

Abstract
Excised outside-out patches from HEK293 cells stably transfected with the human (h) 5-HT3A receptor cDNA were used to determine the effects of cannabinoid receptor ligands on the 5-HT-induced current using the patch clamp technique. In addition, binding studies with radioligands for 5-HT3 as well as for cannabinoid CB1 and CB2 receptors were carried out.
The 5-HT-induced current was inhibited by the following cannabinoid receptor agonists (at decreasing order of potency): Δ9-THC, WIN55,212-2, anandamide, JWH-015 and CP55940. The WIN55,212-2-induced inhibition was not altered by SR141716A, a CB1 receptor antagonist. WIN55,212-3, an enantiomer of WIN55,212-2, did not affect the 5-HT-induced current.
WIN55,212-2 did not change the EC50 value of 5-HT in stimulating current, but reduced the maximum effect.
The CB1 receptor ligand [3H]-SR141716A and the CB1/CB2 receptor ligand [3H]-CP55940 did not specifically bind to parental HEK293 cells. In competition experiments on membranes of HEK293 cells transfected with the h5-HT3A receptor cDNA, WIN55,212-2, CP55940, anandamide and SR141716A did not affect [3H]-GR65630 binding, but 5-HT caused a concentration dependent-inhibition.
In conclusion, cannabinoids stereoselectively inhibit currents through recombinant h5-HT3A receptors independently of cannabinoid receptors. Probably the cannabinoids act allosterically at a modulatory site of the h5-HT3A receptor. Thus the functional state of the receptor can be controlled by the endogenous ligand anandamide. This site is a potential target for new analgesic and antiemetic drugs.
Keywords: Allosteric site, anandamide, cannabinoid, CB1 receptor, excised patch, 5-HT3 receptor, voltage-clamp
Introduction
It is generally accepted that the effect of Δ9-THC and other exogenous and endogenous cannabinoids are mediated via cannabinoid receptors. Two cannabinoid receptor subtypes CB1 and CB2 have so far been identified, characterized and cloned (for reviews, see Pertwee, 1997; 1999; 2000; Ameri, 1999). The CB1 receptor occurs in brain and nervous tissue (Herkenham et al., 1990; Matsuda et al., 1990; Mailleux et al., 1992) whereas the CB2 receptor is expressed mainly extraneuronally by immune cells (Munro et al., 1993). Accordingly, the effects of cannabinoids in the nervous system are generally believed to be mediated almost exclusively by CB1 receptors. Cannabinoid receptor agonists do not only exert psychotropic effects (see reviews quoted above and Abood & Martin, 1992), but they also play a role in the control of pain and emesis (reviews quoted above and Noyes et al., 1975; Martin et al., 1999; Darmani, 2001; Tramer et al., 2001). The latter property is shared by serotonin3 (5-HT3) receptor antagonists.
The 5-HT3 receptor is a ligand-gated ion channel (Peters et al., 1992; Boess & Martin, 1994; Costall & Naylor, 1997) putatively endowed with allosteric modulatory sites. Until now, two human (h) 5-HT3 receptor subunits, h5-HT3A (Miyake et al., 1995) and h5-HT3B (Davies et al., 1999) and two h5-HT3A subunit splice variants (Brüss et al., 2000) have been described. The h5-HT3A subunit and its splice variants contain the 5-HT recognition site, whereas the h5-HT3B subunit does not. Only the h5-HT3A subunit can form homopentameric functional receptor channels, but its splice variants and the h5-HT3B subunit increase or decrease the current through the channel when coexpressed with the h5-HT3A subunit (Davies et al., 1999; Brüss et al., 2000). The pharmacological properties of heteromeric 5-HT3 receptors consisting of the 5-HT3A and 5-HT3B subunits or of the 5-HT3A subunit and one or both of its splice variants are not qualitatively modified (Davies et al., 1999; Brüss et al., 2000; Brady et al., 2001); at most, minor differences in affinity or potency of 5-HT3 receptor ligands may exist (Dubin et al., 1999).
According to the involvement of both cannabinoid (for references, see above) and 5-HT3 receptors (reviews quoted above and Aapro, 1991; Mitchelson, 1992; Karim et al., 1996; Voog et al., 2000; Simpson et al., 2000) in pain and emesis it is conceivable that cannabinoids act not only via CB1 receptors, but also at an allosteric modulatory site of the 5-HT3 receptor. This possibility has been supported by experiments on the rat nodose ganglion neurone (Fan, 1995) which suggested that neuronal cannabinoid receptors interact with the 5-HT3 receptor ion channel and that the latter may be a direct target of cannabinoid receptor agonists. Cannabinoid receptor-independent effects of cannabinoids, in particular the endogenous agonist anandamide, have been reported for other ion channels such as vanilloid VR1 receptors (Zygmunt et al., 1999; Malinowska et al., 2001), TASK-1 K+ channels (Maingret et al., 2001), Shaker-related Kv1.2 K+ channels (Poling et al., 1996) and T-type calcium channels (Chemin et al., 2001).
In view of the putative physiological and therapeutic significance in humans, the aim of the present study was to clarify whether cannabinoids directly inhibit 5-HT-induced currents through the h5-HT3A receptor channels and, if so, to analyse the underlying site and mechanism of action. The experiments were carried out on HEK 293 cells stably transfected with the h5-HT3A receptor cDNA. The patch-clamp technique on excised outside-out patches and standard radioligand binding were used. The recombinant homomeric h5-HT3A receptor channels in this system have previously been characterized (Barann et al., 2000). Preliminary results of the present investigation have been published elsewhere in abstract form (Barann et al., 2001; Godlewski et al., 2002).
Methods
Cell culture
HEK 293 cells were grown as monolayers on culture plates (Nunc) in DMEM Nutrient Mix F12 (1 : 1; v v−1) medium containing 10% heat inactivated foetal calf serum, penicillin (100 IU ml−1), streptomycin (100 μg ml−1), geneticine (0.75 mg ml−1) and glutamine (292 μg ml−1). The cells were cultured at 37°C in a humidified atmosphere (5% CO2). Two days before an experiment cells were subcultured in monodishes (NUNC, 35 mm diameter).
Transfection
For stable transfection 20% confluent HEK 293 cells were transfected by the modified calcium phosphate method with the h5-HT3A receptor cDNA (according to Chen & Okayama, 1987) subcloned into the mammalian expression vector pcDNA3 (Invitrogen) under control of the human cytomegalo virus promoter. Two days after transfection, stably transfected cells were selected by the addition of geneticine (800 μg ml−1) to the culture medium. The medium was changed every 2 days. After occurrence of single cell colonies these were separated by usage of cloning cylinders (Sigma) and further subcultured in 24 well plates (Falcon) until confluence. About 20 to 40 colonies from each transfection experiment were tested for stable expression of the particular cDNA by 5-HT-induced [14C]-guanidinium influx and binding of the selective 5-HT3 receptor antagonist [3H]-GR65630. Colonies with the highest receptor expression were used for further experiments.
Electrophysiology
In a standard voltage-clamp experiment, 30 μM 5-HT was applied for 1–2 s at −100 mV on an excised outside-out patch using a fast solution exchange system (exchange time 1–2 ms; patch resistance: 1–10 gigaohm; Liu & Dilger, 1991). The external solution applied on the patch was of the following composition (mM): NaCl 150, KCl 5.6, CaCl2 1.8, MgCl2 1.0, HEPES 10, pH 7.4. Patch pipettes with resistances of 3–6 MΩ were filled with ‘intracellular' solution containing (mM) KCl 140, EGTA 10, MgCl2 5, HEPES 10, pH 7.4. For current measurements, we used a patch-clamp amplifier (EPC-7; List Electronic, Darmstadt, Germany) with the output filter set at 1 kHz and pClamp software (Axon Instruments, Foster City, CA, U.S.A.). The sampling rate varied between 200 and 2000 Hz; within this range, no significant differences were seen with regard to fitted time constants. The drug application system was equipped with inert materials like teflon tubing and glass to avoid losses of lipophilic drugs.
Radioligand binding experiments
All steps of the preparation procedure were performed on ice. Freshly harvested HEK 293 cells (parental or stably transfected with the human 5-HT3A cDNA) were suspended in 40 ml of a buffer solution containing (mM): HEPES-Na+ 5, EGTA 0.5, MgCl2 0.5, ascorbic acid 0.1, and phenylmethylsulphonylfluoride 0.3, pH 7.4 (buffer I), and homogenized using a glass-Teflon homogenizer (3×30 s). The homogenates were centrifuged (5 min, 1200×g, 4°C). The supernatant was poured off, diluted to 420 ml with buffer I and recentrifuged (20 min, 40,000×g, 4°C). The pellet was washed twice, resuspended in buffer II (mM): (HEPES-Na+ 5, EGTA 0.5, MgCl2 0.5, ascorbic acid 0.1, pH 7.4), homogenized, and diluted to give a protein concentration of about 1 mg ml−1 and stored at −80°C until use. Before the membranes were added to the incubation assay, they were centrifuged (20 min, 40,000×g, 4°C), resuspended in the incubation buffer (buffer II), homogenized by ultrasonication and diluted to a final protein concentration of about 0.6 mg ml−1.
A 400 μl aliquot of membranes was incubated for 60 min with [3H]-GR65630 (1 nM) or [3H]-SR141716A (3 nM) at room temperature in a final volume of 0.5 ml of a solution containing buffer II. The reaction was stopped by rapid vacuum filtration with a Brandel cell harvester through Whatman GF/C glass fibre filters presoaked with polyethyleneimine 0.5 M followed by rapid washing of the incubation tubes and filters with 10 ml ice-cold buffer II. The radioactivity retained in the filters was determined by liquid scintillation counting. Non-specific binding of [3H]-GR65630 was defined by addition of 100 μM MDL72222 and as [3H]-SR141716A binding in the presence of 3 μM CP55940. Competition studies were carried out using [3H]-GR65630 (10 nM) and up to 13 different concentrations of the unlabelled ligand under study, ranging from 1 nM up to 100 μM. Protein concentration was measured by the method of Bradford (1976) using bovine serum albumin as standard.
To test parental HEK 293 cells for the expression of endogenous CB1-/CB2-receptors, radioligand binding was performed as described above with 5 nM [3H]-CP55940 and specific binding was determined by competition with 3 μM unlabelled CP55,940.
Data analysis
The currents were digitized with an interface (Digidata 1200, Axon Instruments) and stored on an IBM 586-compatible PC. Data analysis was performed with pClampR 7 software (Axon). Graph Pad PrismR software (Graph Pad, CA, U.S.A.) was used to create graphics. The concentration-response curves for 5-HT were fitted by the Hill equation, i=cn/(cn+ECn50): i is the immediate peak current as fraction of the maximal (control) current, c is the 5-HT concentration, n is the Hill coefficient and EC50 is the 5-HT concentration inducing the half-maximal effect. This equation was analogously applied to determine the cannabinoid inhibition of the 5-HT response.
Drugs used
3-Tropanyl-3,5-dichlorobenzoate (MDL72222) was from RBI (Natick, U.S.A.). (N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbox-amide hydrochloride (SR141716A) from Sanofi (Montpellier, France); 5-hydroxytryptamine creatine sulphate (5-HT), ([2-methyl-1-propyl-1H-indol-3-yl]-1-naphthalen-ylmethanone) (JWH-015), Δ9-tetrahydrocannabinol (Δ9-THC), (S)-(−)-[2,3-dihydro-5-methyl-3-(4-morpholinyl methyl) pyrrolo [1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone-mesylate (WIN55,212-3) were obtained from Sigma (Munich, Germany); (all Z)-N-(2-hydroxyethyl)-5,8,11,14-eicosa-tetraenamide (anandamide), (−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol (CP55940) and ((R)-(+)-2,3-dihydro-5-methyl-3-(4-morpholinylmethyl) pyrrolo [1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate (WIN55,212-2) were obtained from Tocris/Biotrend (Cologne, Germany); [6-methoxy-2-(4-methoxyphenyl)benzo[b]furane-3-yl][4-cyanophenyl]methanone(LY320135) was a present from Lilly, Indianapolis, Indiana, U.S.A.; [3H](−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl) cyclohexanol ([3H]-CP55940 specific activity 101 Ci mmol−1) from NEN, Zaventem, Belgium; [3H]-(3-(5-methyl-1H-imidazol-4-yl)-1-(1-methyl-1H-indol-3-yl)-1-propanone) ([3H]-GR65630) specific activity 64.8 Ci mmol−1 was obtained from NEN DuPont (Dreieich, Germany); [3H]-(N-piperidine-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbox-amide hydrochloride ([3H]-SR141716A, specific activity 44.0 Ci nmol−1) from Amersham (Braunschweig, Germany).
Solutions
5-HT solutions were prepared daily from 50 mM aqueous stocks (stored at −20°C). Cannabinoid solutions were prepared daily from 10 mM stocks in DMSO (stored at −20°C; final DMSO concentration ⩽0.01%). MDL72222 was dissolved daily in methanesulphonic acid (final concentration ⩽0.001%). The vehicles in the concentrations used, had no effect on radioligand binding or current response.
Results
Effects of cannabinoid receptor ligands on 5-HT-induced currents
In excised outside-out patches of HEK 293 cells stably transfected with h5-HT3A receptors, 5-HT (present for 1–2 s) evoked currents in a concentration dependent manner (Figure 1; EC50=8.9 μM, Hill coefficient=1.4; n=3–6; see also Figure 2 for original traces).
Figure 1.
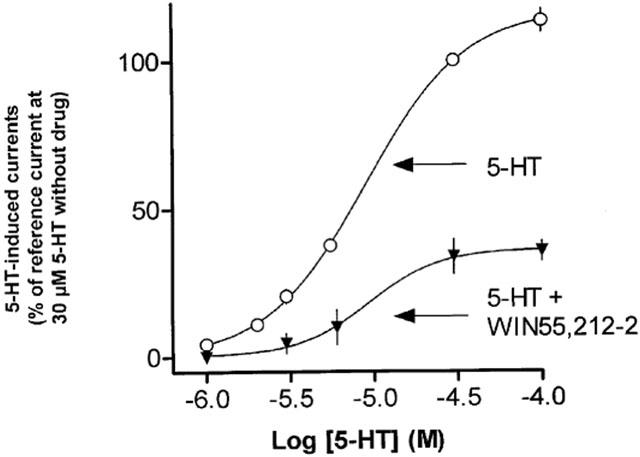
Influence of WIN55,212-2 on the 5-HT-induced currents (−100 mV holding potential) in excised outside-out patches of HEK 293 cells stably transfected with the h5-HT3A receptor cDNA. Concentration-response curve of 5-HT in the absence (open symbols) and in the presence (filled symbols) of WIN55,212-2 (300 nM; present 3 min before, during and after the 2 s 5-HT pulse). The currents in the absence and presence of the drug, respectively, are expressed as percentages of the currents evoked by the standard concentration of 30 μM 5-HT without additional drug (determined in all experiments as a reference effect). Shown are means±s.e.m. of 3–10 different patches.
Figure 2.
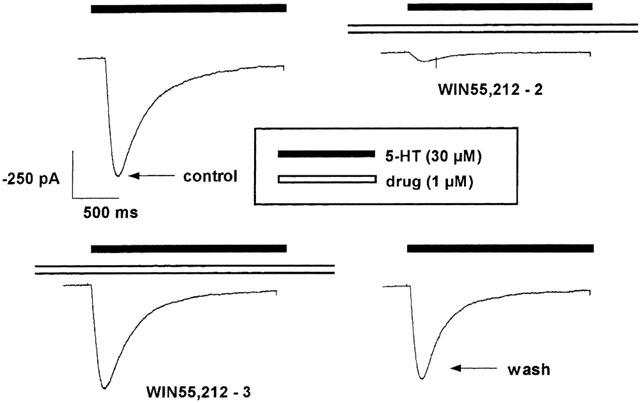
Effects of the enantiomers WIN55,212-2 (upper right panel) and WIN55,212-3 (lower left panel), both at 1 μM, on 5-HT (30 μM; −100 mV)-induced currents obtained in one outside-out patch of a HEK 293 cell stably transfected with the h5-HT3A receptor cDNA. Drugs were applied for 3 min before, during and after stimulation with 5-HT (upper right and lower left panels). 5-HT (30 μM; −100 mV)-induced currents under control conditions (i.e. absence of a further drug; upper left panel); ‘wash' refers to the control response to 30 μM 5-HT after omission of the drug for 3 min at the end of the experiment (lower right panel).
In most of the experiments in which the effects of cannabinoid receptor ligands were determined, 5-HT was applied at a standard concentration of 30 μM and the cannabinoids were present 3 min before and during (and after) the 5-HT stimulus. In experiments with all cannabinoid receptor ligands investigated (at concentrations approximately corresponding to the respective EC50 values n=3–6), this preexposure time has been shown to be sufficient to obtain an equilibrium effect (results not shown).
WIN55,212-2 inhibited the 5-HT-induced current (Figure 2). This inhibition was concentration-dependent (Figure 3a) and not voltage-dependent between −100 mV and +100 mV (n=3, not shown). The CB1 receptor antagonist SR141716A at the concentration of 1 μM did not modify the WIN55,212-2-induced inhibition (Figure 3a). In contrast to WIN55,212-2, its enantiomer WIN55,212-3 even at the high concentration of 1 μM did not change the 5-HT-evoked current (Figures 2 and 3a); at this concentration WIN55,212-2 almost abolished the 5-HT-induced current.
Figure 3.
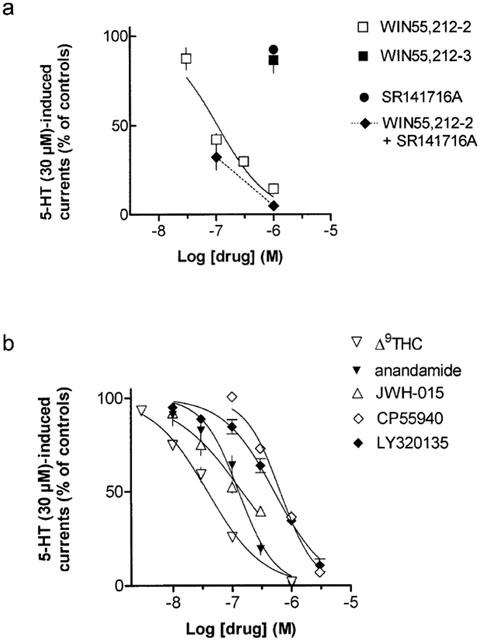
Influence of cannabinoid receptor ligands on 5-HT (30 μM; −100 mV)-induced currents in patches of HEK 293 cells stably transfected with the h5-HT3A receptor cDNA. (a) Concentration-dependent inhibition by the cannabinoid receptor agonist WIN55,212-2 in the absence and presence of the CB1 receptor antagonist SR141716A (at 1 μM) and lack of inhibition by the enantiomer WIN55,212-3 and by SR141716A alone. (b) Concentration-dependent inhibition of 5-HT (30 μM; −100 mV)-induced currents by various cannabinoid receptor ligands. Drugs were present 3 min before, during and after the 2 s 5-HT pulse. Shown are means±s.e.m. of 3–6 different patches.
The cannabinoid receptor agonists Δ9-THC, JWH-015 and CP55940, the endogenous cannabinoid receptor agonist anandamide and the CB1 receptor antagonist LY320135 shared the concentration-dependent inhibitory effect of WIN55,212-2 (Figure 3b). The IC50 values of all compounds are listed in Table 1: The rank order of potency derived from the concentration-response curves shown in Figures 3a,b was as follows Δ9-THC>WIN55,212-2⩾anandamide⩾JWH-015 >LY320135⩾CP55940.
Table 1.
Inhibition of h5-HT3A receptor-mediated currents and affinities for hCB1 and hCB2 receptors
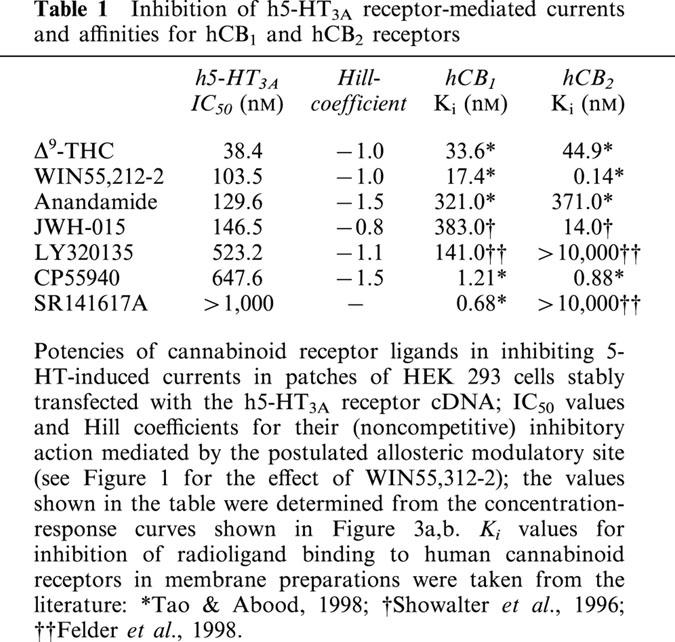
In a further series of experiments, the effect of WIN55,212-2 on the concentration-response curve of 5-HT was determined. When applied at a concentration (300 nM) three times higher than that which produced half maximal inhibition of 5-HT-induced currents through h5-HT3A receptors (compare Table 1), WIN55,212-2 did not change the EC50 value of 5-HT in stimulating currents (8.9 versus 9.3 μM), but reduced the maximum of the 5-HT concentration-response curve (Figure 1).
Finally the effect of 1 μM WIN55,212-2 on the 5-HT-evoked current was studied at different time schedules of application (Figure 4). Under the standard conditions, i.e. application of the drug 3 min before, during (and after) the 2 s stimulation with 5-HT, WIN55,212-2 produced an almost complete inhibition of current (lower left panel). Virtually the same degree of inhibition of 5-HT-induced current was obtained when the cannabinoid was present only 3 min before (and after), but not during stimulation with 5-HT (lower right panel). In contrast, application of the drug exclusively during the 2 s 5-HT pulse (upper right panel) was ineffective. Analogous results were obtained with all cannabinoids investigated (at least n=3 in each series of experiments; results not shown).
Figure 4.
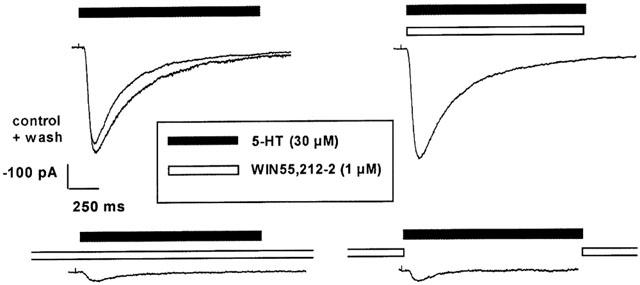
Effects of the cannabinoid receptor agonist WIN55,212-2 (1 μM) on 5-HT (30 μM, −100 mV)-induced currents obtained in one outside-out patch of a HEK 293 cell stably transfected with the h5-HT3A receptor cDNA at three different modes of drug application: exclusive co-application of the drug with 5-HT (without preexposure to the drug; upper right panel); application of the drug 3 min before, during and after stimulation with 5-HT (lower left panel); application exclusively for 3 min prior to and after, but not together with, 5-HT (lower right panel); 5-HT-induced current under control conditions (upper left panel); ‘wash' refers to the response to 30 μM 5-HT after omission of the drug for 3 min at the end of the experiment.
Radioligand binding experiments
In contrast to the clear-cut specific binding of the 5-HT3 receptor radioligand [3H]-GR65630 to membranes of transfected HEK 293 cells (Figure 5), no such binding was detectable in parental cells (n=5, not shown). In membranes of parental HEK 293 cells, we did not find any specific binding of the CB1 receptor ligand [3H]-SR141716A nor of the CB1/CB2 receptor ligand [3H]-CP55940 at concentrations of 3 and 5 nM, respectively, which are sufficient to label cannabinoid receptors (n=8 and 3, respectively; results not shown).
Figure 5.
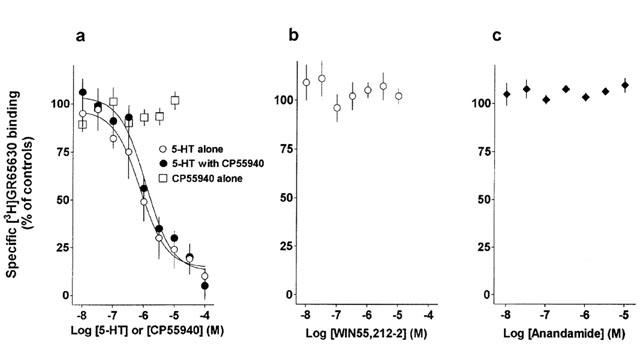
Influence of 5-HT and cannabinoid receptor agonists on specific binding of the selective 5-HT3 receptor ligand [3H-GR65630 to membranes of HEK 293 cells stably transfected with the h5-HT3A receptor cDNA. (a) Concentration-dependent inhibition by 5-HT of specific [3H-GR65630 binding in the absence (open symbols) and presence of the cannabinoid receptor agonist CP55940 and lack of inhibition by CP55940 alone. (b) No inhibition by WIN55212-2 and (c) no inhibition by anandamide. All points are means±s.e.m., n=3–6 different binding experiments.
In competition experiments, 5-HT concentration-dependently inhibited the specific binding of the selective 5-HT3A receptor ligand [3H]-GR65630 (Figure 5a), whereas the cannabinoid receptor agonists WIN55,212-2, CP55940 and anandamide (Figures 5a–c) as well as the CB1 receptor antagonist SR141716A (results not shown) did not reduce specific [3H]-GR65630 binding to membranes of HEK 293 cells stably transfected with the h5-HT3A receptor cDNA. The concentration-dependent inhibition by 5-HT of specific [3H]-GR65630 binding was not altered by CP55940 (Figure 5a). Furthermore, CP55940 (10 μM, 1 h incubation time) did also neither affect the association of [3H]-GR65630 to (n=5, compared to five controls), nor its dissociation from (n=4, compared to four controls) membranes of transfected HEK 293 cells (results not shown).
Discussion
The aim of the present study was to examine whether cannabinoid receptor ligands produce an inhibition of currents through recombinant homomeric h5-HT3A receptors by an action at the receptor itself. The possibility that cannabinoid receptor agonists might directly influence the 5-HT3 receptor was derived from an electrophysiological study on rat nodose ganglion neurones (Fan, 1995). In that investigation on whole cells, Δ9-THC, anandamide, WIN55,212-2, CP55940 and CP56667 inhibited the 5-HT-evoked current. CP55940 and CP56667 are enantiomers, the latter compound being clearly less potent (by a factor of more than 10). The anandamide-induced inhibition was noncompetitive and was not affected by nonhydrolyzable cAMP and GTP analogues. These results were compatible with an action at the 5-HT3 receptor itself, but in view of the putative presence of CB1 receptors in addition to 5-HT3 receptors in these cells, an interaction of both receptors could not be ruled out, in particular because interaction experiments with cannabinoid receptor antagonists were not performed.
The present investigation with cannabinoid receptor ligands on excised outside-out patches from HEK 293 cells expressing the human 5-HT3A receptor, which also revealed an inhibition of 5-HT-induced currents, provides an answer to this open question. The rank order of potencies of cannabinoids in inhibiting h5-HT3A receptors was clearly different from the affinities of these drugs for hCB1 or hCB2 receptors (Table 1), arguing against an involvement of the latter receptor types in this effect. This view is strongly supported by our finding that the selective CB1 receptor antagonist SR141716A (Thomas et al., 1990) did not antagonize the inhibitory effect of WIN55,212-2 on the 5-HT-evoked current (Figure 3a). In addition, the inhibition was stereoselective, which argues against the possibility that the inhibitory effect of the cannabinoids might be exclusively related to their lipophilicity, although a hydrophobic component in the mechanism of action cannot be excluded.
The effect of WIN55,212-2 on the concentration-response curve of 5-HT was typical for a non-competitive inhibitor. This makes a direct interaction with the agonist recognition site unlikely. This conclusion is supported by our radioligand binding experiments, since (1) three cannabinoid receptor agonists did not reduce the specific binding of a selective 5-HT3A receptor ligand and (2) CP55940 did not alter the concentration-dependent inhibition of specific [3H]-GR65630 binding by 5-HT.
Since, furthermore, there was no specific binding of the CB1 receptor ligand [3H]-SR141716A (Rinaldi-Carmona et al., 1996; for reviews, see Pertwee, 1997; 2000) nor of the CB1/CB2 receptor ligand [3H]-CP55940 (for reviews, see Pertwee, 1997; 2000) to membranes of HEK 293 cells, the possibility that such receptors are expressed in the cell membrane of HEK 293 cells is excluded. The most plausible alternative is that WIN55,212-2 and the other cannabinoid receptor agonists act at a particular modulatory site of the h5-HT3A receptor itself. Since the possibility that this site may modify ligand association to, or dissociation from, the 5-HT recognition site of the h5-HT3A receptor was excluded by our binding experiments, the alternative was considered that cannabinoid binding to this site may inhibit currents through the receptor pore, e.g., either by an open channel block or by an inhibitory allosteric interaction leading to a change in channel gating or closure mechanisms.
Evidence as to whether or not the drugs produce an open channel block could be derived from experiments in which the time schedule of drug application was modified and in which the voltage-dependence of the inhibitory effect of WIN55,212-2 was investigated: obviously, the site involved in the inhibition of channel function is not easily accessible, as can be derived from the rather long equilibrium period of 3 min before stimulation which is necessary to establish the effect and from the failure of the drugs to produce an inhibition when applied exclusively during, but not before, stimulation with 5-HT. However, a location in transmembrane or cytosolic domains of the receptor protein rather than within the open pore would be compatible with the necessity of the presence of the cannabinoids before stimulation at such an equilibrium time and with the lack of voltage-dependence of the WIN55,212-2-induced inhibition of 5-HT-evoked current.
The location in the transmembrane domain or its immediate proximity would make this site easily accessible to the endogenous ligand anandamide (Devane et al., 1992; Fride & Mechoulam, 1993); this compound is produced by enzymatic cleavage from membrane lipid precursors (reviewed by Piomelli et al., 2000; Porter & Felder, 2001) and it inhibits 5-HT-evoked currents with remarkable potency (Table 1). It is an attractive hypothesis to postulate that the 5-HT3 receptor is endowed with a motif which is recognized not only by all CB1 and CB2 receptor agonists, but also by the CB1 receptor antagonist LY320135 (Felder et al., 1998). This drug mimics the inhibitory effect of the cannabinoid receptor agonists at the allosteric modulatory site of the 5-HT3 receptor.
At this modulatory site which, thus, shares pharmacological properties with hCB1 and hCB2 receptors without being identical with them, the phytocannabinoid Δ9-THC acts at a potency similar to its affinity for hCB1 and hCB2 receptors, whereas synthetic cannabinoids are less potent at this site compared to their affinity for hCB1 or hCB2 or both receptors (Table 1). This property of Δ9THC is compatible with the view that the inhibition of h5-HT3A receptor function found here may contribute to the behavioural effects of this compound.
As already mentioned, the potency of the endogenous ligand anandamide at this site is rather high (Table 1). Accordingly, it is conceivable that the allosteric site of the h5-HT3A receptor may be tonically activated by anandamide and, hence may play a physiological role by mediating a regulatory effect on the functional state of the 5-HT3 receptor. The allosteric modulatory site of the 5-HT3 receptor may be considered as a target for a new class of drugs used, e.g., for the control of pain and emesis.
A prerequisite for such effects is that the cannabinoid receptor-independent inhibitory effect of cannabinoids is also operative in vivo. Evidence for this can be derived from preliminary results obtained in a study which has been performed in anaesthetized rats pretreated with the CB1 receptor antagonist SR141716A (Godlewski et al., 2002). In such animals, activation of the 5-HT3 receptors on cardiac afferent vagal nerves by bolus injection of phenylbiguanide induced the Bezold-Jarisch reflex, i.e. a decrease in heart rate. This effect was inhibited by CP55940 and WIN55,212-2 but not by WIN55,212-3, whereas the vanilloid VR1 receptor-mediated Bezold-Jarisch reflex was unaffected.
Taken together, the present patch-clamp study on excised outside-out patches of HEK 293 cells expressing recombinant homomeric 5-HT3A receptors revealed that cannabinoid receptor ligands including the phytocannabinoid Δ9THC, the endocannabinoid anandamide and the CB1 receptor antagonist LY320135 inhibit the 5-HT-induced currents. The synthetic cannabinoid WIN55,212-2, but not its enantiomer WIN55,212-3, shared the inhibitory effect. Since radioligand binding experiments excluded both the involvement of CB1 and CB2 receptors in this effect and an action of the cannabinoids at the 5-HT recognition site of the 5-HT3 receptor, the most plausible site and mechanism of action is a direct modulatory effect on the h5-HT3A receptor channel via an allosteric site.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft (Go 343/6-3 and SFB 400). We thank Zita Dorner and Marlies Hartwig for expert technical support during experiments. We are also grateful to Lilly (Indianapolis, U.S.A.) and Sanofi (Montpellier, France) for the gifts of LY320135 and SR 141716A, respectively.
Abbreviations
- Anandamide
((all Z)-N-(2-hydroxyethyl)-5,8,11,14-eicosa-tetraenamide)
- CP55940
(−)-cis -3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol
- GR65630
(3-5-methyl-1H-imidazol-4-yl)-1-(1-methyl-1H-indol-3-yl)-1-propanone)
- JWH-015
([2-methyl-1-propyl-1H-indol-3-yl]-1-naphthalen-ylmethanone)
- 5-HT
5-hydroxytryptamine creatinine sulphate
- SR141716A
(N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbox-amide hydrochloride)
- Δ9THC
Δ9-tetrahydrocannabinol; WIN55,212-2, ((R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl) pyrrolo [1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone) mesyalte
- WIN55,212-3
((S)-(−)-[2,3-Dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxaxin-6-yl]-1-naphthalenylmethanone) mesylate
References
- AAPRO M.S. 5-HT3 receptor antagonists. An overview of their present status and future potential in cancer therapy-induced emesis. Drugs. 1991;42:551–568. doi: 10.2165/00003495-199142040-00002. [DOI] [PubMed] [Google Scholar]
- ABOOD M.E., MARTIN B.R. Neurobiology of marijuana abuse. Trends Pharmacol. Sci. 1992;13:201–206. doi: 10.1016/0165-6147(92)90064-d. [DOI] [PubMed] [Google Scholar]
- AMERI A. The effects of cannabinoid on the brain. Prog. Neurobiol. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- BARANN M., MOLDERINGS G., URBAN B.W., GÖTHERT M.Inhibition by cannabinoid1 receptor agonists of currents through human 5-HT3A receptors Soc. Neurosci. 2001272126abstract [Google Scholar]
- BARANN M., MEDER W., DORNER Z., BRÜSS M., BÖNISCH H., GÖTHERT M., URBAN B.W. Recombinant human 5-HT3A receptors in outside-out patches of HEK293 cells: basic properties and barbiturate effects. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000;362:255–265. doi: 10.1007/s002100000288. [DOI] [PubMed] [Google Scholar]
- BOESS F.G., MARTIN I.L. Molecular biology of 5-HT receptors. Neuropharmacology. 1994;33:275–317. doi: 10.1016/0028-3908(94)90059-0. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- BRADY C.A., STANFORD I.M., ALI I., LIN L., WILLIAMS J.M., DUBIN A.E., HOPE A.G., BARNES N.M. Pharmacological comparison of human homomeric 5-HT3A receptors versus heteromeric 5-HT3A/3B receptors. Neuropharmacology. 2001;41:282–284. doi: 10.1016/s0028-3908(01)00074-0. [DOI] [PubMed] [Google Scholar]
- BRÜSS M., BARANN M., HAYER-ZILLGEN M., EUCKER T., GÖTHERT M., BÖNISCH H. Modified 5-HT3 receptor function by co-expression of alternatively spliced human 5-HT3A receptor isoforms. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000;362:393–401. doi: 10.1007/s002100000342. [DOI] [PubMed] [Google Scholar]
- CHEMIN J., MONTEIL A., PEREZ-REYES E., NARGEOT J., LORY P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. EMBO J. 2001;20:7033–7040. doi: 10.1093/emboj/20.24.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN C., OKAYAMA H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSTALL B., NAYLOR R.J.Neuropharmacology of 5-HT3 receptor ligands Handbook of Experimental Pharmacology 1997Heidelberg: Springer-Verlag Berlin; 409–438.eds. Baumgarten, H.G. and Göthert, M [Google Scholar]
- DARMANI N.A. Delta-9-tetrahydrocannabinol differentially suppresses cisplatin induced emesis and indices of motor function via cannabinoid CB(1) receptors in the least shrew. Pharmacol. Biochem. Behav. 2001;69:239–249. doi: 10.1016/s0091-3057(01)00531-7. [DOI] [PubMed] [Google Scholar]
- DAVIES P.A., PISTIS M., HANNA M.C., PETERS J.A., LAMBERT J.J., HALES T.G., KIRKNESS E.F. The 5-HT3B subunit is a major determinant of serotonin receptor function. Nature. 1999;397:359–363. doi: 10.1038/16941. [DOI] [PubMed] [Google Scholar]
- DEVANE W.A., HANUS L., BREUER A., PERTWEE R.G., STEVENSON L.A., GRIFFIN G., GIBSON D., MANDELBAUM A., ETINGER A., MECHOULAM R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- DUBIN A.E., HUVAR R., ANDREA M.R.D., PYATI J., ZHU J.Y., JOY K.C., WILSON S.J., GALINDO J.E., GLASS C.A., LUO L., JACKSON M.R., LOVENBERG T.W., ERLANDER M.G. The pharmacological and functional characteristics of the serotonin 5-HT3A receptor are specifically modified by a 5-HT3B receptor subunit. J. Biol. Chem. 1999;43:30799–30810. doi: 10.1074/jbc.274.43.30799. [DOI] [PubMed] [Google Scholar]
- FAN P. Cannabinoid agonists inhibit the activation of 5-HT3 receptors in rat nodose ganglion neurones. J. Neurophysiol. 1995;73:907–910. doi: 10.1152/jn.1995.73.2.907. [DOI] [PubMed] [Google Scholar]
- FELDER C.C., JOYCE K.E., BRILEY E.M., GLASS M., MACKIE K.P., FAHEY K.J., CULLINAN G.J., HUNDEN D.C., JOHNSON D.W., CHANEY M.0., KOPPEL G.A., BROWNSTEIN M. LY320135, a novel cannabinoid CB1 receptor antagonist, unmasks coupling of the CB1 receptor to stimulation of cAMP accumulation. J. Pharmacol. Exp. Ther. 1998;284:291–297. [PubMed] [Google Scholar]
- FRIDE E., MECHOULAM R. Pharmacological activity of the cannabinoid receptor agonist anandamide, a brain constituent. Eur. J. Pharmacol. 1993;231:313–314. doi: 10.1016/0014-2999(93)90468-w. [DOI] [PubMed] [Google Scholar]
- GODLEWSKI G., KWOLEK G., MALINOWSKA B., BARANN M., MOLDERINGS G.J., URBAN B.W., GÖTHERT M. Inhibitory effects of cannabinoid receptor agonists on the 5-HT3 receptor-mediated Bezold-Jarisch Reflex in rats and human 5-HT3A receptors in HEK 293 cells. Naunyn Schmiedeberg's Arch. Pharmacol. 2002;365 Suppl. 1:R28. [Google Scholar]
- HERKENHAM M., LYNN A.B., LITTLE M.D., JOHNSON M.R., MELVIN L.S., DE COSTA B.R., RICE K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARIM F., ROERIG S.C., SAPHIER D. Role of 5-hydroxytryptamine3 (5-HT3) antagonists in the prevention of emesis caused by anticancer therapy. Biochemical Pharmacol. 1996;52:685–692. doi: 10.1016/0006-2952(96)00346-2. [DOI] [PubMed] [Google Scholar]
- LIU Y., DILGER J.P. Opening rate of acetylcholine receptor channels. Biophys. J. 1991;60:424–432. doi: 10.1016/S0006-3495(91)82068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAILLEUX P., PARMENTIERT M., VANDERHAEGHEN J.J. Distribution of cannabinoid receptor messenger RNA in the human brain: an in situ hybridization histochemistry with oligonucleotides. Neurosci. Lett. 1992;143:200–204. doi: 10.1016/0304-3940(92)90265-9. [DOI] [PubMed] [Google Scholar]
- MALINOWSKA B., KWOLEK G., GÖTHERT M. Anandamide and methanandamide induce both vanilloid VR1- and cannabinoid CB1 receptor-mediated changes in heart rate and blood pressure in anaesthetized rats. Naunyln Schmiedeberg's Arch. Pharmacol. 2001;364:562–569. doi: 10.1007/s00210-001-0498-6. [DOI] [PubMed] [Google Scholar]
- MARTIN W.J., COFFIN P.O., ATTIAS E., BALINSKY M., TSOU K., WALKER J.M. Anatomical basis for cannabinoid-induced antinociception as revealed by intracerebral microinjections. Brain Res. 1999;822:237–242. doi: 10.1016/s0006-8993(98)01368-7. [DOI] [PubMed] [Google Scholar]
- MATSUDA L.A., LOLAIT S.J., BROWNSTEIN M.J., YOUNG A.C., BONNER T.I. Structure of an cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- MAINGRET F., PATEL A.J., LAZDUNSKI M., HONORE E. The endocannabinoid anandamide is a direct and selective blocker of the background K(+) channel TASK-1. EMBO J. 2001;20:47–54. doi: 10.1093/emboj/20.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIYAKE A., MOCHIZUKI S., TAKEMOTO Y., AKUZAWA S. Molecular cloning of human 5-hydroxytryptamine3 receptor: heterogeneity in distribution and function among species. Mol. Pharmacol. 1995;48:407–416. [PubMed] [Google Scholar]
- MITCHELSON F.Pharmacological agents affecting emesis Drugs 199243295–315.A Review (Part I) [DOI] [PubMed] [Google Scholar]
- MUNRO S., THOMAS K.L., ABU-SHAAR M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- NOYES R., JR, BRUNK S.F., BARAM D.A., CANTER A. Analgesic effect of delta-9-tetrahydrocannabinol. J. Clin. Pharmacol. 1975;15:139–143. doi: 10.1002/j.1552-4604.1975.tb02348.x. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G. Cannabinoid receptor ligands: clinical and neuropharmacological considerations, relevant to future drug discovery and development. Expert Opin. Investig. Drugs. 2000;9:1553–1571. doi: 10.1517/13543784.9.7.1553. [DOI] [PubMed] [Google Scholar]
- PETERS J.A., MALONE H.M., LAMBERT J.J. Recent advance in the electrophysiological characterization of 5-HT3 receptors. Trends Pharmac. Sci. 1992;13:391–397. doi: 10.1016/0165-6147(92)90119-q. [DOI] [PubMed] [Google Scholar]
- PIOMELLI D., GIUFFRIDA A., CALIGNANO A., DE FONSECA F.R. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmac. Sci. 2000;21:218–224. doi: 10.1016/s0165-6147(00)01482-6. [DOI] [PubMed] [Google Scholar]
- POLING J.S., ROGAWSKI M.A., SALEM N., JR, VICINI S. Anandamide, an endogenous cannabinoid, inhibits Shaker-related voltage-gated K+ channels. Neuropharmacology. 1996;35:983–991. doi: 10.1016/0028-3908(96)00130-x. [DOI] [PubMed] [Google Scholar]
- PORTER A.C., FELDER C.C. The endocannabinoid nervous system: unique opportunities for therapeutic intervention. Pharmacol. Ther. 2001;90:45–60. doi: 10.1016/s0163-7258(01)00130-9. [DOI] [PubMed] [Google Scholar]
- RINALDI-CARMONA M., PIALOT F., CONGY C., REDON E., BARTH F., BACHY A., BRELIÈRE J.-C., SOUBRIÈ P., LE FUR G. Characterization and distribution of binding sites for [3H]-SR 141716A, a selective brain (CB1) cannabinoid receptor antagonist, in rodent brain. Life Sci. 1996;58:1239–1247. doi: 10.1016/0024-3205(96)00085-9. [DOI] [PubMed] [Google Scholar]
- SHOWALTER V.M., COMPTON D.R., MARTIN B.R., ABOOD M.E. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J. Pharmacol. Exp. Ther. 1996;278:989–999. [PubMed] [Google Scholar]
- SIMPSON K., SPENCER C.M., MCCLELLAN K.J. Tropisetron: an update of its use in the prevention of chemotherapy-induced nausea and vomiting. Drugs. 2000;59:1297–1315. doi: 10.2165/00003495-200059060-00008. [DOI] [PubMed] [Google Scholar]
- TAO Q., ABOOD M.E. Mutation of a highly conserved aspartate residue in the second transmembrane domain of the cannabinoid receptors, CB1 and CB2, disrupts G-protein coupling. J. Pharmacol. Exp. Ther. 1998;285:651–658. [PubMed] [Google Scholar]
- THOMAS B.F., COMPTON D.R., MARTIN B.R. Characterization of the lipophilicity of natural and synthetic analogs of delta-9-tetrahydrocannabinol and its relationship to pharmacological potency. J. Pharmacol. Exp. Ther. 1990;255:624–630. [PubMed] [Google Scholar]
- TRAMER M.R., CARROLL D., CAMPBELL F.A., REYNOLDS D.J., MOORE R.A., MCQUAY H.J. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. B.M.J. 2001;323:16–21. doi: 10.1136/bmj.323.7303.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOOG O., ALSTERGEN P., LEIBUR E., KALLIKORM R., KOPP S. Immediate effects of the serotonin antagonist granisetron on temporomandibular joint pain in patients with systemic inflammatory disorders. Life Sci. 2000;68:591–602. doi: 10.1016/s0024-3205(00)00965-6. [DOI] [PubMed] [Google Scholar]
- ZYGMUNT P.M., PETERSSON J., ANDERSSON D.A., CHUANG H., SORGARD M., DI MARZO V., JULIUS D., HOGESTATT E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]