

Abstract
C-C chemokine receptor-1 (CCR1) has been implicated in mediating a variety of inflammatory conditions including multiple sclerosis and organ rejection. Although originally referred to as the MIP-1α/RANTES receptor, CCR1 is quite promiscuous and can be activated by numerous chemokines.
We used radioligand binding and [35S]-GTPγS exchange assays in membranes from a cell line transfected to express CCR1 (Ba/F3-hCCR1) to characterize a panel of chemokines (HCC-1, MIP-1α, MIP-1β, MIP-1δ, MPIF-1, MCP-2, MCP-3, and RANTES) as CCR1 ligands. In this recombinant model, these chemokines displaced 125I-MIP-1α with a wide range of potencies and, with the exception of MCP-2, acted as full agonists in stimulating [35S]-GTPγS exchange.
We then assessed the utility of HL-60 cells cultured with known differentiating agents (PMA, DMSO, dibutyryl-cAMP or retinoic acid) for investigating CCR1 pharmacology. In [35S]-GTPγS exchange assays, membranes from cells cultured with retinoic acid (4–6 days) were the most responsive to activation by MIP-1α and MPIF-1. FACS analysis and comparative pharmacology confirmed that these activities were mediated by CCR1.
Using [35S]-GTPγS exchange assays, intracellular calcium flux and/or whole cell chemotaxis assays in HL-60(Rx) cells, we validated that MIP-1α was the most potent CCR1 ligand (MIP-1α>MPIF-1>RANTES⩾MIP-1β) although the ligands differed in their efficacy as agonists. MPIF-1 was the more efficacious (MPIF-1>RANTES=MIP-1α>>MIP-1β). 125I-MIP-1β binding in Ba/F3-hCCR1 and HL-60(Rx) membranes was competitively displaced by MIP-1α, MPIF-1 and MIP-1β. The binding Ki for these chemokines with 125I-MIP-1β were essentially identical in the two membrane systems.
Lastly, MIP-1β antagonized [35S]-GTPγS exchange, Ca2+ flux and chemotaxis in HL-60(Rx) cells in response to robust agonists such as MIP-1α, RANTES and MPIF-1. Based on our results, we propose that MIP-1β could function as an endogenous inhibitor of CCR1 function.
Keywords: Chemokine receptor CCR1, MIP-1β, HL-60, [35S]-GTPγS, chemotaxis
Introduction
Chemoattractant cytokines (chemokines) are important mediators of inflammatory responses, stimulating leukocyte function via activation of G protein-coupled receptors. Different classes of chemokines have been defined by the arrangement of conserved cysteine (C) residues within the mature proteins. The C-X-C chemokines have one amino acid residue separating the first two conserved cysteine residues which are adjacent in C-C chemokines. Chemokine receptors bind multiple ligands with the C-C chemokine receptors tending to be more indiscriminate than the C-X-C receptors.
CCR1 was cloned from human promyelocytic leukaemia HL-60 cells differentiated to a monocytic phenotype with the phorbol ester, PMA (Neote et al., 1993). CCR1 has been implicated in mediating a variety of inflammatory conditions including multiple sclerosis and organ rejection (Trebst et al., 2001; Rottman et al., 2000; Horuk et al., 2001a) through the use of genetic knockouts (Rottman et al., 2000) and receptor antagonism with small molecules (Horuk et al., 2001b) or modified chemokines such as Met-RANTES (Ajuebor et al., 2001; Elsner et al., 1997). CCR1 expression overlaps with that of another C-C receptor, CCR5 on a variety of cells including monocytes and T cells although coexpression of the receptors is not absolute. For example, dendritic cells express only CCR5 while neutrophils and eosinophils express CCR1 (Rot et al., 1992; Lee et al., 2000; Sarau et al., 1997). As CCR1 was cloned in differentiated HL-60 cells, several ensuing studies on CCR1 expression and function utilized this cell model (Van Riper et al., 1994; Tiffany et al., 1998). The latter study by Tiffany et al. (1998) showed that both CCR1 and CCR3 were upregulated when HL-60 cells were differentiated into eosinophilic cells by culture with butyric acid. The authors reported extremely high numbers of binding sites for the CCR1 ligand, macrophage inflammatory protein-1α (MIP-1α/CCL3) (650,000 receptors/cell) though the binding affinity was quite low (42 nM).
Originally described as the MIP-1α/regulated upon activation, normal T cells expressed and secreted (RANTES/CCL5) receptor, CCR1 is in fact quite promiscuous. Published ligands include the MIP-1α homologue (LD78β), haemofiltrate CC chemokine 1 (HCC-1/CCL14) and its proteolytic fragment HCC-1(9–74), monocyte chemotactic protein-2 and 3 (MCP-2/CCL8, MCP-3/CCL7), MIP-1δ (CCL15) and various forms of myeloid progenitor inhibitor factor 1 (MPIF-1; CCL23) (for review see Murdoch & Finn, 2000). CCR5 is also activated by multiple ligands including RANTES, MIP-1α and macrophage inflammatory protein-1β (MIP-1β/CCL4). MIP-1β has been widely viewed as CCR5-specific (Murdoch & Finn, 2000; Ward & Westwick, 1998). Nonetheless, there are reports that MIP-1β binds to CCR1 without inducing significant receptor activation (Neote et al., 1993; Ben-Baruch et al., 1995; Sarau et al., 1997). Interestingly, while MIP-1β alone was inactive, it attenuated the proliferative effects of MIP-1α in co-incubations with myeloid progenitor cells and macrophages (Broxmeyer et al., 1991; 1993; Fahey et al., 1992). The mechanism of this inhibition was not elucidated in these studies.
Currently, it remains to be seen which of the chemokines identified as CCR1 ligands are physiologically relevant and many have neither been extensively nor comparatively described. In this paper, we define retinoic acid-differentiated HL-60 cells as a convenient model for elucidating CCR1 pharmacology. We used radioligand binding, [35S]-GTPγS exchange, intracellular calcium flux and whole cell chemotaxis to characterize an extensive panel of chemokines as to their affinity, potency and efficacy as CCR1 ligands. We observed that MIP-1β was pharmacologically very comparable to the receptor antagonist, Met-RANTES at CCR1. MIP-1β was a very weak agonist and as such could antagonize CCR1 activation by MIP-1α, MPIF-1 and RANTES.
Methods
Cells and cell culture
Murine IL-3 dependent pro-B cells Ba/F3 were maintained in RPMI 1640 medium (Life Technologies, Gaithersburg, MD, U.S.A.) supplemented with 10% foetal bovine serum, 2 mM L-glutamine, 100 μg ml−1 streptomycin, and 100 u ml−1 penicillin, 50 μM 2-mercaptoethanol and 2 μg ml−1 of recombinant mouse IL-3 (Biosource International, Camarillo, CA, U.S.A.). HL-60 cells are a human promyelocytic leukaemia cell line which can be differentiated into neutrophilic/eosinophilic/monocytic/macrophagic by altering their culture conditions (Collins, 1987). HL-60 cells were maintained in DMEM medium supplemented with 10% foetal bovine serum, 2 mM L-glutamine, 100 μg ml−1 streptomycin and 100 u ml−1 penicillin (Life Technologies, Gaithersburg, MD, U.S.A.). HL-60 cells were induced to differentiate into either macrophagic cells by culturing in the presence of 20 nM phorbol 12-myristate 13-acetate (PMA), or granulocytic cells by co-culture with 1.2% DMSO, 750 μM dibutyryl-cAMP or 1 μM 13-cis-retinoic acid (Sigma, St. Louis, MO, U.S.A.) (Breitman et al., 1980; Dufer et al., 1989).
Stable expression of recombinant CCR1 and CCR5
Full-length human CCR1 cDNA was generated from DMSO-treated HL-60 cells by PCR using oligonucleotide sequence derived from a published sequence (Genbank accession number L09230). Full-length CCR5 cDNA originally cloned from PBMC (Deng et al., 1996) was obtained from Dr Nathaniel Landau (Aaron Diamond Research Institute, New York, NY, U.S.A.). The cDNAs were each sub-cloned cloned into the mammalian expression vector pME18Sneo, a derivative of the SRα expression vector (Takebe et al., 1988) and sequenced. Ba/F3 were transfected by electroporation and stable clones selected by resistance to G418 (1 mg ml−1, Life Technologies, Gaithersburg, MD, U.S.A.).
Cell membrane preparation
Ba/F3-CCR5 membranes were prepared as previously described (Cox et al., 2001). Briefly, cells were pelleted, resuspended in a lysis buffer (10 mM HEPES, pH 7.5 and Complete® protease inhibitors (Boehringer Mannheim, Indianapolis, IN, U.S.A.) and incubated on ice for 5 min. The cells were transferred to a 4639 cell disruption bomb (Parr Instrument, Moline, IL, U.S.A.) and disrupted with 1500 psi nitrogen for 30 min on ice. Following removal of large cellular debris by centrifugation at 500×g for 5 min, cell membranes in the supernatant were pelleted by centrifugation at 100,000×g for 30 min. Membranes were resuspended in lysis buffer containing 10% sucrose and stored at −80°C.
Ba/F3-CCR1 and HL-60 cell membranes were prepared as previously described (Hipkin et al., 1997). Cells were pelleted by centrifugation, incubated in homogenization buffer (mM): Tris-HCl 10, EDTA 5, EGTA 3, pH 7.6) and 1 mM PMSF for 30 min on ice. The cells were then lysed with a Dounce homogenizer using stirrer type RZR3 polytron homogenizer (Caframo, Wiarton, Ontario, Canada) with 12 strokes at 900 r.p.m. The intact cells and nuclei were removed by centrifugation at 500×g for 5 min. The cell membranes in the supernatant were then pelleted by centrifugation at 100,000×g for 30 min. The membranes were then resuspended in glygly buffer (mM): glycylglycine 20, MgCl2 1, sucrose 250, pH 7.2), aliquoted, quick frozen and stored at −80°C. Protein concentration in membrane preparations was determined using the method of Bradford (1976).
[35S]GTPγS binding assay
The exchange of guanosine 5′-[γ-35S]-triphosphate ([35S]GTPγS, triethylammonium salt; specific activity=1250 Ci mmol−1; NEN Boston, MA, U.S.A.) was measured using a scintillation proximity assay (SPA) as previously described (Cox et al., 2001). For each assay point, 2–4 μg of membrane was preincubated for 30 min at room temperature with 300 μg wheat germ agglutinin-coated SPA beads (WGA-SPA; Amersham, Arlington Heights, IL, U.S.A.) in SPA binding buffer (mM): HEPES 50, CaCl2 1, MgCl2 5, NaCl 125, 0.002% NaN3, 1.0% BSA). The beads and membranes were transferred to a 96-well Isoplate (Wallac, Gaithersburg, MD, U.S.A.) and incubated with the indicated concentrations of guanosine 5′-diphosphate (GDP), 0.3 nM [35S]GTPγS in the presence or absence of various chemokines for 60 min at 30°C. Membrane-bound [35S]-GTPγS was measured using a 1450 Microbeta Trilux counter (Wallac, Gaithersburg, MD, U.S.A.).
Radioligand binding assay
Carrier-free 125I-MIP-1α and 125I-MIP-1β (specific activity=2200 Ci mmol−1) were obtained from New England Nuclear (Boston, MA, U.S.A.). Radioligand competition and saturation binding assays were done using SPA technology (as described above). Membranes (0.5–6 μg per assay point) in SPA binding buffer were preincubated for 30 min at room temperature with 300 μg WGA-SPA (450 μg for 6 μg membranes), transferred to a 96-well Isoplate and further incubated at room temperature with the radioligand and the indicated concentrations of chemokines for 3–6 h. Unless otherwise indicated, the incubation buffer contained 3 μM GDP and 0.3 nM GTPγS. Where indicated, a mouse anti-CCR5 monoclonal antibody (clone 45531.111; R&D Systems, Minneapolis, MN, U.S.A.) was added 30 min prior to addition of chemokines and radioligand. Ligand affinities from competition binding experiments were calculated from binding IC50 using the Cheng-Prusoff equation (Cheng & Prusoff, 1973).
Flow cytometric analysis of surface chemokine receptor expression
HL-60 cells were harvested, washed once in Dulbecco's phosphate-buffered saline (Life Technologies) containing 1% bovine serum albumin (w v−1) and 0.01% sodium azide (Sigma, St. Louis, MO, U.S.A.). Cells were incubated with normal mouse serum (to block nonspecific antibody binding) followed by saturating amounts of fluorochrome-conjugated anti-chemokine receptor antibodies (R&D Systems, Minneapolis, MN, U.S.A.) for 30 min at 4°C. Background fluorescence was determined by staining cells with irrelevant, fluorochrome-conjugated isotype control antibodies. Cells were then washed and the samples analysed with a FACSCalibur (Becton-Dickinson Immunocytometry Systems, Mountain View, CA, U.S.A.).
Chemotaxis
Chemokines were reconstituted in Dulbecco's phosphate-buffered saline (10–25 μM stock). Chemotactic responses were determined by modification (Schwarz et al., in preparation) of a protocol described by Frevert et al. (1998). The buffer for both chemokine dilution and cell resuspension consisted of HL-60 culture medium (as described above) diluted at 1 : 10 ratio into Iscove's DMEM. The assays were performed as previously described (Frevert et al., 1998) using 96-well ChemoTx microplates (NeuroProbe®, Inc., Gaithersburg, MD, U.S.A.) with a 5 μM filter as per manufacturer's instructions with 125,000 cells per well. Cells were allowed to migrate towards the indicated concentrations of chemokines for 2 h at 37°C in a humidified CO2 (5%) chamber. For agonist assays, the chemokine was present in the lower wells. For antagonist assays, the chemokine used as antagonist (e.g., MIP-1β) was present in both the upper and lower wells. The number of cells which migrated into the lower wells was quantitated using a CellTiter96® AQueous Non-Radioactive Cell Proliferation Assay kit (Promega, Madison, WI, U.S.A.) according to the manufacturer's instructions.
Calcium flux
Cells were suspended in Dulbecco's phosphate-buffered saline (2×107 ml−1) and incubated at room temperature with 3 μg ml−1 FURA-2-AM (Sigma, St. Louis, MO, U.S.A.) in the dark for 1 h. The cells were then washed and resuspended in Hank's balanced salt buffer (Life Technologies, Gaithersburg, MD, U.S.A.) with 1% foetal calf serum (v v−1). The cell suspension (4×106 cells in 200 μl) was added to 1.6 ml of pre-warmed assay buffer (Hanks' balanced salt solution with 10 mM HEPES and 1.6 mM CaCl2) in a cuvette with continuous stirring at 37°C. Chemokines were added as 10× concentrated stock solutions to reach indicated final concentrations. Calcium mobilization was measured in a Perkin-Elmer LS-50B Luminescence Fluorometer (Norwalk, CT, U.S.A.). Wavelengths for excitation 1, excitation 2, and emission are 340, 380 and 510 nm, respectively. The emission was calculated as a ratio of fluorescence intensities measured at 340 and 380 nm.
Materials
Chemokines were purchased from R&D Systems Inc. (Minneapolis, MN, U.S.A.). The Act-2 variant of MIP-1β (Cat no. 271-BME) and the 66 amino acid LD78α form of MIP-1α (Cat no. 270-LD) were used in these studies. Nonlinear regression analysis of the data and calculation of EC50 and Ki was performed using Prism 2.0c (GraphPad Software, San Diego, CA, U.S.A.). All other reagents were of the best grade available and purchased from common suppliers.
Results
CCR1 binding and activation in Ba/F3-CCR1 membranes
Our initial studies were performed using a murine ProB cell line, Ba/F3, stably transfected to express human CCR1 (Ba/F3-CCR1). To measure receptor expression and affinity, Ba/F3-CCR1 membranes were incubated in binding buffer (as described in Methods) containing the indicated concentrations of 125I-MIP-1α in the absence or presence of excess unlabelled chemokine. Receptor-bound radioligand was measured using Scintillation Proximity Assay (SPA) technology (as described in Methods). No radioligand binding was detectable in membranes from parental Ba/F3 cells (data not shown). Saturation analysis demonstrated that CCR1 was highly expressed in Ba/F3-CCR1 membranes (7.1±1.2 pmol mg−1; n=2; data not shown) and bound 125I-MIP-1α with expected high affinity (KD=32±7 pM, n=2; data not shown). A [35S]GTPγS exchange assay was developed to measure CCR1 activation using the same incubation conditions (see above) with the addition of 3 μM GDP and 0.3 nM [35S]-GTPγS. 125I-MIP-1α binding affinity decreased slightly in the presence of GDP and unlabelled GTPγS with no effect on Bmax (KD=85±35 pM, 7.8±0.4 pmol receptor mg−1; n=2; data not shown). In 125I-MIP-1α competition binding assays, MIP-1α bound with the highest affinity (Figure 1 and Table 1) followed by MPIF-1, MCP-3 and MIP-1β (Ki<20 nM). HCC-1 and MIP-1δ bound with intermediate affinity (25–50 nM). The apparent binding affinity of MCP-2 was much lower than has been reported (Gong et al., 1997) (Figure 1b; Ki>300 nM). However, MCP-2 did not inhibit 125I-MIP-1α binding to nonspecific levels (possibly due to ligand aggregation). This was also the case for RANTES (Figure 1a), which can self-aggregate at higher concentrations (Proudfoot et al., 1999). As a result, calculation of binding Ki from binding IC50 by the Cheng-Prusoff equation (Cheng & Prusoff, 1973) is not appropriate for MCP-2 and RANTES.
Figure 1.
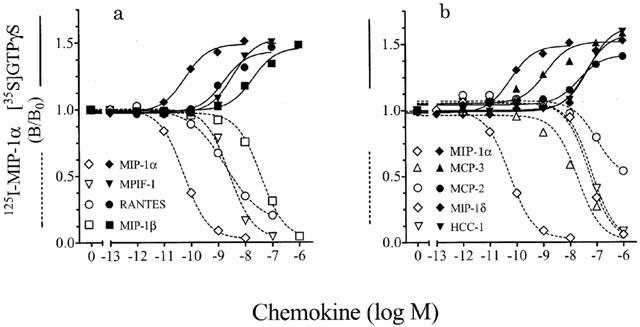
Competition and [35S]-GTPγS bindings in Ba/F3-CCR1 membranes. Membranes (2–4 μg/well) from Ba/F3-CCR1 cells were incubated in binding buffer at 30°C (as described in Methods) with the indicated concentrations of various chemokines and 50–100 pM 125I-MIP-1α, 3 μM GDP and 0.3 nM GTPγS (open symbols, broken lines) or 3 μM GDP and 0.3 nM [35S]-GTPγS (closed symbols, solid lines). Radioligand binding to the membranes was measured by WGA-SPA scintillation. Data represent the mean±s.e.mean of triplicate determinations from 2–7 independent experiments and are expressed relative to binding in the absence of chemokine (B/B0). Ligand affinities from competition bindings were calculated from binding IC50 using the Cheng-Prusoff equation.
Table 1.
Effect of chemokines on 125I-MIP-1α and [35S]-GTPγS binding in Ba/F3-CCR1 membranes

Functionally, chemokine potency in stimulating [35S]-GTPγS exchange varied considerably (Figure 1, Table 1). MIP-1α was very potent (EC50=15–25 pM) while RANTES, MPIF-1, and MCP-3 stimulated a half-maximal response in the low nanomolar range. Both MIP-1β, and MCP-2 were reasonably potent (EC50=10–20 nM) while MIP-1δ and HCC-1 were less so (EC50=39–50 nM). In this recombinant system, all the chemokines tested with the exception of MCP-2 appeared fully effective in stimulating [35S]-GTPγS binding. There was no measurable stimulation of [35S]-GTPγS binding with the chemokines examined in this study in membranes from untransfected Ba/F3 cells (data not shown).
The effect of HL-60 cell differentiation upon receptor activation with MIP-1α and MPIF-1
Distinguishing partial from full agonists in transfectants can be problematic as partial agonists can behave as full agonists with higher receptor expression (Whaley et al., 1994; Gonsiorek et al., 2000). Therefore, we attempted to identify a cell which endogenously expresses CCR1 at more relevant levels for further characterization of the more potent CCR1 ligands (MIP-1α, RANTES, MIP-1β and MPIF-1). To this end, we assessed putative CCR1 expression and functionality in differentiated HL-60 cells. HL-60 cells are promeylocytic leukaemia cells which can be differentiated into macrophagic or granulocytic cells by varying their culture conditions (Breitman et al., 1980; Dufer et al., 1989). HL-60 cells were grown in the presence or absence of 20 nM PMA for 2 days to stimulate macrophagic differentiation or with 1.2% DMSO (3 days), 750 μM dibutyryl-cAMP (3 days) or 1 μM 13-cis-retinoic acid (5 days) to encourage granulocytic differentiation. Membranes were then prepared for the initial assessment of CCR1 expression by analysis of [35S]-GTPγS exchange in response to MIP-1α and MPIF-1. As can be seen in Figure 2a, membranes from HL-60 cultured for 5 days with 1 μM retinoic acid were the most responsive to MIP-1α. As MIP-1α could activate other chemokine receptors such as CCR5, we also assessed the response to MPIF-1 which is more CCR1-selective (Nardelli et al., 1999). Stimulation with MPIF-1 was also more striking upon cell differentiation with retinoic acid (Figure 2b). The time course of retinoic acid differentiation is shown in Figure 2c. MIP-1α was ineffective in stimulating GTPγS exchange in membranes from undifferentiated cells (day 0) although a significant stimulation was evident upon 24 h differentiation. The response to MIP-1α was maximal by day 4 and maintained through day 6. In competition bindings, there was a parallel increase in 125I-MIP-1α binding in these membranes with no apparent change in binding affinity (data not shown). Saturation binding isotherms in day 5 membranes confirmed expression of high affinity binding sites for 125I-MIP-1α (Figure 3a; KD=13±4 pM, 1.08±0.02 pmol mg−1 membrane; n=2). As MIP-1β displaced 125I-MIP-1α with good affinity, we initiated competitive binding assays using the same conditions using 125I-MIP-1β in both Ba/F3-hCCR1 and HL-60(Rx) membranes. Measurable 125I-MIP-1β binding in both systems was inhibited in a concentration-dependent manner by MIP-1β, MIP-1α and MPIF-1 (Figure 3b). Chemokine binding Ki with 125I-MIP-1β were equivalent in HL-60(Rx) and Ba/F3-hCCR1 cell membranes (Ba/F3-MIP-1β=6.0±0.35 nM, MIP-1α=0.023±0.007 nM, MPIF-1=0.73±0.12 nM; HL-60(Rx)-MIP-1β=13±6 nM, MIP-1α=0.026±0.001 nM, MPIF-1=0.80±0.10 nM, n=3) and were essentially identical to their binding Ki with 125I-MIP-1α (see Table 1).
Figure 2.

Effect of MIP-1α and MPIF-1 on [35S]-GTPγS exchange in differentiated HL-60(Rx) membranes. (a, b) Membranes (2–4 μg/well) from HL-60 cells pretreated with retinoic acid, DMSO, PMA or dibutyryl-cAMP or (c) from HL-60 cells pretreated with retinoic acid for 0, 1, 4, 5 or 6 days were incubated in binding buffer containing 3 μM GDP with 0.3 nM [35S]-GTPγS and the indicated concentrations of MIP-1α (a, c) or MPIF-1 (b). Incubations were performed at 30°C for 60 min. [35S]-GTPγS binding to the membranes was measured by WGA-SPA scintillation. Data represents the mean±s.e.mean binding of triplicate determinations from a representative experiment (n=2–3) expressed relative to binding in the absence of chemokine (B/B0).
Figure 3.

Radioligand binding analysis in HL-60(Rx) membranes. (a) Membranes (1 μg/well) were incubated in binding buffer (as described in Methods) with the indicated concentrations of 125I-MIP-1α in the absence (total binding) or presence of 100 nM unlabelled MIP-1β (nonspecific binding). (b) Membranes from Ba/F3-hCCR1 (4 μg/well; closed symbols) and HL-60(Rx) cells (6 μg/well; open symbols) were incubated in binding buffer (as described in Methods) with 0.2 nM 125I-MIP-1β and the indicated concentrations of MIP-1β (squares), MIP-1α (circles) or MPIF-1 (diamonds). Radioligand binding to the membranes was measured by WGA-SPA scintillation. Data represents (a) the mean±s.e.mean specific binding of triplicate determinations from a representative experiment (n=2) or (b) the mean±s.e.mean binding of triplicate determinations from a representative experiment (n=3) expressed relative to binding in the absence of chemokine (B/B0).
Characterization of chemokine receptor expression in differentiated HL-60 cells
Based on functional and binding data, HL-60 cells differentiated with retinoic acid for 5 days (defined as HL-60(Rx)) were used in subsequent experiments. We tested the effect of MIP-1α, MPIF-1, RANTES and MIP-1β on [35S]GTPγS exchange in HL-60(Rx) membranes. The rank order of potencies (Figure 4; MIP-1α>>MPIF-1≅RANTES⩾MIP-1β) was consistent with that measured in Ba/F3-CCR1, strongly suggesting that CCR1 also mediated the response in the HL-60(Rx) membranes). Under these assay conditions, MIP-1α, MPIF-1 and RANTES were equally efficacious in stimulating [35S]-GTPγS binding in the HL-60(Rx) membranes. Contrary to the Ba/F3-CCR1 data (Figure 1), MIP-1β exhibited only partial agonism.
Figure 4.
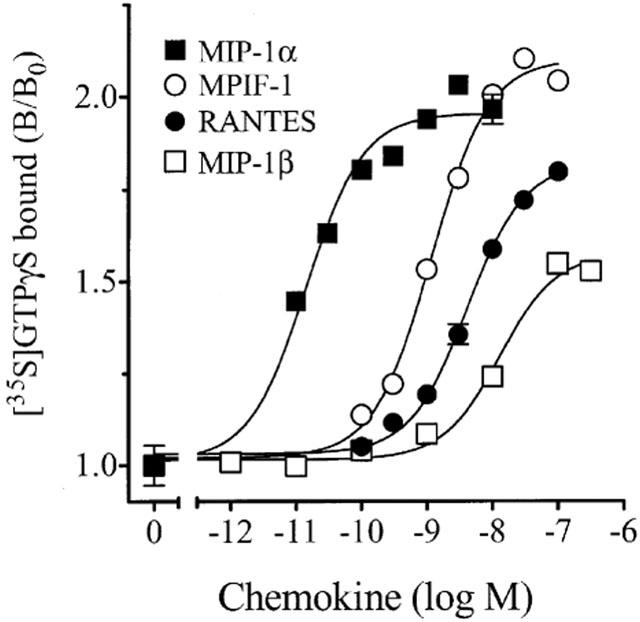
Effect of chemokines on [35S]GTPγS exchange in HL-60(Rx) membranes. Membranes (4 μg/well) from retinoic acid-pretreated HL-60 cells were incubated in binding buffer containing 3 μM GDP with 0.3 nM [35S]GTPγS and the indicated concentrations of MIP-1α, MIP-1β, RANTES or MPIF-1. Incubations were performed at 30°C for 60 min. [35S]GTPγS binding to the membranes was measured by WGA-SPA scintillation. Data represents the mean±s.e.mean binding of triplicate determinations from a representative experiment (n=2–3) expressed relative to binding in the absence of chemokine (B/B0).
MPIF-1 appears selective for CCR1 (Nardelli et al., 1999) and the rank order of chemokine potencies in the HL-60(Rx) membranes were consistent with CCR1 activation. Nonetheless, MIP-1α, MIP-1β and RANTES could activate receptors other than CCR1 such as CCR5 (Ward & Westwick, 1998; Murdoch & Finn, 2000). Consequently, we initiated flow cytometric and functional analysis to determine chemokine receptor expression on HL-60(Rx) cells. Using chemokine receptor antibodies, we found that undifferentiated HL-60 cells had no discernable expression of CCR1 (Figure 5, top left) consistent with our observation that these cells do not respond to MIP-1α (Figure 2c). There was no demonstrable surface expression of CCR6 and a small amount of CCR2 expression (data not shown). There is little, if any, detectable CCR5 expression (Figure 5, bottom left). Following differentiation with retinoic acid, there was a selective upregulation of CCR1 expression (Figure 5, top right), consistent with our functional data (Figures 2 and 4). Again, CCR6 expression was not evident whilst CCR2 levels remained constant (data not shown). CCR5 expression remained negligible (Figure 5, bottom right). Functionally, MCP-1 (CCL2) and eotaxin (CCL11) were ineffective in stimulating [35S]-GTPγS exchange in the HL-60(Rx) membranes (data not shown) precluding the involvement of CCR2 and CCR3. IL-8 (CXCL8) and GRO-α (CXCL1) stimulated [35S]-GTPγS binding (data not shown) suggesting the expression of CXCR1 and CXCR2, consistent with a granulocytic phenotype.
Figure 5.

Fluorescence-activated cell sorting for chemokine receptor expression on HL-60(Rx) cells. CCR1 (top) and CCR5 (bottom) expression on undifferentiated (left) and retinoic acid-differentiated (right) HL60 cells.
Although HL-60(Rx) cells seemed devoid of surface CCR5 expression (Figure 5), we wanted to categorically exclude CCR5 participation in the stimulation of cellular activation by MIP-1α, RANTES and MIP-1β in these cells. We compared the chemokine pharmacology in the HL-60(Rx) cells versus that in a Ba/F3 cell line transfected to express hCCR5 (Ba/F3-CCR5). Predictably, RANTES (EC50=0.27±0.13 nM), MIP-1α (EC50=3.9±2.4 nM) and MIP-1β (EC50=5.8±2.7 nM) stimulated [35S]-GTPγS exchange in Ba/F3-CCR5 membranes (n=2, Figure 6). In contrast to the response in the HL-60(Rx) membranes, MIP-1β was the most efficacious agonist in Ba/F3-CCR5 membranes, RANTES (rather than MIP-1α) was the most potent and MPIF-1 was inactive. Thus, chemokine potency and efficacy in HL-60(Rx) membranes is consistent with the pharmacology of CCR1 but not CCR5. Nevertheless, we assessed the effect of a neutralizing anti-CCR5 monoclonal antibody (α-CCR5; clone 45531.111) on [35S]-GTPγS exchange in the HL-60(Rx) and Ba/F3-CCR5 membranes. In Ba/F3-CCR5 membranes, α-CCR5 inhibited [35S]-GTPγS exchange stimulated by MIP-1β (Figure 7), RANTES and MIP-1α; (data not shown). Co-incubation of HL-60(Rx) membranes with up to 50 μg ml−1 α-CCR5 had no measurable effect on the stimulation of [35S]-GTPγS exchange by MIP-1α, MIP-1β, MPIF-1 (Figure 7) or RANTES (data not shown). Analogous studies using a neutralizing anti-CCR1 antibody would be the more direct approach to test the involvement of CCR1 in mediating the functional response to these chemokines. Unfortunately, thorough review of the literature and following our own studies (data not shown), we concluded that a CCR1-neutralizing antibody is currently unavailable.
Figure 6.

Chemokine stimulation of [35S]-GTPγS exchange in HL-60(Rx) and Ba/F3-CCR5 membranes. Membranes (3 μg/well) from retinoic acid-pretreated HL-60 (open symbols) or Ba/F3-CCR5 cells (closed symbols) were incubated in binding buffer containing 3 μM GDP and 0.3 nM [35S]-GTPγS in the presence or absence of MIP-1α (circles), MIP-1β (squares), RANTES (triangles) or MPIF-1 (diamonds). Incubations were performed at 30°C for 60 min. [35S]-GTPγS binding to the membranes was measured by WGA-SPA scintillation. Data represents the mean±s.e.mean binding of triplicate determinations from a representative experiment (n=2) expressed relative to binding in the absence of chemokine (B/B0).
Figure 7.

Effect of anti-CCR5 antibody on [35S]-GTPγS exchange in Ba/F3-CCR5 and HL-60(Rx) membranes. Membranes (2–4 μg/point) from Ba/F3-CCR5 (open symbol) or retinoic acid-pretreated HL-60 cells (closed symbol) were incubated with MIP-1β (10 nM, Ba/F3-CCR5; 30 nM, HL-60), 1 nM MIP-1α or 10 nM MPIF-1 and the indicated concentrations of an anti-CCR5 monoclonal antibody (α-CCR5). All incubations were at 30°C for 60 min in binding buffer with 3 μM GDP, 0.3 nM [35S]-GTPγS. [35S]-GTPγS binding to the membranes was measured by WGA-SPA scintillation. Data represent the mean±s.e.mean specific binding of triplicate determinations from a representative experiment (n=2) expressed relative to binding in the absence of antibody (B/B0).
Taken together, the data exclude CCR5 and implicate CCR1 as mediating the stimulation of [35S]-GTPγS exchange by MIP-1α, MIP-1β, RANTES and MPIF-1 in HL-60(Rx) cells.
Characterization of CCR1 pharmacology in HL-60(Rx) cell membranes
Breivogel et al. (1998) demonstrated that relative to full agonists, the efficacy of partial agonists at the cannabinoid CB1 receptor to stimulate [35S]-GTPγS exchange was more susceptible to inhibition with elevated GDP concentrations. As can be seen in Figure 8, receptor activation stimulated by 100 nM MIP-1β (expressed as fold of basal) plateaued at 3 μM GDP and declined as the GDP concentration increased. The efficacy of 100 nM RANTES and 10 nM MPIF-1 to stimulate [35S]-GTPγS exchange were roughly equivalent up to 3 μM GDP whereupon the greater efficacy of MPIF-1 became apparent. The response to both RANTES and MPIF-1 plateaued at 30 μM GDP and then declined. From these data, we initiated competition bindings with 125I-MIP-1α and repeated the [35S]-GTPγS binding assays in HL-60(Rx) membranes (see Figure 4) in the presence of 30 μM GDP The more potent chemokines identified in the Ba/F3-CCR1 studies were tested (MIP-1α, MIP-1β, RANTES, MCP-3 and MPIF-1). We also characterized methionylated RANTES (Met-RANTES) which has been used in vivo as a CCR1/CCR3 antagonist (Proudfoot et al., 1999). As was the case in the Ba/F3-CCR1 membranes, the addition of GDP and GTPγS reduced the binding affinity of 125I-MIP-1α without inhibiting total binding (KD=175±55 pM; Bmax=1026±74 fmol mg−1, n=2; data not shown). In the presence of 30 μM GDP, the binding affinity of the chemokines in HL-60(Rx) membranes (see Table 2; Figure 9a,b) was lower than that measured in Ba/F3-CCR1 in the presence of 3 μM GDP (see Table 1). This decrease in binding affinities can be attributed to the increased GDP concentration as co-incubation with GDP decreased the chemokine binding affinity in competition binding assays with 125I-MIP-1α and 125I-MIP-1β in both Ba/F3-CCR1 and HL-60(Rx) membranes in a concentration-dependent manner (data not shown). The RANTES binding Ki was not assessed (see above). It should be noted that the Met-RANTES Ki was considerably lower than previously reported (Proudfoot et al., 1999). Therefore, we tested it in parallel competition bindings using Ba/F3-CCR1 membranes and found that it displaced 125I-MIP-1α with the same apparent low affinity (Figure 9b).
Figure 8.
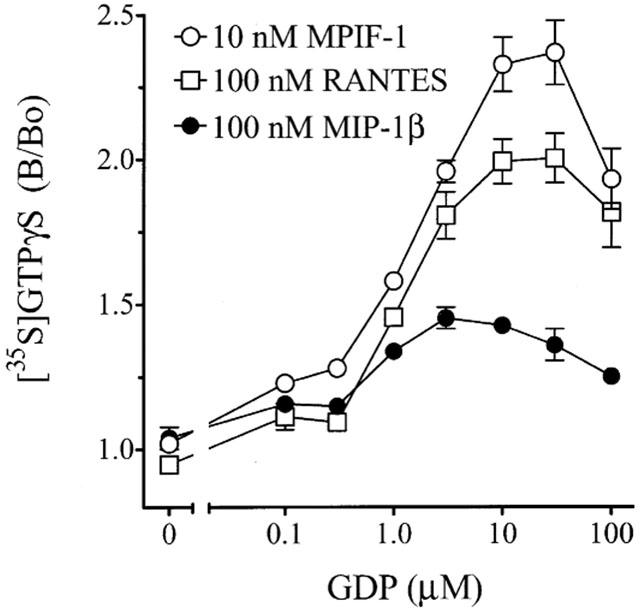
Effect of GDP concentration on chemokine-stimulated [35S]-GTPγS exchange in HL-60(Rx) membranes. Membranes (3 μg/well) from retinoic acid-pretreated HL-60 were incubated in binding buffer containing 0.3 nM [35S]-GTPγS and the indicated concentration of GDP in the presence or absence of 100 nM MIP-1β, 100 nM RANTES or 10 nM MPIF-1. Data represent the mean±range specific binding of triplicate determinations from duplicate experiments expressed relative to binding in the absence of GDP (B/B0).
Table 2.
Effect of chemokines on 125I-MIP-1α and [35S]-GTPγS binding in HL-60(Rx) membranes

Figure 9.

Effect of chemokines on 125I-MIP-1α binding and [35S]-GTPγS exchange. Membranes (2–3 μg/well) from retinoic acid-pretreated HL-60 or Ba/F3-CCR1 cells were incubated in binding buffer containing 0.3 nM [35S]-GTPγS (closed symbols, solid lines) or 200 pM 125I-MIP-1α (open symbols, broken lines), 30 μM GDP and the indicated concentrations of (a) MIP-1α (circles), MPIF-1 (diamonds), RANTES (triangles) or MIP-1β (squares or (b) MPIF-1 (diamonds), MCP-3 (squares) or Met-RANTES (circles or triangle in Ba/F3-CCR1). Incubations were performed at 30°C for 60 min. Radioligand binding to the membranes was measured by WGA-SPA scintillation. Data (expressed as B/B0) represent the mean±range specific binding of triplicate determinations from a representative experiment (n=2–4) expressed relative to binding in the absence of chemokine (B/B0).
Functionally, MIP-1α was again the most potent chemokine tested in stimulating [35S]-GTPγS (MIP-1α>MPIF-1≅RANTES>MCP-3=MIP-1β>>Met-RANTES; Figure 9, Table 2). MPIF-1 was slightly but consistently more efficacious in stimulating [35S]-GTPγS than the other chemokines and therefore would be characterized as a full agonist (Figure 9; MPIF-1>MIP-1α=RANTES=MCP-3>>Met-RANTES⩾MIP-1β).
Effect of MIP-1β on chemokine-stimulated [35S]-GTPγS exchange, Ca2+ flux and chemotaxis in HL-60(Rx) cells
We tested the effect of MIP-1β on MIP-1α- and MPIF-1-stimulated [35S]-GTPγS exchange in HL-60(Rx) membranes. Membranes were incubated with 30 μM GDP and increasing concentrations of MIP-1α or MPIF-1 in the presence or absence of 300 nM MIP-1β. MIP-1β decreased the potency of both MPIF-1 (Control; 4.4±0.05 nM; MIP-1β-8.2±0.8 nM) and especially MIP-1α (Control-47±1 pM; MIP-1β-5.2±1.5 nM) to stimulate GTPγS exchange through CCR1 without altering the maximal response (data not shown; n=2).
We next assessed the effect of chemokines alone or in combination, on Ca2+ mobilization in HL-60(Rx) cells. Cells were preloaded with the FURA-2 for 60 min, washed and incubated with the indicated concentrations of chemokines. As was the case in the [35S]-GTPγS exchange assays (Figure 9a), MIP-1α and MPIF-1 stimulated an increase in intracellular Ca2+ in a dose-responsive manner (EC50=0.1–0.3 and 3–10 nM, respectively) (Figure 10a,b) while MIP-1β was all but inactive (Figure 10c). In some experiments, a small Ca2+ flux was elicited with 100 nM MIP-1β (data not shown and Figure 10d) consistent with the weak agonism defined in [35S]-GTPγS exchange assays (Figure 9). RANTES also stimulated an increase in intracellular Ca2+ in a concentration-dependent manner (data not shown). Chemokine potency in stimulating Ca2+ flux mirrored that seen in [35S]-GTPγS exchange assays (MIP-1α>MPIF-1≅RANTES). As MIP-1β bound CCR1 without significant receptor activation, we tested if MIP-1β would antagonize CCR1 activation by more effective agonists. FURA-2-loaded HL-60(Rx) cells were exposed to MIP-1β (3–100 nM) for 60 s prior to stimulation with 0.1 nM MIP-1α (≈EC50; Figure 10d). MIP-1β suppressed the Ca2+ flux in response to MIP-1α in a concentration-dependent manner with an EC50=10–30 nM. Ca2+ flux stimulated by 3 nM RANTES or MPIF-1 were similarly reduced by MIP-1β (data not shown). In co-incubations with MIP-1α, MIP-1β blunted the cellular response to MIP-1α (Figure 10e) albeit to a lesser extent than that seen in the pretreatment paradigm (Figure 10d).
Figure 10.

Effect of chemokines on intracellular calcium in HL-60(Rx) cells. Retinoic acid-pretreated HL-60 cells were incubated with FURA 2-AM for 60 min, washed, incubated at 37°C and stimulated with the indicated concentrations of MIP-1α (a), MPIF-1 (b) or MIP-1β (c) or pre-incubated with the indicated concentrations of MIP-1β followed by 0.1 nM MIP-1α (d) or co-incubated with the indicated concentrations of MIP-1β and 0.1 nM MIP-1α (e). Chemokine additions are marked with an arrowhead. Data are of two independent experiments. The fluorescence was monitored as ratio of fluorescence intensities measured at 340 and 380 nm.
We assessed the stability of MIP-1β, MIP-1α and MPIF-1 to stimulate HL-60 (Rx) cell chemotaxis. HL-60(Rx) cells were placed in the top chamber of a 96-well chemotaxis plate and incubated for 2 h at 37°C with the indicated concentrations of chemokine in the bottom chamber. Cells which migrated through a 5 μM filter into the bottom well were quantitated by MTS uptake (as described in Methods). Cells migrated to the chemokines with classic bell-shaped response curve. MIP-1α was again the most potent chemoattractant (EC50=0.1 nM vs MPIF-1 EC50=10–30 nM), MPIF-1 was the most efficacious and MIP-1β was inactive (Figure 11a). To see if MIP-1β would also antagonize chemotaxis, cell migration towards MIP-1α (0, 0.1 or 0.3 nM) was assessed in the absence or presence of the indicated concentrations of MIP-1β (Figure 11b). Consistent with the Ca2+ flux experiments (Figure 10d,e), MIP-1β attenuated MIP-1α-stimulated chemotaxis in a concentration-dependent manner. Cell movement to 0.1 nM MIP-1α was inhibited approximately 50% with 100 nM MIP-1β (⩾3 fold MIP-1β Ki; Table 2). With greater receptor occupancy (300–1000 nM=10–30 fold MIP-1β; Ki; Table 2), MIP-1β completely inhibited chemotaxis to MIP-1α. As was the case in the GTPγS exchange assay with Ba/F3-hCCR1 membranes (Figure 1), MIP-1β was an effective agonist in stimulating chemotaxis of the Ba/F3-hCCR1 cells (data not shown). Therefore, experiments analogous to those shown in Figure 11b were not undertaken using the transfected cell line.
Figure 11.

Chemokine stimulation of HL-60(Rx) cell chemotaxis. Retinoic acid-pretreated HL-60 cells in the upper wells of a 96-well chemotaxis apparatus were allowed to migrate towards (a) the indicated concentrations of MIP-1β, MIP-1α or MPIF-1 or (b) 0, 0.1 or 0.3 nM MIP-1α in the absence or presence of the indicated concentrations of MIP-1β. After 2 h at 37°C, the cells in the bottom chamber were quantified. Data (expressed in absorbance units at 490 nm) represent duplicate determinations from a representative experiment (n=2).
Discussion
The studies presented herein characterize chemokine activation of hCCR1 expressed both in transfected cells and endogenously in differentiated HL-60 cells. MIP-1α, MPIF-1, RANTES, MCP-3 and MIP-1β were potent ligands of CCR1 although their efficacy as agonists varied greatly. MIP-1β was a very weak agonist which behaved very similarly to the CCR1 agonists, methionylated hRANTES (Met-RANTES; Proudfoot et al., 1996; 1999; Elsner et al., 1997; Grone et al., 1999; Ajuebor et al., 2001). Concentrations of MIP-1β corresponding to 3–30 fold its CCR1 Ki antagonized the activation of the receptor by more efficacious chemokines such as MIP-1α, RANTES or MPIF-1.
For the most part, our studies were carried out using HL-60 cells differentiated with retinoic acid (HL-60(Rx)). Eosinophilic differentiation by culture with butyric acid was shown to induce both CCR1 and CCR3 expression in HL-60 cells (Tiffany et al., 1998). Retinoic acid induces a more general granulocytic differentiation with little if any functional CCR3 expression (see above). Butyric acid differentiation stimulated very high CCR1 expression (650,000 receptors cell−1=5–6 pmol mg−1 assuming 106 cells≈200 μg membrane protein; Tiffany et al., 1998). This level of expression rivaled that in our Ba/F3-CCR1 cells (7 pmol mg−1; Figure 3) and exceeded CCR1 expression measured in the HL-60(Rx) cells (1 pmol mg−1). In the Ba/F3-CCR1 membranes, all the chemokine tested which bound with any affinity at CCR1 acted as a full agonist in stimulating [35S]-GTPγS exchange and effectively stimulated chemotaxis in whole cells (data not shown). However, with decreased endogenous receptor expression in the HL-60(Rx) cells, it was possible to fully delineate the chemokine pharmacology at CCR1 ligands both in terms of potency and efficacy. We found that MPIF-1 rather than MIP-1α or RANTES was the most efficacious agonist although MIP-1α was 25–55 fold more potent. Other chemokines were as efficacious as MIP-1α but considerably less potent (i.e. RANTES and MCP-3). The observation that MPIF-1 was a full agonist bolsters the finding by MacPhee et al. (1998) that MPIF-1 was more effective than MIP-1α in stimulating chemotaxis and Ca2+ flux in human eosinophils (MacPhee et al., 1998). Recently, a truncated form of MPIF-1 (MPIF-1(24–99)) was reported to be considerably more active than the long form (MPIF-1(1–99)) used in our studies (Berkhout et al., 2000). Experiments on the pharmacology of MPIF-1(24–99) are ongoing.
In the literature, MIP-1β is often cited as a CCR5-specific chemokine (Murdoch & Finn, 2000; Ward & Westwick, 1998) although MIP-1β binding to CCR1 was noted in other studies (Neote et al., 1993; Ben-Baruch et al., 1995; Sarau et al., 1997). Indeed, we found that both MIP-1β and 125I-MIP-1β bound CCR1 with a higher or similar affinity than did a number of more recognized CCR1 ligands such as MCP-3, MIP-1δ or HCC-1. That being said, the affinity MIP-1β for CCR1 was 5–10 fold lower than what is reported for CCR5 (1–2 nM; (Raport et al., 1996; Blanpain et al., 2000)). In [35S]GTPγS exchange assays in HL-60(Rx) membranes, MIP-1β was a very weak agonist and was essentially inactive in stimulating calcium signalling or chemotaxis in whole cells. As such, MIP-1β acted as a CCR1 antagonist in blocking Ca2+ flux or cell migration in response to the more effective CCR1 ligands. In calcium flux experiments, the stimulation of intracellular Ca2+ in response to 0.1 nM MIP-1α (≅EC50) in the HL-60 cells was significantly blunted with pre-exposure to 10–30 nM MIP-1β and completely blocked with 100 nM MIP-1β. These concentrations of MIP-1β are quite high relative to the effective concentrations of MIP-1α and MPIF-1 required to activate CCR1 and it is not known if these levels of MIP-1β are physiologically relevant. Nevertheless, these concentrations of MIP-1β are in the range required to maximally activate CCR5 signalling pathways including Ca2+ flux (Blanpain et al., 2000), tyrosine kinase activation (Ganju et al., 1998) and stimulation of GTPγS exchange (Figure 6).
Chemokine receptor antagonism by chemokines has been described before. For example, MCP-3 was proposed to function as a CCR5 antagonist (Blanpain et al., 1999) and Martinelli et al. (2001) showed that eotaxin (CCL11) was weak agonist at CCR2b and as such antagonized cell chemotaxis to 1 nM MCP-1 (CCL2). In that study, significant antagonism was seen with 1 nM eotaxin. This result was surprising as receptor occupancy by eotaxin based on its affinity for CCR2 (8–12 nM) would be relatively low. The CXCR3 ligands, IP-10, Mig and I-TAC, were characterized as antagonists at CCR3 (Loetscher et al., 2000). I-TAC bound CCR3 with the highest affinity of the three (Ki=70–80 nM) and inhibited eotaxin-stimulated Ca2+ flux and chemotaxis, albeit with elevated concentrations (100–1000 nM). Again, the chemokine levels required to block CCR3 activation are very high but appropriate given the affinity of these ligands for the receptor. Indeed, high levels of certain chemokines are required for activation of their cognate receptors. Low micromolar concentrations of BCA-1 (CXCL13) and Mig (CXCL9) are required to maximally activate CXCR5 and CXCR3, respectively (Legler et al., 1998; Weng et al., 1998; Trentin et al., 1999) while chemotaxis of cells expressing CCR10 was maximal with 300 nM CTACK (CCL27) and VIC (CCL28) (Pan et al., 2000).
If chemokine receptor antagonism by chemokines is possible in situ, is it physiologically relevant? The high concentrations of MIP-1β required to block CCR1 activation indicates that interference in receptor signalling could only occur in close proximity to the inflammatory site. This would be the only location at which MIP-1β expression could be high enough to be functionally relevant as a CCR1 antagonist. Moreover, MIP-1β antagonism of CCR1-mediated chemotaxis could only be pertinent in cells that do not co-express CCR5 such as activated neutrophils (Bonecchi et al., 1999; Lee et al., 2000; Cheng et al., 2001; Combadiere et al., 1996), eosinophils and natural killer cells (Rot et al., 1992; Combadiere et al., 1996; Inngjerdingen et al., 2001). Functionally, these cells respond to MIP-1α but not to MIP-1β (Cheng et al., 2001; Maghazachi et al., 1994). Macrophages and myeloid progenitor cells, express CCR1 and possibly CCR5 although they are relatively unresponsive to MIP-1β. Broxmeyer et al. (1991) showed that MIP-1α inhibited myeloid progenitor cell proliferation. MIP-1β had no effect alone but suppressed the effects of MIP-1α. In analogous studies, Fahey et al. (1992) demonstrated that mMIP-1β alone was ineffective in stimulating tumour necrosis factor (TNF) secretion by macrophages but attenuated the stimulatory effects of MIP-1α on TNF secretion in co-incubations. In these prior studies, the mechanism(s) by which MIP-1β moderated the actions of MIP-1α were not resolved.
In conclusion, we characterized a differentiation protocol for HL-60 myeloid cells as a convenient and prototypical model for defining the pharmacology of endogenously expressed hCCR1. We classify MPIF-1 as a full agonist on CCR1 and MIP-1β as a potent but weak agonist at this receptor. As such, MIP-1β could inhibit Ca2+ flux and chemotaxis in HL-60(Rx) cells in response to MIP-1α, RANTES and MPIF-1. Based on our results, we propose that MIP-1β may have the capacity to serve as an endogenous inhibitor of CCR1 function in certain inflammatory settings.
Acknowledgments
We would like to thank Drs Joe Hedrick and Phillip Murphy for their helpful discussions on these studies.
Abbreviations
- ENA-78
endothelial-cell derived neutrophil activating protein 78
- GRO-α
growth related oncogene alpha
- HCC-1
haemofiltrate CC chemokine 1
- IP-10
interferon-inducible protein of 10 kDa
- I-TAC
interferon-inducible T cell α chemoattractant
- IL-8
interleukin-8
- MIP-1α
macrophage inflammatory protein-1α
- MIP-1β
macrophage inflammatory protein-1β
- MIP-1δ
macrophage inflammatory protein-1δ
- MCP-1, MCP-2, MCP-3
monocyte chemotactic protein-1, -2 and -3
- Met-RANTES
methionylated RANTES
- Mig
monokine-induced by human interferon γ
- MPIF-1
myeloid progenitor inhibitor factor-1
- NAP-2
neutrophil activating peptide-2
- RANTES
regulated upon activation, normal T cell expressed and secreted
- WGA-SPA
wheat germ agglutinin bead-scintillation proximity assay
References
- AJUEBOR M.N., HOGABOAM C.M., KUNKEL S.L., PROUDFOOT A.E., WALLACE J.E. The chemokine RANTES is a crucial mediator of the progression from acute to chronic colitis in the rat. J. Immunol. 2001;166:552–558. doi: 10.4049/jimmunol.166.1.552. [DOI] [PubMed] [Google Scholar]
- BEN-BARUCH A., XU L., YOUNG P.R., BENGALI K., OPPENHEIM J.J., WANG J.M. Monocyte chemotactic protein-3 (MCP3) interacts with multiple leukocyte receptors. C-C CKR1, a receptor for macrophage inflammatory protein-1 alpha/Rantes, is also a functional receptor for MCP3. J. Biol. Chem. 1995;270:22123–22128. doi: 10.1074/jbc.270.38.22123. [DOI] [PubMed] [Google Scholar]
- BERKHOUT T.A., GOHIL J., GONZALEZ P., NICOLS C.L., MOORES K.E., MACPHEE C.H., WHITE J.R., GROOT P.H. Selective binding of the truncated form of the chemokine CKbeta8 (25–99) to CC chemokine receptor 1 (CCR1) Biochem. Pharmacol. 2000;59:591–596. doi: 10.1016/s0006-2952(99)00354-8. [DOI] [PubMed] [Google Scholar]
- BLANPAIN C., LEE B., TACKOEN M., PUFFER B., BOOM A., LIBERT F., SHARRON M., WITTAMER V., VASSART G., DOMS R.W., PARMENTIER M. Multiple nonfunctional alleles of CCR5 are frequent in various human populations. Blood. 2000;96:1638–1645. [PubMed] [Google Scholar]
- BLANPAIN C., MIGEOTTE I., LEE B., VAKILI J., DORANZ B.J., GOVAERTS C., VASSART G., DOMS R.W., PARMENTIER M. CCR5 binds multiple CC-chemokines: MCP-3 acts as a natural antagonist. Blood. 1999;94:1899–1905. [PubMed] [Google Scholar]
- BONECCHI R., POLENTARUTTI N., LUINI W., BORSATTI A., BERNASCONI S., LOCATI M., POWER C., PROUDFOOT A., WELLS T.N., MACKAY C., MANTOVANI A., SOZZANI S. Up-regulation of CCR1 and CCR3 and induction of chemotaxis to CC chemokines by IFN-gamma in human neutrophils. J. Immunol. 1999;162:474–479. [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- BREITMAN T.R., SELONICK S.E., COLLINS S.J. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. U.S.A. 1980;77:2936–2940. doi: 10.1073/pnas.77.5.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BREIVOGEL C.S., SELLEY D.E., CHILDERS S.R. Cannabinoid receptor agonist efficacy for stimulating [35S]GTPgammaS binding to rat cerebellar membranes correlates with agonist-induced decreases in GDP affinity. J. Biol. Chem. 1998;273:16865–16873. doi: 10.1074/jbc.273.27.16865. [DOI] [PubMed] [Google Scholar]
- BROXMEYER H.E., SHERRY B., COOPER S., LU L., MAZE R., BECKMANN M.P., CERAMI A., RALPH P. Comparative analysis of the human macrophage inflammatory protein family of cytokines (chemokines) on proliferation of human myeloid progenitor cells. Interacting effects involving suppression, synergistic suppression, and blocking of suppression. J. Immunol. 1993;150:3448–3458. [PubMed] [Google Scholar]
- BROXMEYER H.E., SHERRY B., COOPER S., RUSCETTI F.W., WILLIAMS D.E., AROSIO P., KWON B.S., CERAMI A. Macrophage inflammatory protein (MIP)-1 beta abrogates the capacity of MIP-1 alpha to suppress myeloid progenitor cell growth. J. Immunol. 1991;147:2586–2594. [PubMed] [Google Scholar]
- CHENG S.S., LAI J.J., LUKACS N.W., KUNKEL S.L. Granulocyte-macrophage colony stimulating factor up-regulates CCR1 in human neutrophils. J. Immunol. 2001;166:1178–1184. doi: 10.4049/jimmunol.166.2.1178. [DOI] [PubMed] [Google Scholar]
- CHENG Y., PRUSOFF W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- COLLINS S.J. The HL-60 promyelocytic leukemia cell line: proliferation, differentiation, and cellular oncogene expression. Blood. 1987;70:1233–1244. [PubMed] [Google Scholar]
- COMBADIERE C., AHUJA S.K., TIFFANY H.L., MURPHY P.M. Cloning and functional expression of CC CKR5, a human monocyte CC chemokine receptor selective for MIP-1(alpha), MIP-1(beta), and RANTES. J. Leukoc. Biol. 1996;60:147–152. doi: 10.1002/jlb.60.1.147. [DOI] [PubMed] [Google Scholar]
- COX M.A., JENH C.H., GONSIOREK W., FINE J., NARULA S.K., ZAVODNY P.J., HIPKIN R.W. Human interferon-inducible 10-kDa protein and human interferon-inducible T cell alpha chemoattractant are allotopic ligands for human CXCR3: differential binding to receptor states. Mol. Pharmacol. 2001;59:707–715. doi: 10.1124/mol.59.4.707. [DOI] [PubMed] [Google Scholar]
- DENG H., LIU R., ELLMEIER W., CHOE S., UNUTMAZ D., BURKHART M., DI MARZIO P., MARMON S., SUTTON R.E., HILL C.M., DAVIS C.B., PEIPER S.C., SCHALL T.J., LITTMAN D.R., LANDAU N.R. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- DUFER J., BIAKOU D., JOLY P., BENOIST H., CARPENTIER Y., DESPLACES A. Quantitative morphological aspects of granulocytic differentiation induced in HL-60 cells by dimethylsulfoxide and retinoic acid. Leuk. Res. 1989;13:621–627. doi: 10.1016/0145-2126(89)90131-8. [DOI] [PubMed] [Google Scholar]
- ELSNER J., PETERING H., HOCHSTETTER R., KIMMIG D., WELLS T.N., KAPP A., PROUDFOOR A.E. The CC chemokine antagonist Met-RANTES inhibits eosinophil effector functions through the chemokine receptors CCR1 and CCR3. Eur. J. Immunol. 1997;27:2892–2898. doi: 10.1002/eji.1830271122. [DOI] [PubMed] [Google Scholar]
- FAHEY T.J., TRACEY K.J., TEKAMP-OLSON P., COUSENS L.S., JONES W.G., SHIRES G.T., CERAMI A., SHERRY B. Macrophage inflammatory protein 1 modulates macrophage function. J. Immunol. 1992;148:2764–2769. [PubMed] [Google Scholar]
- FREVERT C.W., WONG V.A., GOODMAN R.B., GOODWIN R., MARTIN T.R. Rapid fluorescence-based measurement of neutrophil migration in vitro. J. Immunol. Methods. 1998;213:41–52. doi: 10.1016/s0022-1759(98)00016-7. [DOI] [PubMed] [Google Scholar]
- GANJU R.K., DUTT P., WU L., NEWMAN W., AVRAHAM H., AVRAHAM S., GROOPMAN J.E. Beta-chemokine receptor CCR5 signals via the novel tyrosine kinase RAFTK. Blood. 1998;91:791–797. [PubMed] [Google Scholar]
- GONG X., GONG W., KUHNS D.B., BEN-BARUCH A., HOWARD O.M., WANG J.M. Monocyte chemotactic protein-2 (MCP-2) uses CCR1 and CCR2B as its functional receptors. J. Biol. Chem. 1997;272:11682–11685. doi: 10.1074/jbc.272.18.11682. [DOI] [PubMed] [Google Scholar]
- GONSIOREK W., LUNN C., FAN X., NARULA S., LUNDELL D., HIPKIN R.W. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: antagonism by anandamide. Mol. Pharmacol. 2000;57:1045–1050. [PubMed] [Google Scholar]
- GRONE H.J., WEBER C., WEBER K.S., GRONE E.F., RABELINK T., KLIER C.M., WELLS T.N., PROUDFOOD A.E., SCHLONDORFF D., NELSON P.J. Met-RANTES reduces vascular and tubular damage during acute renal transplant rejection: blocking monocyte arrest and recruitment. Faseb J. 1999;13:1371–1383. [PubMed] [Google Scholar]
- HIPKIN R.W., FRIEDMAN J., CLARK R.B., EPPLER C.M., SCHONBRUNN A. Agonist-induced desensitization, internalization, and phosphorylation of the sst2A somatostatin receptor. J. Biol. Chem. 1997;272:13869–13876. doi: 10.1074/jbc.272.21.13869. [DOI] [PubMed] [Google Scholar]
- HORUK R., CLAYBERGER C., KRENSKY A.M., WANG Z., GRONE H.J., WEBER C., WEBER K.S., NELSON P.J., MAY K., ROSSER M., DUNNING L., LIANG M., BUCKMAN B., GHANNAM A., NG H.P., ISLAM I., BAUMAN J.G., WEI G.P., MONAHAN S., XU W., SNIDER R.M., MORRISSEY M.M., HESSELGESSER J., PEREZ H.D. A non-peptide functional antagonist of the CCR1 chemokine receptor is effective in rat heart transplant rejection. J. Biol. Chem. 2001a;276:4199–4204. doi: 10.1074/jbc.M007457200. [DOI] [PubMed] [Google Scholar]
- HORUK R., SHUREY S., NG H.P., MAY K., BAUMAN J.G., ISLAM I., GHANNAM A., BUCKMAN B., WEI G.P., XU W., LIANG M., ROSSER M., DUNNING L., HESSELGESSER J., SNIDER R.M., MORRISSEY M.M., PEREZ H.D., GREEN C. CCR1-specific non-peptide antagonist: efficacy in a rabbit allograft rejection model. Immunol. Lett. 2001b;76:193–201. doi: 10.1016/s0165-2478(01)00172-9. [DOI] [PubMed] [Google Scholar]
- INNGJERDINGEN M., DAMAJ B., MAGHAZACHI A.A. Expression and regulation of chemokine receptors in human natural killer cells. Blood. 2001;97:367–375. doi: 10.1182/blood.v97.2.367. [DOI] [PubMed] [Google Scholar]
- LEE S.C., BRUMMET M.E., SHAHABUDDIN S., WOODWORTH T.G., GEORAS S.N., LEIFERMAN K.M., GILMAN S.C., STELLATO C., GLADUE R.P., SCHLEIMER R.P., BECK L.A. Cutaneous injection of human subjects with macrophage inflammatory protein-1 alpha induces significant recruitment of neutrophils and monocytes. J. Immunol. 2000;164:3392–3401. doi: 10.4049/jimmunol.164.6.3392. [DOI] [PubMed] [Google Scholar]
- LEGLER D.F., LOETSCHER M., ROOS R.S., CLARK-LEWIS I., BAGGIOLINI M., MOSER B. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J. Exp. Med. 1998;187:655–660. doi: 10.1084/jem.187.4.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOETSCHER P., PELLEGRINO A., GONG J.H., MATTIOLI I., LOETSCHER M., BARDI G., BAGGIOLINI M., CLARK-LEWIS I. The ligands of CXC chemokine receptor 3, I-TAC, Mig and IP10, are natural antagonists for CCR3. J. Biol. Chem. 2000;10:10. doi: 10.1074/jbc.M005652200. [DOI] [PubMed] [Google Scholar]
- MACPHEE C.H., APPELBAUM E.R., JOHANSON K., MOORES K.E., IMBURGIA C.S., FORNWALD J., BERKHOUT T., BRAWNER M., GROOT P.H., O'DONNELL K., O'SHANNESSY D., SCOTT G., WHITE J.R. Identification of a truncated form of the CC chemokine CK beta-8 demonstrating greatly enhanced biological activity. J. Immunol. 1998;161:6273–6279. [PubMed] [Google Scholar]
- MAGHAZACHI A.A., AL-AOUKATY A., SCHALL T.J. C-C chemokines induce the chemotaxis of NK and IL-2-activated NK cells. Role for G proteins. J. Immunol. 1994;153:4969–4977. [PubMed] [Google Scholar]
- MARTINELLI R., SABROE I., LAROSA G., WILLIAMS T.J., PEASE J.E. The CC chemokine eotaxin (CCL11) is a partial agonist of CC chemokine receptor 2b. J. Biol. Chem. 2001;276:42957–42964. doi: 10.1074/jbc.M103933200. [DOI] [PubMed] [Google Scholar]
- MURDOCH C., FINN A. Chemokine receptors and their role in vascular biology. J. Vasc. Res. 2000;37:1–7. doi: 10.1159/000025707. [DOI] [PubMed] [Google Scholar]
- NARDELLI B., TIFFANY H.L., BONG G.W., YOUREY P.A., MORAHAN D.K., LI Y., MURPHY P.M., ALDERSON R.F. Characterization of the signal transduction pathway activated in human monocytes and dendritic cells by MPIF-1, a specific ligand for CC chemokine receptor 1. J. Immunol. 1999;162:435–444. [PubMed] [Google Scholar]
- NEOTE K., DIGREGORIO D., MAK J.Y., HORUK R., SCHALL T.J. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell. 1993;72:415–425. doi: 10.1016/0092-8674(93)90118-a. [DOI] [PubMed] [Google Scholar]
- PAN J., KUNKEL E.J., GOSSLAR U., LAZARUS N., LANGDON P., BROADWELL K., VIERRA M.A., GENOVESE M.C., BUTCHER E.C., SOLER D. A novel chemokine ligand for CCR10 and CCR3 expressed by epithelial cells in mucosal tissues. J. Immunol. 2000;165:2943–2949. doi: 10.4049/jimmunol.165.6.2943. [DOI] [PubMed] [Google Scholar]
- PROUDFOOT A.E., BUSER R., BORLAT F., ALOUANI S., SOLER D., OFFORD R.E., SCHRODER J.M., POWER C.A., WELLS T.N. Amino-terminally modified RANTES analogues demonstrate differential effects on RANTES receptors. J. Biol. Chem. 1999;274:32478–32485. doi: 10.1074/jbc.274.45.32478. [DOI] [PubMed] [Google Scholar]
- PROUDFOOT A.E., POWER C.A., HOOGEWERF A.J., MONTJOVENT M.O., BORLAT F., OFFORD R.E., WELLS T.N. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J. Biol. Chem. 1996;271:2599–2603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- RAPORT C.J., GOSLING J., SCHWEICKART V.L., GRAY P.W., CHARO I.F. Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCR5) for RANTES, MIP-1beta, and MIP-1alpha. J. Biol. Chem. 1996;271:17161–17166. doi: 10.1074/jbc.271.29.17161. [DOI] [PubMed] [Google Scholar]
- ROT A., KRIEGER M., BRUNNER T., BISCHOFF S.C., SCHALL T.J., DAHINDEN C.A. RANTES and macrophage inflammatory protein 1 alpha induce the migration and activation of normal human eosinophil granulocytes. J. Exp. Med. 1992;176:1489–1495. doi: 10.1084/jem.176.6.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROTTMAN J.B., SLAVIN A.J., SILVA R., WEINER H.L., GERARD C.G., HANCOCK W.W. Leukocyte recruitment during onset of experimental allergic encephalomyelitis is CCR1 dependent. Eur. J. Immunol. 2000;30:2372–2377. doi: 10.1002/1521-4141(2000)30:8<2372::AID-IMMU2372>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- SARAU H.M., RUSH J.A., FOLEY J.J., BRAWNER M.E., SCHMIDT D.B., WHITE J.R., BARNETTE M.S. Characterization of functional chemokine receptors (CCR1 and CCR2) on EoL-3 cells: a model system to examine the role of chemokines in cell function. J. Pharmacol. Exp. Ther. 1997;283:411–418. [PubMed] [Google Scholar]
- TAKEBE Y., SEIKI M., FUJISAWA J., HOY P., YOKOTA K., ARAI K., YOSHIDA M., ARAI N. SR alpha promoter: an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol. Cell. Biol. 1988;8:466–472. doi: 10.1128/mcb.8.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIFFANY H.L., ALKHATIB G., COMBADIERE C., BERGER E.A., MURPHY P.M. CC chemokine receptors 1 and 3 are differentially regulated by IL-5 during maturation of eosinophilic HL-60 cells. J. Immunol. 1998;160:1385–1392. [PubMed] [Google Scholar]
- TREBST C., SORENSEN T.L., KIVISAKK P., CATHCART M.K., HESSELGESSER J., HORUK R., SELLEBJERG F., LASSMANN H., RANSOHOFF R.M. CCR1+/CCR5+ mononuclear phagocytes accumulate in the central nervous system of patients with multiple sclerosis. Am. J. Pathol. 2001;159:1701–1710. doi: 10.1016/s0002-9440(10)63017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRENTIN L., AGOSTINI C., FACCO M., PIAZZA F., PERIN A., SIVIERO M., GURRIERI C., GALVAN S., ADAMI F., ZAMBELLO R., SEMENZATO G. The chemokine receptor CXCR3 is expressed on malignant B cells and mediates chemotaxis. J. Clin. Invest. 1999;104:115–121. doi: 10.1172/JCI7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN RIPER G., NICHOLSON D.W., SCHEID M.P., FISCHER P.A., SPRINGER M.S., ROSEN H. Induction, characterization, and functional coupling of the high affinity chemokine receptor for RANTES and macrophage inflammatory protein-1 alpha upon differentiation of an eosinophilic HL-60 cell line. J. Immunol. 1994;152:4055–4061. [PubMed] [Google Scholar]
- WARD S.G., WESTWICK J. Chemokines: understanding their role in T-lymphocyte biology. Biochem. J. 1998;333:457–470. doi: 10.1042/bj3330457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WENG Y., SICILIANO S.J., WALDBURGER K.E., SIROTINA-MEISHER A., STARUCH M.J., DAUGHERTY B.L., GOULD S.L., SPRINGER M.S., DEMARTINO J.A. Binding and functional properties of recombinant and endogenous CXCR3 chemokine receptors. J. Biol. Chem. 1998;273:18288–18291. doi: 10.1074/jbc.273.29.18288. [DOI] [PubMed] [Google Scholar]
- WHALEY B.S., YUAN N., BIRNBAUMER L., CLARK R.B., BARBER R. Differential expression of the beta-adrenergic receptor modifies agonist stimulation of adenylyl cyclase: a quantitative evaluation. Mol. Pharmacol. 1994;45:481–489. [PubMed] [Google Scholar]