a Cl− gradient, CPT–cAMP elicited a dose dependent Isc increase that was blocked by glibenclamide but largely insensitive to 500 μM 4,4′-diisothiocyanato-stilbene-2,2′-disulfonic acid (DIDS). The maximal response was 986.9±56.6 μA cm−2 in N8 and 67.2±3.9 μA cm−2 in N10. These values were in good agreement with the level of CFTR expression determined by Northern blot analysis. Therefore, we normalized CFTR activity by dividing the Isc to the relative CFTR expression (CFTR/G3PDH ratio). The maximal response obtained pooling normalized Isc from both clones was equal to 532.3±37.2 μA cm−2 and the concentration that produced half of the maximal effect was 84±8.7 μM (n=8). On monolayers expressing G551D-CFTR cells, CPT–cAMP activated a Isc that was considerably lower. After normalization for CFTR expression, the maximal cAMP-dependent Isc was 7.7±0.6 μA cm−2 (n=9, see Figure 2d). Despite the difference in the amplitude of Isc, the concentration that gave half of the maximal effect in G551D clones was 68.6±13.4 μM, not statistically different from that measured in wt-CFTR monolayers.
Figure 2.
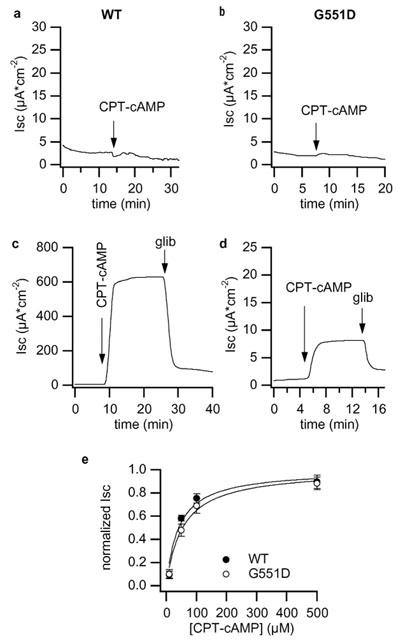
Isc response to CPT-cAMP. Representative traces depicting the response of wt-CFTR (a and c, clone N10) and of G551D-CFTR (b and d, clone 6E) monolayers to 500 μM CPT–cAMP and the inhibition caused by 500 μM glibenclamide. (a,b) Isc in non-permeabilized epithelia and with symmetrical Cl−. (c,d) Isc after basolateral membrane permeabilization with amphotericin B and in asymmetrical Cl−. (e) Dose-response relationships to CPT–cAMP of wild type and G551D-CFTR monolayers. Each point is the mean of 8 (wt) or 9 (G551D) experiments and vertical bars are s.e.
Response of wt- and G551D-CFTR monolayers to genistein
Apical application of genistein to wt-CFTR monolayers produced a sustained Isc increase (Figure 3). The maximal response was 107.4±11.7 μA cm−2 and was obtained with 50 μM. Genistein was more effective if given to cells pre-treated with a low concentration of CPT–cAMP (not shown). Genistein concentrations higher than 50 μM were less effective so that the dose-response relationship was bell-shaped as already observed by others (Illek et al., 1995; Wang et al., 1998). Genistein had an inhibitory effect if applied to cells maximally stimulated with 500 μM CPT–cAMP concentrations (Figure 3c). Untransfected FRT did not respond to genistein and CPT–cAMP even after permeabilization (not shown). In contrast to wild type CFTR, the G551D mutant did not respond to genistein (10–200 μM). However, subsequent addition of CPT–cAMP evoked a response that was more than 20 fold that of CPT–cAMP alone (compare Figure 3b with Figure 2d). In the reverse experiment, addition of genistein to cells previously stimulated with maximal CPT–cAMP concentration produced a vigorous dose dependent Isc increase (Figure 3d). For instance, 200 μM genistein increased Isc from 4.2±0.5 to 168.1±23.1 μA cm−2 (n=9; P<0.0001). Lower concentrations of genistein, like 50 and 100 μM, also potentiated CPT–cAMP effect (P<0.05 and P<0.005, respectively). Genistein concentrations higher than 300 μM were instead inhibitory. After preactivation with low CPT–cAMP concentrations (10–30 μM) the response to genistein was significantly smaller. The currents activated by CPT–cAMP and/or genistein in wild type and G551D-CFTR cells were totally blocked by 500 μM glibenclamide. A high DIDS concentration (500 μM) was largely ineffective (see Figure 3a).
Figure 3.
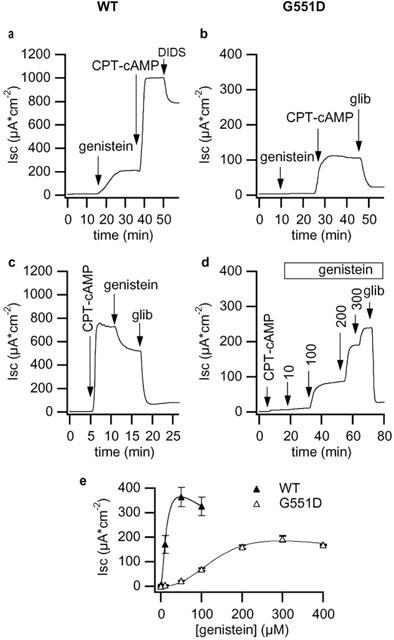
Isc response to genistein. Representative traces depicting the response of wt-CFTR (a and c, clone N10) and of G551D-CFTR (b and d, clone 6E) permeabilized monolayers to apical application of genistein. If not otherwise stated, the concentration of genistein was 50 μM, and that of CPT–cAMP, glibenclamide and DIDS was 500 μM. (e) Dose-response relationship to genistein of wild type and G551D-CFTR epithelia after preactivation with 30 μM or 500 μM CPT–cAMP, respectively. Each point is the mean of four to seven different experiments and vertical bars are s.e.
Response of monolayers to 8-cyclopentyl-1,3-dipropylxanthine (CPX)
Apical application of CPX (10–100 μM) to wt-CFTR monolayers elicited a very small Isc increase corresponding to 5.9±0.5 μA cm−2 (Figure 4a). However, when CPX was added after 30 μM CPT–cAMP the response was dramatically larger (194.1±22.9 μA cm−2, n=4, Figure 4e). A small Isc inhibition was instead observed when CPX was added after 500 μM CPT–cAMP (Figure 4). CPX alone was unable to activate G551D-CFTR. However, stimulation with 500 μM CPT–cAMP of cells previously treated with 100 μM CPX resulted in a response significantly larger than that obtained with CPT–cAMP alone (compare Figure 4b with Figure 2d). The synergy between CPX and CPT–cAMP was evident also when the two compounds were applied in the reverse order. Indeed, when CPX was given after 500 μM CPT–cAMP it stimulated a significant apical membrane Isc increase from 4.2±0.5 to 9.2±1.5 μA cm−2 (P<0.005). No synergy was observed instead between CPX and genistein on CPT–cAMP treated monolayers.
Figure 4.
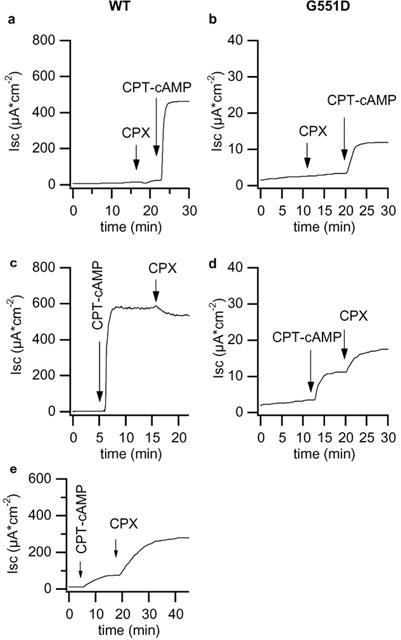
Isc response to CPX. Traces depict the response of wt-CFTR (a and c, clone N8) and of G551D-CFTR (b and d, clone A1) permeabilized monolayers to apical addition of 100 μM CPX. The concentration of CPT–cAMP was 500 μM. In (e) the response of wild type monolayers to CPX after 30 μM CPT–cAMP.
Response of wt- and G551D-CFTR to the benzo[c]quinolizinium MPB-07
Apical MPB-07 added to wt-CFTR monolayers after a low CPT–cAMP concentration (30 μM) evoked a sustained Isc increase (126.3±17.4 μA cm−2, n=3, see Figure 5e). Nevertheless, when MPB-07 was added without prestimulation it had no significant effect on wt- or on G551D-CFTR monolayers. Similarly, after the maximal effective concentration of CPT–cAMP, MPB-07 was ineffective on both types of monolayers (Figure 5c,d).
Figure 5.
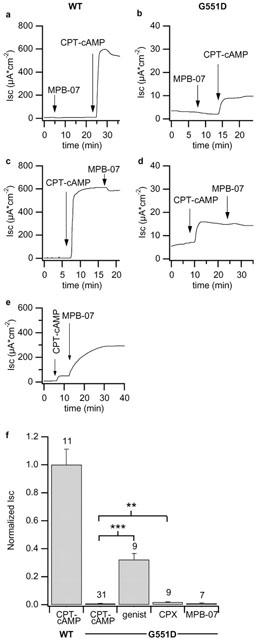
Isc response to MPB-07. Traces depict the response of wt-CFTR (a and c, clone N10) and of G551D-CFTR (b and d, clone 6E) permeabilized monolayers to apical application of 200 μM MPB-07. The concentration of CPT–cAMP was 500 μM. In (e) the response of wild type monolayers to MPB-07 after preactivation with 30 μM CPT–cAMP. The normalized response of FRT monolayers to the different CFTR activators is represented in (f). Mean of wt-CFTR epithelia were obtained pooling data from clones N8 and N10, and mean of G551D-CFTR epithelia were obtaining pooling data from clones 6E and A1. Above columns are the numbers of experiments in each condition. Vertical bars are SE. Asterisks indicate statistically different values (**P<0.0005, ***P<0.0001).
Relative efficacy of genistein, CPX and MPB-07 on G551D FRT cells
To determine the relative effect of CFTR openers on the mutant CFTR, Isc activated on G551D cells in response to the different compounds in the presence of 500 μM CPT–cAMP was compared with the response of wild type CFTR monolayers to CPT–cAMP alone, which we assume as maximal CFTR activity. Genistein (200 μM) increased the G551D-CFTR Isc from 0.8±0.3% to 32.2±4.4% of the wild type value (from 4.2±0.5 to 168.1±23.1 μA cm−2, see Figure 5e). The effect of CPX was low, although significant. In fact, it increased Isc to 9.2±1.5 μA cm−2 that is 1.8±0.2% of wt-CFTR. MPB-07 was instead ineffective.
Response to CFTR openers of nasal G551D and non-CF primary cultures
To strengthen the results obtained with transfected FRT monolayers, we decided to test the openers on human airway epithelial cells. Primary cultures of nasal polyps were obtained from four non-CF subjects and from one G551D CF patient. Application of 500 μM CPT–cAMP to amiloride treated G551D epithelia elicited a small Isc increase of 0.20±0.05 μA cm−2 (n=13). The subsequent addition of 200 μM genistein further increased Isc by about 10 fold (1.85±0.53 μA cm−2, n=7, P<0.0005, Figure 6). The half-effective concentration of CPT–cAMP was 116.6±23.2 μM (n=3), similar to those obtained in wild type and mutant CFTR transfected FRT cells. In nasal G551D epithelia, genistein effect was also dose-dependent and the dose-response relationship was bell-shaped. As in FRT cells the maximal effective concentration was near 200 μM (Figure 6d). cAMP- and genistein-dependent currents were blocked by glibenclamide. The response of G551D nasal epithelia to genistein required the presence of the cAMP analogue (see Figure 6d). Neither CPX nor MPB07 elicited a significant stimulatory effect on G551D epithelia despite a good effect on non-CF nasal monolayers (not shown). The response of non-CF nasal epithelia to CPT–cAMP was strong, reaching values of 9.47±1.6 μA cm−2 and the activated Isc was completely blocked by glibenclamide (Figure 6e). Figure 6f depicts mean responses of non-CF and G551D-CF epithelia to cAMP and the correction of G551D monolayers obtained with 200 μM genistein.
Figure 6.
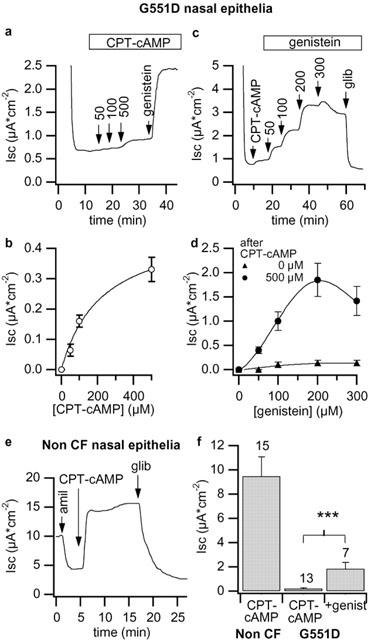
G551D and non-CF nasal polyps. Representative traces showing the response of G551D nasal epithelia to CPT–cAMP and genistein (a and c, respectively). The dose-response relationship of CPT–cAMP in (b) was constructed with the mean of eight different experiments similar to that of panel (a). Dose-response relationships in (d) were constructed with the mean of five experiments and depicted the response to increasing genistein in the absence of preactivation or after 500 μM CPT–cAMP. In (e) the response to 500 μM CPT–cAMP and to glibenclamide of non CF nasal epithelia. In (f) mean data of different experiments. Above columns are the numbers of experiments in each condition. Vertical bars are s.e. Asterisks indicate statistically different values (***P<0.0001).
Chronic stimulation with genistein
To determine if chronic stimulation with genistein alters the electrophysiological characteristics and viability of epithelia, wt- and G551D-FRT monolayers and CF and non-CF nasal epithelia were incubated for 48 h with genistein (50 or 200 μM). Transepithelial potential difference (PD) and electrical resistance of genistein-treated preparations were not statistically different from controls. For example, resistance and PD were 4.388±0.192 and 4.200±0.236 kΩ cm2 and −6.4±0.5 and −5±0.7 mV for control and genistein treated G551D-CFTR FRT monolayers. For control and genistein treated G551D nasal epithelia, resistance and PD were 1.910±0.103 and 2.092±0.149 kΩ cm2 and −58±1 and −57±4 mV, respectively.
Discussion
The stimulatory effect of some drugs on wild type CFTR has suggested that it may be possible to use a pharmacological approach to address the primary functional defect in CF patients. However, little is known of the ability of such compounds to activate the different CFTR mutants and how much correction can be obtained. Other concerns are related to possible adverse effects that these compounds may have on transepithelial ion transport. For example it has been found that genistein inhibits K+ channels and therefore may affect the basolateral K+ conductance that is required to support Cl− secretion in epithelia (Mall et al., 2000). Therefore, we decided to perform a comparative study using a heterologous expression system, FRT cells, and primary cultures of nasal epithelial cells. The FRT model is particularly useful since it has no endogenous CFTR-like current and therefore allows the study of the activity of wild type and mutant CFTR after transfection with the appropriate plasmid. Results obtained in FRT cells were then validated on human airway epithelia, which is the gold standard for studies related to CF. In the present study we compare the pharmacological properties of three major CFTR openers, namely genistein, CPX, and MPB-07. Although various studies report the effects of CFTR openers, each of them usually deal with a single compound in a different cell preparation and with different assays and conditions (single channel and whole cell Cl− currents, transepithelial Cl− transport, I− efflux, room temperature and 37°C, etc.). To our knowledge this is the first study comparing the effects of CFTR openers in homogeneous experimental conditions on polarized epithelial preparations.
G551D is a relatively common CF mutation with the characteristic of being correctly processed and inserted in apical membrane, therefore a good target for a comparative pharmacological study. We found that permeabilized FRT monolayers expressing wild type CFTR responded vigorously to apical genistein, CPX and MPB-07 when cells were pre-treated with a low CPT–cAMP concentration. On the other hand, genistein was the only compound that was active in the absence of CPT–cAMP. This behaviour suggests that in FRT cells, wild type CFTR is phosphorylated at very low levels and that some openers, such as CPX and MPB-07, require more CFTR phosphorylation to be effective. In contrast, the ability of genistein to activate CFTR even without prestimulation suggests that its action is less dependent on CFTR phosphorylation status.
The normalization of Isc by CFTR expression allowed us to compare the responses of cells expressing the wild type and the mutant sequence. We found that the maximal cAMP-dependent Isc in permeabilised G551D-CFTR was less than 1% of the response of wt monolayers. Only genistein and CPX increased this response significantly, but with substantially different potency. Indeed, while CPX only doubled the conductance of G551D monolayers, genistein potentiated it to values that correspond to 7, 15 and 30% of wild type monolayers (with 50, 100 and 200 μM genistein, respectively). Previous studies suggested that only 5–10% of cells expressing CFTR are required in a monolayer to have a normal phenotype (Johnson et al., 1992) and mutations that allow 4–5% of residual CFTR activity show a mild phenotype. Therefore, the results obtained in our FRT cell model indicate that genistein, but not CPX or MPB-07, might correct the G551D CF phenotype. We recently found that MPB-91, an improved derivative of MPB-07, is able to increase the conductance of G551D transfected FRT monolayers. However, its correction corresponds to 5% of that of wild type CFTR (Derand et al., 2001). Therefore, flavonoids are still the most interesting class of substances for the correction of class III and class IV mutants.
The data obtained on human nasal epithelia confirm quite well the conclusions from FRT cells. Given that only one allele carries the G551D mutation in the nasal epithelia that we analysed, the recovery of 20% of CFTR function by 200 μM genistein is in good agreement with the 30% correction found in FRT cells which carry exclusively the G551D mutant. We cannot exclude that part of the genistein effect is due to activation of the unknown allele. Nevertheless, the pattern of response to genistein, including the shift of the dose-response relationship to higher concentrations, and to CPX and MPB-07, is similar in G551D nasal polyp and in G551D-transfected FRT cells. This suggests that the response to openers in the nasal polyp is mainly due to stimulation of G551D-CFTR. The good concordance of results between the two cell preparations indicate that FRT cells are a suitable model for CF related studies and particularly for drug discovery approaches such as high throughput screenings. The meaningful correction of G551D monolayers conductance with genistein supports recent data by Illek et al. (1999). These authors observed with genistein an increase of isoproterenol-dependent nasal PD of G551D CF patients and calculated that the hyperpolarization obtained corresponded to 17% of the response of normal subjects.
The maximal effective genistein concentration on G551D-CFTR was far higher than that observed on wild type CFTR. Indeed, it was 200–300 μM in our experiments on G551D FRT and nasal epithelia while 30–50 μM genistein is the maximal effective concentration observed on wild type CFTR in our experiments and in single cell measurements by other authors (Illek et al., 1995; Wang et al., 1998). Recent studies (Howell et al., 2000; Randak et al., 1999) suggested that genistein activated CFTR by inhibiting the ATPase activity of the second nucleotide binding domain. Inhibition of the ATPase activity at NBD1 is probably responsible for the inhibition of CFTR at higher genistein concentrations. The most appealing explanation for the difference between wild type and G551D-CFTR data, is that G551D mutant is displaying genistein-dependent activation, but the typical inhibition is shifted to much higher opener concentrations due to a difficult interaction with mutated NBD1.
Recently, Mall et al. (2000) have described an inhibitory effect of genistein on basolateral K+ channels, the activity of which is required to support Cl− secretion in epithelia. If genistein is inhibiting K+ channels on our airway epithelia, this action seems much less important than the stimulatory effect on G551D-CFTR since the net result is a substantial increase on Cl− secretion. We have no explanation for the discrepancy between the data of these authors and ours, but an incomplete block of K+ channels by genistein can be speculated. It is also possible that other types of K+ channels, insensitive to genistein, may provide the driving force required for Cl− secretion.
Our experiments of chronic treatment of epithelial cells indicate that genistein has no significant cytotoxic effects on these cells, as evident from the lack of changes in electrical resistance and transepithelial ion transport. These measurements also confirm that genistein alone does not activate G551D-CFTR in the absence of an intracellular cAMP increase (see Figure 3b) since CFTR activation would have reduced resistance of the monolayers.
In conclusion, our study shows that genistein is a particularly potent CFTR agonist able to correct the G551D functional defect to therapeutic levels in polarized preparations. However, the concentrations needed for this effect are very high so that local and systemic adverse effects might be expected in vivo. It is therefore desirable to develop new derivatives with higher affinity. This search can be helped by high-throughput screening analysis of lead-based libraries of organic compounds. Our study suggests that flavonoid-based combinatorial libraries will be a promising source of potent and more selective derivatives that could show a significant effect on G551D and similar CFTR mutants.
Acknowledgments
We thank Prof Lucio Nitsch for providing us with FRT cells. This work was supported by AFLM and by TeleThon Italy (GP0296Y01).
Abbreviations
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- CPT-cAMP
8-(4-chlorophenylthio) adenosine-3′-5′-cyclic monophosphate
- CPX
8-cyclopentyl-1,3-dipropylxanthine
- GFP
green fluorescent protein
- G3PDH
glyceraldehyde 3-phosphate dehydrogenase
- DIDS
4,4′-diisothiocyanato-stilbene-2,2′-disulfonic acid
- FRT
Fisher rat thyroid
- NBD
nucleotide binding domain
References
- ARISPE N., MA J., JACOBSON K.A., POLLARD H.B. Direct activation of cystic fibrosis trasmembrane conductance regulator channels by 8-cyclopentyl-1,3-dipropylxanthine (CPX) and 1,3-diallyl-8-cyclohexylxanthine (DAX) J. Biol. Chem. 1998;273:5727–5734. doi: 10.1074/jbc.273.10.5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARNS S., SFAKIANOS J., COWARD L., KIRK M. Soy isoflavones and cancer prevention: underlying biochemical and pharmacological issues. Adv. Exp. Med. Biol. 1996;401:87–100. doi: 10.1007/978-1-4613-0399-2_7. [DOI] [PubMed] [Google Scholar]
- BECQ F., METTEY Y., GRAY M.A., GALIETTA L.J., DORMER R.L., MERTEN M., METAYE T., CHAPPE V., MARVINGT-MOUNIR C., ZEGARRA-MORAN O., TARRAN R., BULTEAU L., DERAND R., PEREIRA M.M., MCPHERSON M.A., ROGIER C., JOFFRE M., ARGENT B.E., SARROUILHE D., KAMMOUNI W., FIGARELLA C., VERRIER B., GOLA M., VIERFOND J.-M. Development of substituted Benzo[c]quinolizinium compounds as novel activators of the cystic fibrosis chloride channel. J. Biol. Chem. 1999;274:27415–27425. doi: 10.1074/jbc.274.39.27415. [DOI] [PubMed] [Google Scholar]
- BULTEAU L., DERAND R., METTEY Y., METAYE T., MORRIS M.R., MCNEILLY C.M., FOLLI C., GALIETTA L.J.V., ZEGARRA-MORAN O., PEREIRA M.M.C., JOUGLA C., DORMER R.L., VIERFOND J.-M., JOFFRE M., BECQ F. Properties of CFTR activated by the xanthine derivative X-33 in human airway Calu-3 cells. Am. J. Physiol. Cell Physiol. 2000;279:C1925–C1937. doi: 10.1152/ajpcell.2000.279.6.C1925. [DOI] [PubMed] [Google Scholar]
- CASHMAN S.M., PATINO A., DELGADO M.G., BYRNE L., DENHAM B., DEARCE M. The irish cystic fibrosis database. J. Med. Genet. 1995;32:972–975. doi: 10.1136/jmg.32.12.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN B.E., LEE G., JACOBSON K.A., KIM Y.C., HUANG Z., SORSCHER E.J., POLLARD H.B. 8-cyclopentyl-1,3-dipropylxanthine and other xanthines differentially bind to the wild-type and delta F508 first nucleotide binding fold (NBF-1) domains of the cystic fibrosis transmembrane conductance regulator. Biochemistry. 1997;36:6455–6461. doi: 10.1021/bi970150v. [DOI] [PubMed] [Google Scholar]
- DERAND R., BULTEAU L., METTEY Y., ZEGARRA-MORAN O., HOWELL D., RANDAK C., GALIETTA L.J.V., COHN J.A., NOREZ C., ROMIO L., VIERFOND J.-M., JOFFRE M., BECQ F. Activation of G551D-CFTR channel with the benzo[c]quinolizinium compound MPB-91: regulation by ATPase activity and phosphorylation. Am. J. Physiol. 2001;281:C1657–C1666. doi: 10.1152/ajpcell.2001.281.5.C1657. [DOI] [PubMed] [Google Scholar]
- DIENER M., HUG F. Modulation of Cl− secretion in rat distal colon by genistein, a protein tyrosine kinase inhibitor. Eur. J. Pharmacol. 1996;299:161–170. doi: 10.1016/0014-2999(95)00832-2. [DOI] [PubMed] [Google Scholar]
- EIDELMAN O., GUAY-BRODER C., VAN GALEN P.J., JACOBSON K.A., FOX C., TURNER R.J., CABANTCHIK Z.I., POLLARD H.B. A1 adenosine-receptor antagonists activate chloride efflux from cystic fibrosis cells. Proc. Natl. Acad. Sci. U.S.A. 1992;89:5562–5566. doi: 10.1073/pnas.89.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRENCH P.J., BIJMAN J., BOT A.G., BOOMAARS W.E.M., SCHOLTE B.J., DE JONGE, H.R. Genistein activates CFTR Cl- channels via a tyrosine kinase- and protein phosphatase-independent mechanism. Am. J. Physiol. 1997;273:C747–C753. doi: 10.1152/ajpcell.1997.273.2.C747. [DOI] [PubMed] [Google Scholar]
- GONDOR M., NIXON P.A., DEVOR D.C., WINNIE G.B., SCHULTZ B.D., SINGH A.K., BRIDGES R.J., WALCZAK S., FRIZZELL R.A., PILEWSKI J.M. Genistein stimulates chloride secretion in normal volunteers and CF patients with a G551D mutation. Pediatr. Pulmon. 1998;S17:253. [Google Scholar]
- HAMOSH A., KING T.M., ROSENSTEIN B.J., COREY M., LEVISON H., DURIE P., TSUI L.-C., MCINTOSH I., KESTON M., BROCK D.J.H., MACEK M., Jr, ZEMKOVA D., KRASNICANOVA H., VAVROVA V., MACEK M., SR, GOLDER N., SCHWARTZ M.J., SUPER M., WATSON E.K., WILLIAMS C., BUSH A., O'MAHONEY S.M., HUMPHRIES P., DEARCE M.A., REIS A., BURGER J., STUHRMANN M., SCHMIDTKE J., WULBRAND U., DORK T., TUMMLER B., CUTTING G.R. Cystic Fibrosis patients bearing the common missense mutation Gly–Asp at codon 551 and the ΔF508 mutation are clinically indistinguishable from ΔF508 homozygotes, except for a decreased risk of meconium ileus. Am. J. Hum. Genet. 1992;51:245–250. [PMC free article] [PubMed] [Google Scholar]
- HAWS C.M., NEPOMUCENO I.B., KROUSE M.E., WAKELEE H., LAW T., XIA Y., NGUYEN H., WINE J.J. ΔF508-CFTR channels: kinetics, activation by forskolin, and potentiation by xanthines. Am. J. Physiol. 1996;270:C1544–C1555. doi: 10.1152/ajpcell.1996.270.5.C1544. [DOI] [PubMed] [Google Scholar]
- HOWELL D., BORCHARDT R., COHN J.A. ATP hydrolysis by a CFTR domain: pharmacology and effects of G551D mutation. Biochem. Biophys. Res. Comm. 2000;271:518–525. doi: 10.1006/bbrc.2000.2659. [DOI] [PubMed] [Google Scholar]
- HUANG J., NASR M., KIM Y., MATHEWS H.R. Genistein inhibits protein histidine kinase. J. Biol. Chem. 1992;267:15511–15515. [PubMed] [Google Scholar]
- ILLEK B., FISCHER H., SANTOS G.F., WIDDICOMBE J.H., MACHEN T.E., REENSTRA W.W. cAMP-independent activation of CFTR Cl channels by the tyrosine kinase inhibitor genistein. Am. J. Physiol. 1995;268:C886–C893. doi: 10.1152/ajpcell.1995.268.4.C886. [DOI] [PubMed] [Google Scholar]
- ILLEK B., ZHANG L., LEWIS N.C., MOSS R.B., DONG J.Y., FISCHER H. Defective function of the cystic fibrosis-causing missense mutation G551D is recovered by genistein. Am. J. Physiol. 1999;277:C833–C839. doi: 10.1152/ajpcell.1999.277.4.C833. [DOI] [PubMed] [Google Scholar]
- JOHNSON L.G., OLSEN J.C., SARKADI B., MOORE K.L., SWANSTROM R., BOUCHER R.C. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat. Genet. 1992;2:21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- LI C., RAMJEESINGH M., WANG W., GARAMI E., HEWRYK M., LEE D., ROMMENS J.M., GALLEY K., BEAR C.E. ATPase activity of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1996;271:28463–28468. doi: 10.1074/jbc.271.45.28463. [DOI] [PubMed] [Google Scholar]
- LOGAN J., HIESTAND D., DARAM P., HUANG Z., MUCCIO D.D., HARTMAN J., HALEY B., COOK W.J., SORSCHER E.J. Cystic fibrosis transmembrane conductance regulator mutations that disrupt nucleotide binding. J. Clin. Invest. 1994;94:228–236. doi: 10.1172/JCI117311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALL M., WISSNER A., SEYDEWITZ H.H., HUBNER M., KUEHR J., BRANDIS M., GREGER R., KUNZELMANN K. Effect of genistein on native epithelial tissue from normal individuals and CF patients and on ion channels expressed in Xenopus oocytes. Br. J. Pharmacol. 2000;130:1884–1892. doi: 10.1038/sj.bjp.0703520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARKOVITS J., LINASSIER C., FOSSE P., COUPRIE J., PIERRE J., JACQUEMIN-SABLON A., SAUCIER J.-M., LE PECQ, J.-B., LARSEN A.K. Inhibitory effects of the tyrosine kinase inhibitor genistein on mammalian DNA topoisomerase II. Cancer Res. 1989;49:5111–5117. [PubMed] [Google Scholar]
- RANDAK C., AUERSWALD E.A., ASSFALG-MACHLEIDT I., REENSTRA W.W., MACHLEIDT W. Inhibition of ATPase, GTPase and adenylate kinase activities of the second nucleotide-binding fold of the cystic fibrosis transmembrane conductance regulator by genistein. Biochem. J. 1999;340:227–235. [PMC free article] [PubMed] [Google Scholar]
- SCHULTZ B.D., SINGH A.K., DEVOR D.C., BRIDGES R.J. Pharmacology of CFTR chloride channel activity. Physiological Reviews. 1999;79:S109–S144. doi: 10.1152/physrev.1999.79.1.S109. [DOI] [PubMed] [Google Scholar]
- SHEPPARD D.N., RICH D.P., OSTEDGAARD L.S., GREGORY R.J., SMITH A.E., WELSH M.J. Mutations in CFTR associated with mild-disease-form Cl- channels with altered pore properties. Nature. 1993;362:160–164. doi: 10.1038/362160a0. [DOI] [PubMed] [Google Scholar]
- WANG F., ZELTWANGER S., YANG I.C.-H., NAIRN A.C., HWANG T.-C. Actions of genistein on cystic fibrosis transmembrane conductance regulator channel gating. J. Gen. Physiol. 1998;111:477–490. doi: 10.1085/jgp.111.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEINREICH F., WOOD P.G., RIORDAN J.R., NAGEL G. Direct action of genistein on CFTR. Pflugers Arch. 1997;434:484–491. doi: 10.1007/s004240050424. [DOI] [PubMed] [Google Scholar]
- WELSH M.J., SMITH A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- WELSH M.J., TSUI L.-C., BOAT T.F., BEAUDET A.L.Cystic Fibrosis The Metabolic and Molecular Basis of Inherited Disease 1995New York: McGraw-Hill; 3799–3876.ed. Scriver C.R., Beaudet A.L., Sly W.S. and Valle D pp [Google Scholar]