

Abstract
Zinc is an important trace element in the body and is involved in both the proliferation and growth arrest of many kinds of cells including colorectal epithelial cells. The aim of this study was to identify the molecular mechanism of the growth regulation of colorectal cancer cells by extracellular zinc.
Zinc-stimulated activation of the mitogen-activated protein kinase (MAPK) cascade was measured by immunoblotting and Elk-1 dependent trans-reporter gene expression, and zinc-stimulated p21Cip/WAF1 activation by immunoblotting, Northern blot analysis and immunochemistry. Cell proliferation was measured by thymidine and bromodeoxyuridine (BrdU) incorporation.
By treating colorectal cancer cells with 100 μM ZnCl2, MAPKs were activated in two different phases, the initial weak activation occurred within 5 min and this was followed by a stronger and more prolonged activation.
Zinc concomitantly activated Raf-1-MEK-MAPK kinases, and induced Elk-1 dependent trans-reporter gene expression.
Prolonged activation of MAPKs by 100 μM of ZnCl2 resulted in the induction and nuclear localization of p21Cip/WAF1 and was related to the inhibition of both thymidine and BrdU incorporations.
These results not only suggest the presence of a mechanism for p21Cip/WAF1 dependent negative regulation of colorectal cancer cell growth by zinc but also suggest potential usage of zinc to control the growth of colorectal cancer cells.
Keywords: Zinc, prolonged ERK activation, growth inhibition, cell cycle arrest, p21Cip/WAF1
Introduction
Zinc has been known to be an important trace element in the body and numerous studies have reported zinc's stimulating effect on cell growth (Prasad, 1995; Nishi, 1996). However, it is also known that zinc plays a role in cell growth inhibition and that zinc deficiency often results in aberrant cell proliferation (Lee et al., 1990; Ellwood et al., 1994; Carter et al., 1997). Most significantly, zinc deficiency is one of the major factors associated with the increased risk of developing oesophageal cancer (Barch et al., 1992; Fong & Magee, 1999), and zinc replenishment reduced oesophageal tumour incidence in zinc-deficient rats (Fong et al., 1998). Zinc also inhibits the formation of tumours and dietary zinc deficiency was also found to increase the incidence of tumours (Barch & Iannaccone, 1986; Liang et al., 1999). The levels of zinc in tumours and normal tissues have been shown to be different (Margalioth et al., 1983; Song et al., 1993; Ellwood et al., 1994) and cell growth was shown to be affected by the applied zinc dosage (Prasad, 1979; Ellwood et al., 1994). For example, plasma zinc was significantly reduced in oesophageal carcinoma compared to age matched healthy controls (Mellow et al., 1983; Barch & Iannaccone, 1986). Serum zinc levels were also found to be decreased in patients with sarcoma and lung cancers (Margalioth et al., 1983) and plasma zinc levels have also been associated with squamous cancer of the head and neck (Abdulla et al., 1979; Mellow et al., 1983). It was suggested that zinc is a primary determinant of risk in human colorectal cancer (Martin Mateo & Martin, 1988; Song et al., 1993; Carter et al., 1997) and that low zinc intake may increase the incidence of colorectal cancer (Martin Mateo & Martin, 1988). Furthermore, dietary zinc effected adenomatous polyps (AP) and invasive adenocarcinoma (CA) in mice, with chemically induced colon carcinogenesis (Carter et al., 1997). Therefore, zinc may be involved in the growth regulation of human colorectal cells. However, no mechanism showing the regulation of colorectal cell growth by zinc has been illustrated.
Recent studies have identified zinc as a weak activator of extracellular signal regulated kinases (ERKs) (Hansson, 1996; Samet et al., 1998). The role of the ERK pathway activation in the proliferation of cells has been well elucidated (Pages et al., 1993; Schaeffer & Weber, 1999) and the role of the activation of the MAPK pathway in growth inhibition has also been reported (Sewing et al., 1997; Woods et al., 1997; Pumiglia & Decker, 1997). Therefore, the ERK pathway plays a dual role in the stimulation and inhibition of cell growth, which is dependent on cell type and the characteristics of the signal (Sewing et al., 1997; Woods et al., 1997; Tombes et al., 1998). The negative regulation of cell growth by ERK pathway occurs through regulation of the expression of genes for proteins involving the cell-cycle control such as p21Cip/WAF1 (Sewing et al., 1997; Woods et al., 1997). A recent study identified the inhibition of human prostate carcinoma cell growth by extracellular zinc, and that was mediated by activation of p21Cip/WAF1 (Liang et al., 1999). Currently, a mechanism for the zinc induced p21Cip/WAF1 activation is not known.
In this study, we identified a mechanism for the negative regulation of human colorectal cancer cell growth by extracellular zinc and its relationship with ERK pathway activation. Here, the induction and sub-cellular localization of p21Cip/WAF1, which are dependent upon the activation of the ERK pathway, were identified as important indicators of the negative growth behaviour of the cells by zinc.
Methods
Materials
HT29 (ATCC HTB-38), DLD-1 (ATCC CCL-221), and HCT116 (ATCC CCL-247) human colorectal cell lines were obtained from the American Type Culture Collection (ATCC, Rockville, MD, U.S.A.). McCoy's 5A and RPMI1640 media, foetal bovine serum (FBS), antibiotics and Lipofectamin plus reagent were purchased from Life Technologies, Inc. (Grand Island, NY, U.S.A.). The Tri Reagent used for RNA isolation was purchased from Molecular Research Center (Cincinnati, OH, U.S.A.). Phospho-ERK, phospho-MEK antibodies, and the MAPK assay kit were obtained from New England Biolabs Inc. (Beverly, MA, U.S.A.), and ERK antibody was from Stratagene (La Jolla, CA, U.S.A.). The p21Cip/WAF1 antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, U.S.A.), and Raf-1 antibody, horseradish peroxidase (HRP)-conjugated goat anti-rabbit and anti-mouse IgG antibodies were from Transduction Laboratories (Lexington, KY, U.S.A.). Phospho-Raf-1 (Ser 338) antibody was purchased from Upstate Biotechnology (Lake Placid, N.Y., U.S.A). The Protran nitrocellulose membrane and nylon membrane were purchased from Schleicher and Schuell Co. (Dassel, Germany), and the enhanced chemiluminescence (ECL) system from Genepia (Seoul, Korea). The Radiprime II random prime labelling system were from Amersham Inc. (Buckinghamshire, U.K.), Elk-1-dependent pathdetect plasmids (pFR-Luc and pFA2-Elk1) were from Stratagene (La Jolla, CA, U.S.A.), the Luciferase assay kit and U0126 were from Promega Co. (Madison, WI, U.S.A.), PD98059 was from Calbiochem (La Jolla, CA, U.S.A.), 4′,6′-diamidine-2′-phenylindole dihydrochloride (DAPI) was from Boehringer Mannheim (Mannheim, Germany), the goat anti-mouse-Cy™2- or goat anti-rabbit Rhodamine Red ™-X-conjugated secondary antibodies were purchased from Jackson Immuno Research Laboratories Inc. (West Grove, PA, U.S.A.), and finally the anti-BrdU monoclonal antibody was purchased from DAKO Co. (Carpinteria, CA, U.S.A.).
Cell culture
HT29 and HCT116 colorectal cell lines were maintained in McCoy's 5A medium supplemented with 10% foetal bovine serum (FBS), 100 unit ml−1 of penicillin and 100 μg ml−1 of streptomycin in 5% CO2 at 37°C. The DLD-1 cells were cultured with RPMI1640 medium using the same conditions and supplements. Experiments were performed on the cells at 70% confluence, unless otherwise stated. To observe the effects of zinc, the cells were grown in a media (McCoy's 5A or RPMI1640) containing 1% FBS for 16–20 h and then treated with 100 μM of ZnCl2. Thirty min prior to the ZnCl2 treatment, the MEK inhibitor, PD98059 or U0126, was added to 1–50 μM in the required cases.
Extract preparation
The cells were rinsed twice with phosphate buffered saline (PBS) and harvested by scraping the cells into 500 μl of ice-cold PBS. They were then centrifuged and resuspended in 300 μl of lysis buffer (70 mM β-glycerophosphate pH 7.2, 0.1 mM each meta- and ortho-vanadate, 2 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 0.5% Triton X-100, 0.2 mM phenylmethylsulphonyl fluoride, and 5 μg ml−1 each of pepstatin A, chymostatin, leupeptin and peptin). After incubation on ice for 30 min, the lysate was sonicated for 20 s on ice, unbroken cell debris was removed by centrifugation at 23,000×g for 15 min and proteins were quantified. Samples were aliquoted immediately and stored frozen at −80°C.
Western blot analysis and in vitro MAPK assay
Samples of protein (10–50 μg) from whole-cell extracts were separated by 8–10% sodium dodecyl sulphate (SDS) polyacrylamide gel (acrylamide: bis-acrylamide at a ratio of 29 : 1) and Western blot analysis was performed as described previously (Park et al., 1999). The activations of the endogenous ERKs, MEKs and Raf-1 kinases was determined using phospho-specific ERK, MEK-1 and Raf-1 antibody, respectively. The level of ERKs was analysed using anti-ERK antibody, and the level and modification of Raf-1 was analysed using anti-Raf-1 antibody, and levels of thep21Cip/WAF1 proteins were determined using anti-p21Cip/WAF1 antibody. Blots were probed with HRP-conjugated goat anti-rabbit and anti-mouse IgG secondary antibodies, and bands were visualized using ECL. MAPK assay was performed in vitro using the MAPK assay kit with Elk-1 as a substrate, as described by the manufacturer (New England Biolabs).
PathDetect trans-reporting system, cell transfection and luciferase assay
To determine whether the ERK activation signal was transmitted into the nucleus, we adapted the Elk-1 dependent trans-reporting system (Stratagene, La Jolla, CA, U.S.A.). This incorporated a reporter vector (pFR-Luc) containing five tandem repeats of GAL4 binding elements and a basic promoter element (TATA box), followed by a coding sequence of firefly luciferase, and a fusion trans-activator plasmid (pFA2-Elk-1) expressing a trans-activator protein consistent with the yeast GAL4 binding domain and the activation domain of trans-activator Elk-1.
HT29, HCT116 and DLD-1 cells were plated into 6-well plates at 4×105 cells per well using McCoy's 5A or RPMI1640 medium supplemented with 10% FBS, 100 unit ml−1 of penicillin and 100 μg ml−1 of streptomycin. After allowing 24 h for growth, HT29, HCT116, DLD-1 cells were co-transfected with 0.5 μg of reporter plasmid pFR-Luc along with 25 ng of trans-activator pFA2-Elk-1 using Lipofectamine plus reagent (Grand Island, NY, U.S.A.) according to the manufacturer's instructions. When required, 0.5 μg of pSV-Sport1-Raf-1-dn (gift from Dr J.H. Kim at Korea University, Seoul, Korea), or pSV-Sport1 empty vector was co-transfected to express dominant negative Raf-1. For normalization purposes, the cells were co-transfected with 0.1 μg of pCMV-β-gal reporter (Clonetech) containing the gene for β-galactosidase under the control of a CMV promoter. The transfected cells were serum starved for an additional 16 h, and the medium was replaced with fresh medium with or without 100 μM of ZnCl2. The extracts were prepared 12 h after treatment with the zinc. Luciferase activity was measured using the luciferase assay kit, and normalized using β-galactosidase as an internal control.
Northern blot analysis
Total RNAs were prepared by using Tri Reagent as recommended by the manufacturer. Twenty μg of total RNA was resolved on a 1% denaturing agarose gel containing 6% formaldehyde, and the RNA was transferred onto a nylon membrane and u.v.-cross-linked. The membrane was pre-hybridized with hybridization buffer and then hybridized at 65°C for 2 h with p21Cip/WAF1 probes. The membrane was then washed four times for 15 min with 0.2× SSC, 0.1% SDS at 65°C. Membranes were placed under film and exposed for 4 h at −80°C. As a control, the blot was stripped and re-probed with 32P-labelled cDNA for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). 32P-labelled probes were generated by the Rediprime II random prime labelling system using human p21Cip/WAF1 or GAPDH cDNAs as a template, respectively.
Cell counting and thymidine incorporation
HT29 cells were seeded into 12-well plates at 0.8×105 cells per well. After growing for 24 h in McCoy's 5A, the cells were placed in the same medium containing 1% FBS for 16–20 h. Then 100 μM of ZnCl2 was added into the medium. When required, PD98059 was also added 30 min before the ZnCl2 treatment. The attached cell numbers were counted by mounting 10 μl of the cell mixture (1 : 1 mixture of 0.4% trypan blue and HT29 cells suspension) onto a Tiefe Depth Profondeur 0.0025 mm2 cell counting plate (Superior Co., Germany) under an optical microscope. For thymidine incorporation, the HT29 cells were plated into 12-well plates with 0.8×105 cells per well. After growing overnight, cells were serum starved for 16–20 h. The medium was then replaced with same medium containing 100 μM of ZnCl2 and the cells were further incubated for 48 h. Finally, the incubations were continued in the presence of [3H]-thymidine (0.5 μCi well−1) for 12 h and the DNA-associated [3H]-activity was counted in a liquid scintillation counter.
FACS analysis
HT29 cells were grown in McCoy's 5A medium containing 10% FBS, and further grown for 9 h in an identical medium containing 1% FBS, 10% FBS or 1% FBS plus 100 μM ZnCl2. Cells collected from 6-well plates were rinsed twice with PBS and fixed by adding 70% cold ethanol. Cells were washed with PBS containing 1% horse serum. Subsequently, the DNA was stained with 100 μg ml−1 of propidium iodide for 30 min at 37°C. The cell cycle profile and forward scatter (FSC) were determined using a Becton Dickinson FACS Caliber and the data were analysed using ModFit LT 2.0 (Verity Software House, Inc., ME, U.S.A.) and WinMDI 2.8 (Created by Joseph Trotter, Scripps Research Institute, CA, U.S.A.).
Immunocytochemistry and BrdU incorporation
For immunocytochemistry, HT29 cells were plated onto cover slips at a density of 2×105 cells/coverslip into 6-well plates. The cells were grown overnight and serum starved for 16–20 h and treated with 100 μM ZnCl2 for 9 h before being subjected to immunocytochemistry. In required cases, they were transfected with 0.5 μg of pCMV-MEK-2A (gift from Dr G. Johnson at National Jewish Medical and Research Center, Denver, CO, U.S.A.). After transfection, the cells were serum starved and treated with 100 μM of ZnCl2 as described above. The HT29 cells were then washed twice with PBS, fixed in methanol/formaldehyde (99 : 1) mixture at −20°C for 15 min, permeabilized with PBS containing 0.2% Triton X-100, and finally gently washed five times with PBS for 5 min. After treatment with blocking solution (PBS containing 1% BSA, 0.1% gelatin, and 5% goat serum), the cover slips were further incubated with primary antibody (anti-p21 or anti-MEK antibody at a 1 : 100 dilution) for 2 h, and washed five times with PBS, containing 1% BSA, and 0.1% gelatin. They were then further incubated with goat anti-mouse-Cy™2- or goat anti-rabbit-Rhodamine Red ™-X-conjugated secondary antibody at a 1 : 100 dilution for 1 h, and washed five times with PBS.
For the BrdU incorporation study, HT29 cells were grown in McCoy's 5A media containing 20 μM of BrdU for 12 h before immunocytochemistry. After performing immunocytochemistry, as described above, the cells were fixed in 3.7% formaldehyde for 10 min at room temperature and rinsed with PBS before being incubated for 10 min in 2 N HCl. They were then washed three times with PBS for 5 min. After blocking, the cells were incubated with anti-BrdU monoclonal antibody at a 1 : 20 dilution for 2 h and washed with PBS containing 1% BSA, 0.1% gelatin, and this was followed by incubation with goat anti-mouse-Cy™2-conjugated secondary antibody at a 1 : 100 dilution for 1 h. The cells were then washed five times with PBS. Each experiment was performed at least three times. DAPI was then treated at a final concentration of 1 μg ml−1 in PBS for 10 min, and the cells were extensively washed with PBS and mounted for photography by a Radiance 2000/MP, multi-photon imaging system (Bio-Rad, U.K.).
Results
Zinc-induced prolonged activation of the ERKs through the Raf-1-MEK-ERK module
In this study, we tested whether extracellular zinc can activate the ERK pathway in colorectal cancer cells, and investigated cell growth related with the activation of the ERK pathway by zinc. Phospho-ERK levels, which represent ERK activities, increased in two different phases in the HT29 human colorectal cancer cells treated with 100 μM of ZnCl2 (Figure 1A). Initially, the level of phospho-ERKs increased within 5 min about 4 fold (Figure 1A) as previous reports have observed in other cell types (Hansson, 1996; Samet et al., 1998), however, the activities of ERKs were further strongly increased (8 fold) and prolonged after initial activation in cells treated with 100 μM of ZnCl2 (Figure 1A). The level of prolonged ERK activation by 100 μM of ZnCl2 treatment for 9 h was similar to the level of transient ERK activation induced by treatment with 5 ng ml−1 of epidermal growth factor (EGF) for 10 min (data not shown). The strong activation of ERKs, which peaked 6–9 h after zinc treatment (see also Figure 5A), was also observed in other colorectal cancer cells like DLD-1 and HCT116 (Figure 1B).
Figure 1.
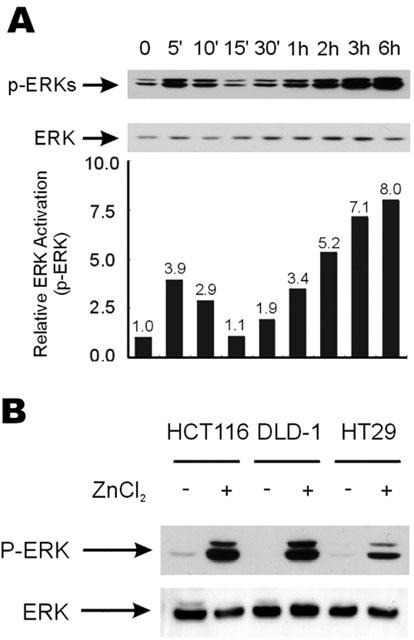
Biphasic activation of ERKs by zinc. (A) The HT29 cells were serum starved for 16 h. Cells were then either treated with 100 μM of ZnCl2 before the cells were harvested at different time points for extract preparations. Western blot analysis was performed using anti-phospho-ERK (Thr202/Tyr204) or anti-ERK antibody, respectively. Relative ERK activations were quantitated by densitometric scanning of phospho-ERK protein bands. (B) The HCT116, DLD-1, and HT29 cells were grown as described in A and treated or not treated with 100 μM ZnCl2 for 9 h before the preparation of cell extracts for Western blot analysis.
Figure 5.
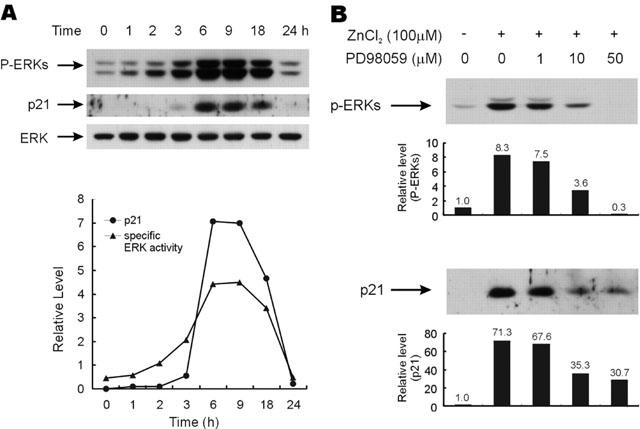
Induction of p21Cip/WAF1 by zinc and its dependence on the activation of ERKs. (A) HT29 cells were serum starved and further grown in the media containing 100 μM of ZnCl2 over the time course. ERK activity and p21Cip/WAF1 level were quantified by densitometric scanning of the protein bands as visualized by Western blot analysis. Specific ERK activities represent densitometric values of phospo-ERK divided by ERK. (B) HT29 cells were serum starved and further grown in the identical medium with or without 100 μM of ZnCl2. In required cases, the cells were pre-incubated with different concentrations of PD98059 (1–50 μM). Cells were harvested 9 h after treatment with ZnCl2. Phospho-ERK or p21Cip/WAF1 protein bands were detected by Western blot, and quantitated by densitometric scanning of the p-ERK and p21Cip/WAF1 protein bands.
To identify the mechanism responsible for the prolonged activation of ERKs by 100 μM ZnCl2, we measured the activation status of the upstream components, i.e., Raf-1 and MEK kinases, when ERK activation was maximal. When levels of phospho-ERKs were maximally increased by 100 μM ZnCl2 treatment, phospho-MEK proteins were also strongly increased and upstream Raf-1 protein was shifted to a higher molecular weight form (Figure 2A). In addition, the level of phospho-Ser-338 Raf-1, which correlated with Raf-1 protein activation (Dhillon et al., 2002), was increased 3–4-fold by zinc treatment (Figure 2A). Therefore, extracellular zinc treatment resulted in the activation of all the MAPK module kinases (Raf-1, MEK and ERK). To determine whether ERK activation was related with target gene expression, Elk-1 dependent trans-reporter gene expression was measured after zinc treatment. Elk-1 dependent trans-reporter gene expression was also found to concomitantly increased in HT29 cells after treatment with 100 μM of ZnCl2 (Figure 2B). In addition, in both HCT116 and DLD-1 cells, Elk-1 dependent trans-reporter gene expression was also increased by treatment with 100 μM of ZnCl2 (data not shown).
Figure 2.
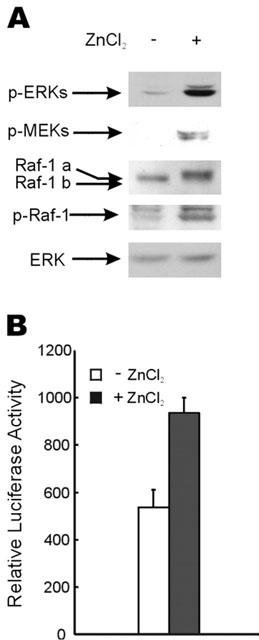
Activation of ERK pathway by zinc. (A) Before treatment with 100 μM of ZnCl2, HT29 cells were grown for 16 h in medium containing 1% FBS. Cells were harvested 9 h after zinc treatment. Phospho-ERK (Thr202/Tyr204), phospho-MEK (Ser217/Ser221), Raf-1, phospho-Raf-1 (Ser338) and ERK were detected by Western blot analysis using anti-phospho-ERK, -phospho-MEK, -Raf-1, -phospho-Raf-1 or -ERK antibody, respectively. A shift in the Raf-1 protein band generated by zinc treatment was identified as Raf-1a, while the un-shifted band was marked as Raf-1b. (B) HT29 cells were co-transfected with 0.5 μg of Luciferase reporter plasmid pFR-Luc and 25 ng of trans-activator pFA2-Elk-1. For normalization, the cells were co-transfected with 0.1 μg of plasmid containing the gene for β-galactosidase under the control of a CMV promoter. Cells were incubated in medium containing 1% FBS for an additional 16 h. The medium was replaced with fresh medium with or without 100 μM ZnCl2, and extracts were made 12 h after the zinc treatment. Luciferase activity was measured and normalized using the β-galactosidase level as an internal control. Each result represents the average value of three independent experiments. Error bars indicate standard deviations.
The zinc induced prolonged activation of the ERK pathway was inhibited by PD98059 and by dominant negative Raf-1
To further characterize the zinc-induced prolonged activation of the ERK pathway, we pre-treated the MEK-specific inhibitor, PD98059, and determined whether the drug could block the zinc-induced prolonged activation of the ERK pathway. In the present study, the zinc-dependent increase of phospho-ERKs levels was reduced by 40% by pre-treatment with 10 μM of PD98059 (Figure 3A). The activation of ERKs by zinc and the blocking of kinase activation by PD98059 were further confirmed by an in vitro kinase assay (Figure 3A). Interestingly, modification of the upstream component Raf-1 by zinc treatment was also blocked by treating cells with PD98059 (Figure 3A, bottom panel; under-shift of Raf-1 band from band ‘a' to band ‘b' in the cells pre-treated with PD98059). Raf-1 protein shifted further to a smaller-molecularly sized band after treating zinc untreated cells with PD98059 (Figure 3A; under-shift of Raf-1 band from ‘b' to ‘c'). The activation of Elk-1 dependent trans-reporter gene expression by 100 μM of ZnCl2 was also blocked PD98059 treatment (Figure 3B) or by the transient transfection of dominant negative Raf-1 (dn-Raf-1) (Figure 3C). We also observed down-regulation of Elk-1 dependent trans-reporter gene expression, which was increased by 100 μM of ZnCl2, by expressing dn-Raf-1 in the other colorectal cancer cells (data not shown). Overall, these results suggest that the prolonged activation of ERKs by zinc could be acquired through the Raf-1-MEK-ERK module via an auto-regulatory loop, and the signal was further transmitted into nucleus to activate target gene expression.
Figure 3.
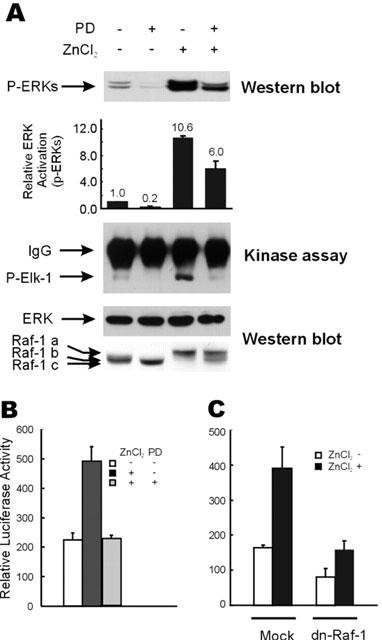
Down-regulation of the zinc-induced activation of the ERK pathway by inhibition of MEK or Raf-1 kinases. (A) HT29 cells were serum starved as described in Figure 1A, and treated with 100 μM of ZnCl2 for 9 h before harvesting for extract preparation. In required cases, cells were incubated with 10 μM of PD98059 for 30 min before zinc treatment. Western blot analysis was performed to visualize the phospho-ERK, Raf-1, and ERK protein bands. Phospho-ERK levels were quantitated by densitometric scanning of phospho-ERK protein bands. Relative ERK activations are average values of the results of three independent Western blot analyses. The in vitro kinase activity of ERKs was measured using purified GST-Elk-1 substrate, as described by the manufacturer (New England Biolab Inc.). (B) HT29 cells were grown and transfected with pFR-Luc and pFA2-Elk-1 vectors as in Figure 2B, except that 10 μM of PD98059 was administered 30 min prior to treatment with 100 μM ZnCl2, in the required cases. Elk-1 dependent trans-reporter gene expression was measured 12 h after treatment with 100 μM of ZnCl2. (C) After transfection with 0.5 μg of pFR-Luc and 25 ng of pFA2-Elk-1 plasmids, HT29 cells were serum starved and treated with 100 μM ZnCl2. In selected cases, 0.5 μg of the pSV-Sport1 vector or pSV-Sport1-Raf-1-dn was cotransfected together with the plasmids mentioned above. Elk-1 dependent trans-reporter gene expression was measured 12 h after treatment with 100 μM of ZnCl2.
The 100 μM of ZnCl2 inhibit cell growth and proliferation
To identify a role for the activation of the ERK pathway by 100 μM of ZnCl2 with respect to colorectal cancer cell growth, we counted cell numbers and measured the level of DNA synthesis after treatment with 100 μM of ZnCl2. Even though 100 μM of ZnCl2 strongly activated the ERK pathway, cell numbers did not increase, and fold increase levels of the cells were decreased after treatment with 100 μM of ZnCl2 during a time course (Figure 4A). [3H]-thymidine incorporation, which represents the cell proliferation status, was also lower in the cells treated with 100 μM of ZnCl2 compared to the cells which were not treated with zinc (Figure 4B). Therefore, 100 μM of ZnCl2 may be involved in the growth inhibition of colorectal cancer cells, although strongly activating ERKs. The G1 to S phase cell cycle progression induced by serum treatment was mostly blocked 9 h after 100 μM ZnCl2 treatment, and no significant level of the sub-G1 fraction of the DNA content was observed (Figure 4C). Therefore, the anti-proliferation of colorectal cancer cells by zinc may occur by G1 cell cycle arrest not by apoptosis.
Figure 4.
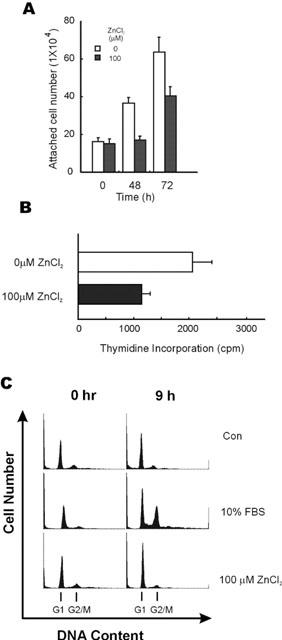
Inhibition of cell growth and [3H]-thymidine incorporation induced by zinc. (A) HT29 cells were seeded at a density of 0.8×105 cells per well in 6-well plates. The cells were serum starved for 16 h and either treated or not treated with 100 μM of ZnCl2. The numbers of attached cells were counted in a hemocytometer at 0, 48, 72 h after zinc treatment. Each data point represents the average value of three independent experiments. Error bars indicate the standard deviations of three independent experiments. (B) HT29 cells were seeded at a density of 0.8×105 cells per well into 6-well plates and serum starved for 16 h. After treatment with 100 μM of ZnCl2, the cells were further cultured for 48 h before addition of [3H]-thymidine (0.5 μCi well−1). The cells were further incubated for 12 h, and the incorporation of [3H]-thymidine was determined. Results represent the average of three independent experiments. Error bars indicate the standard deviations of three independent experiments. (C) Inhibition of serum stimulated G1 to S phase cell cycle progression by zinc. HT29 cells were grown in McCoy's 5A medium containing 1% FBS (Con), 10% FBS or 1% FBS plus 100 μM ZnCl2, as described in Methods. Cells were stained with propidium iodide to show cell-cycle distribution by DNA content. G1 and G2/M phases of the cell cycle are indicated by arrows.
Zinc-induced induction of p21Cip/WAF1 by ERK pathway activation
Although the ERK signalling pathway is a major network for cell proliferation, the role of an identical ERK pathway in cell growth arrest has been reported, and that has been related with the induction of a negative cell cycle regulator, p21Cip/WAF1 (Sewing et al., 1997; Woods et al., 1997). Furthermore, the induction of p21Cip/WAF1 is a good indicator of colorectal cancer cell growth arrest (Archer et al., 1998; Nakano et al., 1997). Therefore, we investigated the expression levels of p21Cip/WAF1 in colorectal cancer cells treated with 100 μM of ZnCl2 in order to identify a relationship between ERK activation and p21Cip/WAF1 induction related to cell growth inhibition. Strong p21Cip/WAF1 induction was also observed after treating 100 μM of ZnCl2, moreover, the profile of this induction was highly correlated with prolonged ERK activation (Figure 5A). In addition, patterns of p21Cip/WAF1 induction and ERK activation were also well correlated in cells treated with different zinc concentrations (data not shown). To identify a direct relationship between p21Cip/WAF1 induction and ERK activation, we examined the effect of PD98059 upon the induction of p21Cip/WAF1. ERK activity inhibition by PD98059 was dependent on the pre-treated PD98059 concentration. Therefore, phospho-ERKs levels were 57% reduced by 10 μM of PD98059 and completely abolished at 50 μM (Figure 5B). However, the inhibition of p21Cip/WAF1 induction by PD98059 was only partial, and the p21Cip/WAF1 level was only slightly reduced (less than 5%) upon increasing PD98059 from 10–50 μM (Figure 5B). Both the ERK activation and p21Cip/WAF1 induction of the cells were similarly reduced by pre-treating the cells with an alternative MEK inhibitor, U0126, which directly inhibit MEK activity. In addition, the p21Cip/WAF1 band also was not completely abolished at the higher U0126 concentration above 10 μM (data not shown). Therefore, extracellular zinc increased the p21Cip/WAF1 protein level by a mechanism, which was dependent on the ERK cascade, although some of the p21Cip/WAF1 may have been induced by ERK independent mechanism(s).
Induction of p21Cip/WAF1 mRNA by 100 μM ZnCl2 depended on the activation of ERKs
Although p21Cip/WAF1 protein level was increased by treating the colorectal cancer cells with zinc, it was unclear whether that was due to the level of protein or the level of mRNA. Therefore, we performed Northern blot analysis to determine the levels of p21Cip/WAF1 mRNA, which was found to be also increased by 100 μM ZnCl2 treatment in cells, and this increase was also blocked by pre-treating with PD98059 (Figure 6A). Therefore, the induction of p21Cip/WAF1 by zinc could be caused by an increased mRNA level induced by the activation of the ERK pathway. It is known that the p21Cip/WAF1 is induced by both p53 dependent and independent mechanisms (Liu et al., 1996; Nakano et al., 1997; Bottazzi et al., 1999). To investigate the p53 dependency of the induction of p21Cip/WAF1, we measured levels of the p21Cip/WAF1 mRNA in several colorectal cancer cells (HT29, DLD-1, HCT116), which contain either the wild type or mutant p53 gene (Bunz et al., 1998). As found for ERK activity, levels of p21Cip/WAF1 mRNA were increased by 100 μM of ZnCl2 treatment in HT29, HCT116 and DLD-1 cells regardless of the mutational status of the p53 gene (Figure 6B). However, the levels of p21Cip/WAF1 mRNA induced by 100 μM of ZnCl2 were not in direct proportion with the levels of ERK activation (compare Figure 1B with 6B) in the different cell types.
Figure 6.
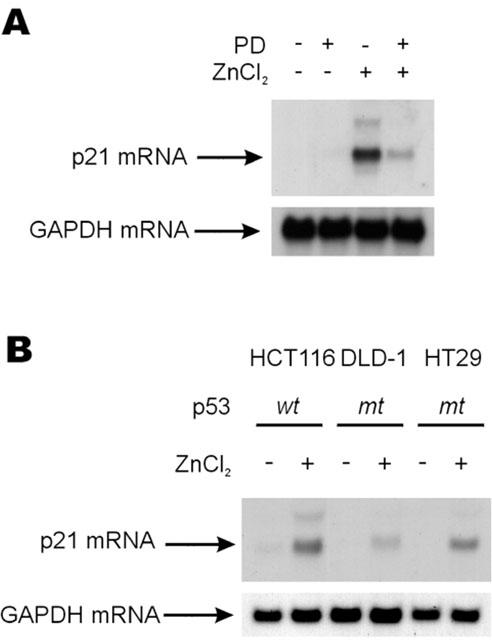
Increase of p21Cip/WAF1 mRNA by a mechanism dependent on ERK pathway. (A) HT29 cells were treated with 100 μM of ZnCl2 for 5 h before harvesting for total RNA preparation. The cells were incubated with 10 μM of PD98059 for 30 min prior to zinc treatment in the required cases. Northern blot analyses were performed using 32P-labelled p21Cip/WAF1 or GAPDH cDNA as probes. (B) The HT29, HCT116 and DLD-1 cells were grown and serum starved as shown in A except that RPMI1640 medium was used in case of the DLD-1 cells.
The induction and nuclear localization of p21Cip/WAF1 by 100 μM ZnCl2 depended on the activation of ERKs, and the p21Cip/WAF1 activation inhibited BrdU incorporation
The cell cycle inhibitory activity of p21Cip/WAF1 is intimately related with its induction and nuclear localization, and these are directly related to cell growth (Halevy et al., 1995; Asada et al., 1999). In this study, we investigated the localization of p21Cip/WAF1 after treatment with 100 μM of ZnCl2 to further characterize its role in colorectal cancer cell growth regulation. In serum starved HT29 cells, the p21Cip/WAF1 protein level was low and the protein was mainly localized to the perinuclear compartment and the cytosol (Figure 7A; upper panel). However, the p21Cip/WAF1 protein level was both increased and localized to the nucleus in 100 μM ZnCl2 treated cells. The induction and nuclear localization of p21Cip/WAF1, caused by 100 μM of ZnCl2, blocked by 10 μM PD98059 pre-treatment (Figure 7A; upper panel). In addition, p21Cip/WAF1 induction and especially its nuclear localization by 100 μM of ZnCl2 were also significantly blocked in cells expressing MEK-2A (Figure 7A; lower panel). These results confirm the ERK dependence in the induction and nuclear localization of the p21Cip/WAF1.
Figure 7.
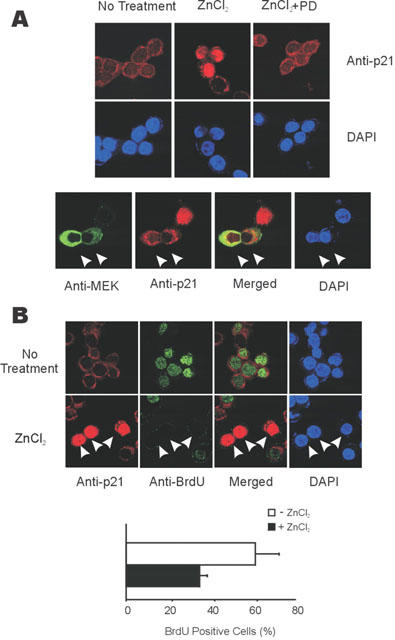
ERK pathway dependent nuclear localization of p21Cip/WAF1 inhibits BrdU incorporation. (A) Nuclear localization of p21Cip/WAF1 induced by zinc and inhibited by PD98059 (upper panel) or by the expression of MEK-2A (lower panel). Upper panel; HT29 cells were treated or not treated with 100 μM of ZnCl2 for 9 h before immunocytochemistry. In the required cases, they were incubated PD98059 (10 μM) for 30 min before treatment with zinc. Lower panel; HT29 cells were grown and transfected with 0.5 μg of pCMV-MEK-2A. The cells were treated with 100 μM of ZnCl2 for 9 h before immunocytochemistry. The expression and localization of p21Cip/WAF1 and MEK-2A was revealed by immunocytochemistry using the anti- p21Cip/WAF1 antibody together with Rhodamin-conjugated goat anti-rabbit IgG, or anti-MEK antibody together with Cy-2-conjugated goat anti-mouse IgG antibody; nuclei were stained by DAPI. The representative cells inhibiting induction and nuclear localization of p21Cip/WAF1 by expressing MEK-2A were marked by arrows. (B) Inhibition of BrdU incorporation by induction and nuclear localization of p21Cip/WAF1 by zinc. Upper panel; Zinc dependent induction and nuclear localization of p21Cip/WAF1 inhibited BrdU incorporation. HT29 cells were serum starved with the media containing 1% FBS, then further grown in the media without or with 100 μM ZnCl2 for 16 h. The cells were labeled with 20 μM of BrdU for last 12 h before assay. BrdU incorporated nuclei were revealed with green colour by immunocytochemistry by using anti-BrdU antibody followed by Cy2-conjugated goat anti-mouse IgG. The representative zinc treated cells blocked the BrdU incorporation by induction and nuclear localization of p21Cip/WAF1 was marked by arrows. Lower panel; The percentage of cells incorporating BrdU was lowered by 100 μM of ZnCl2 treatment. BrdU positive cell data was quantified from the results of upper panel.
To obtain direct evidence upon the anti-proliferative effect at the single cell level by zinc, we also monitored DNA synthesis by measuring BrdU incorporation after treatment of serum starved HT29 cells with 100 μM ZnCl2. In particular, we checked whether BrdU incorporation was blocked in an individual cell containing nuclear localized p21Cip/WAF1, which induced by treatment of 100 μM ZnCl2. In the media containing 1% FBS, around 60.8% of the cells incorporated BrdU, which was decreased to 35.5% upon 100 μM ZnCl2 treatment (Figure 7B). Because cells containing both nuclearly localized and non-nuclearly localized p21Cip/WAF1 were co-present in the total cell population treated with 100 μM ZnCl2, we checked on the effect of nuclear localization of p21Cip/WAF1 on BrdU incorporation in the individual cells. Importantly, no cell induced nuclearly localized p21Cip/WAF1 on incorporating BrdU among around several hundred of cells we counted (representative cases are shown in Figure 7B; upper panel). Therefore, the ERK dependent induction and nuclear localization of the p21Cip/WAF1 can directly be related with anti-proliferation of colorectal cancer cells.
Discussion
Numerous studies have been reported upon the roles of zinc in both the induction and prevention of cancer (Vallee & Falchuk, 1993; Rojas et al., 1999). The epithelium of the colonic epithelial mucosa undergoes continuous renewal, and the control of growth and tumorigenesis of colorectal cells are highly sensitive to zinc levels (Martin Mateo & Martin, 1988; Song et al., 1993; Carter et al., 1997). Low zinc intake may increase the incidence of colorectal tumours and the zinc deprived progression of 1,2-dimethylhydrozine-induced colon tumors (Martin Mateo & Martin, 1988; Carter et al., 1997). Therefore, zinc may be involved in the growth inhibition of human colorectal cells and be related to anti-tumour effects.
In this study, we characterized the role of extracellular zinc on the growth regulation of colorectal cancer cells, and found that ERK pathway dependent p21Cip/WAF1 activation is a route for zinc-induced cellular growth arrest. Treating cells with 100 μM of ZnCl2 caused ERK activities to be transiently increased, which is in agreement with previous observations in several other cell types (Hansson, 1996; Samet et al., 1998). However, we observed a further strong activation of ERKs followed by its initial activation in colorectal cancer cells. The strong and prolonged activation of ERKs induced by the 100 μM of ZnCl2 treatment may have been caused by a positive activation of the Raf-1→MEK→ERK loop after the transient activation of ERKs. Moreover, the presence of the positive activation loop is evidenced by the observed blocking of the upstream Raf-1 modification by the inhibition of downstream MEK using PD98059. Zinc dependent induction of the Elk-1 trans-reporter and its down regulation by PD98059 or dn-Raf-1 further support that the zinc activated the ERK pathway through the Raf-1→MEK→ERK cascade to induce the target gene expression. Currently, it is not clear how extracellular zinc regulates Raf-1-MEK-ERK module kinases. The activation of Raf-1 is highly complex and acquired by both Ras-dependent and independent mechanisms (Sharon et al., 1998). We observed reduction of the zinc dependent induction of the Elk-1 trans-reporter gene by treatment of a dominant negative Ras construct (data not shown), and that suggests the involvement of Ras in the zinc induced activation of the ERK pathway. The strong and prolonged activation of ERKs by 100 μM of ZnCl2 was related to cell growth inhibition, which may have been caused by the activation of p21Cip/WAF1.
A substantial body of evidence supports that the activation of p21Cip/WAF1 is the result of the prolonged activation of ERKs by zinc. First, the profiles of specific ERK activation and p21Cip/WAF1 induction by zinc treatment are highly correlated (Figure 5A). Second, p21Cip/WAF1 induction is inhibited by pretreatment with the MEK inhibitors, PD98059 and U0126 (data not shown). Finally, nuclear localization of p21Cip/WAF1 were blocked at the single cell level by PD98059 pre-treatment or by the transient expression of catalytically inactive MEK-2A. Therefore, zinc activated p21Cip/WAF1 by an ERK pathway dependent mechanism. The zinc-dependent induction of p21Cip/WAF1 mRNA was observed in all colorectal cancer cells containing different status of p53 gene we tested. However, the induced levels of p21Cip/WAF1 mRNA were variable and did not correlate directly with ERK activation levels in different colorectal cancer cell lines (compare Figure 1B and 6B). Therefore, p53 or unknown factor(s) may partly be involved in zinc dependent p21Cip/WAF1 activation. The presence of alternative routes for the induction of p21Cip/WAF1 by zinc was suggested by an experiment upon the effect of zinc upon p21Cip/WAF1 inhibition after pre-treating with PD98059 or U0126 (data not shown) at a concentration higher than 10 μM.
In this study, we did not observe any cells that incorporated BrdU among the several hundred cells that induced and localized p21Cip/WAF1 to the nucleus upon zinc treatment. These results confirm the importance of the p21Cip/WAF1 activation in the anti-proliferation of colorectal cancer cells. Furthermore, it provides an important evidence for the MAPK dependent p21Cip/WAF1 activation in the regulation of colorectal cancer cell proliferation. Although serum stimulated G1 to S phase cell cycle progression was significantly inhibited by zinc, G2 arrest and sub-G1 fractions were not significantly affected. Therefore, growth regulation of colorectal cancer cells by zinc could occur by G1 cell cycle arrest.
The importance of p21Cip/WAF1 in the growth control of the cells, which depends on ERK signalling, is supported by the recent study, which showed the promotion of DNA synthesis by prolonged activation of the ERK pathway in p21Cip/WAF1-null mice (Auer et al., 1998). In addition, a cell cycle arrest caused by induction of p21Cip/WAF1, acquired by high intensity of Raf-1 signals was abolished in p21Cip/WAF1−/− fibroblasts (Woods et al., 1997). Zinc dependent induction of p21Cip/WAF1 was also observed in prostate carcinoma cells (Liang et al., 1999). However, this study did not present a mechanism for the induction of p21Cip/WAF1 by zinc. In the present study, we identified a detailed mechanism for the induction and subcellular localization of p21Cip/WAF1 by zinc, and further related with cellular growth. Our study shows that prolonged ERK pathway dependent activation of p21Cip/WAF1 plays an important role in the negative regulation of colorectal cancer cell proliferation.
It is known that plasma zinc concentrations in patients with various cancers, including sarcoma, lung cancers, head and neck cancers and oesophageal carcinoma tend to be lower than those in matched healthy controls (Abdulla et al., 1979; Mellow et al., 1983; Barch & Iannaccone, 1986). Therefore, it is possible that low zinc levels in the extracellular plasma environment may be related to the growth of tumours (Lee et al., 1989; Endre et al., 1990), and colorectal cell growth is also highly sensitive to zinc levels (Martin Mateo & Martin, 1988; Song et al., 1993; Carter et al., 1997). However, it is also possible that the cancers may result in low zinc levels in affected patients. Our study of growth regulation of colorectal cancer cells by extracellular zinc and other epidemiological studies related with zinc (Song et al., 1993; Fong et al., 1998, 1999) suggest that the maintenance of adequate zinc levels may be important for good health and that at the right dosage zinc usage has the potential to both prevent and inhibit colorectal cancer. The role of calcium in such prevention and therapy has been well illustrated although its detailed mechanism is unknown (Pence & Buddingh, 1988; Rozen et al., 1989), and it is not clear whether calcium uses a similar signalling mechanism with zinc for the growth control of colorectal cancer cells.
Acknowledgments
This work was supported by following grants to K.-Y.Choi by: The ‘1999' Korean National Cancer Control Program, Ministry of Health and Welfare, Korea; The Basic Research Program of the Korean Science and Engineering Foundation (1999-1-212-001-5), and to B.L. Seong by: The Ministry of Commerce, Industry and Energy (IMT 2000 Grant and Clean Technology Grant).
Abbreviations
- BrdU
bromo-deoxyuridine
- DAPI
4′,6′-diamidine-2-phenylindole dihydrochloride
- ERK
extracellular signal-regulated kinase
- FBS
foetal bovine serum
- MEK
MAPK kinase
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate-buffered saline
- PD98059
2-(2′- amino-3′-methoxyphenyl) oxanaphthalen-4-one
- SDS
sodium dodecyl sulphate
- U0126
1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio] butadiene
References
- ABDULLA M., BIORKLUND A., MATHUR A., WALLENIUS K. Zinc and copper levels in whole blood and plasma from patients with squamous cell carcinomas of head and neck. J. Surg. Oncol. 1979;12:107–113. doi: 10.1002/jso.2930120203. [DOI] [PubMed] [Google Scholar]
- ARCHER S.Y., MENG S., SHEI A., HODIN R.A. p21(WAF1) is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6791–6796. doi: 10.1073/pnas.95.12.6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASADA M., YAMADA T., ICHIJO H., DELIA D., MIYAZONO K., FUKUMURO K., MIZUTANI S. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999;18:1223–1234. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AUER K.L., PARK J.S., SETH P., COFFEY R.J., DARLINGTON G., ABO A., MCMAHON M., DEPINHO R.A., FISHER P.B., DENT P. Prolonged activation of the mitogen-activated protein kinase pathway promotes DNA synthesis in primary hepatocytes from p21Cip-1/WAF1-null mice, but not in hepatocytes from p16INK4a-null mice. Biochem. J. 1998;336:551–560. doi: 10.1042/bj3360551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARCH D.H., IANNACCONE P.M. Role of zinc deficiency in carcinogenesis. Adv. Exp. Med. Biol. 1986;206:517–527. doi: 10.1007/978-1-4613-1835-4_36. [DOI] [PubMed] [Google Scholar]
- BARCH D.H., FOX C.C., ROSCHE W.A., RUNDHAUGEN L.M., WRIGHTON S.A. Inhibition of rat methylbenzylnitrosamine metabolism by dietary zinc and zinc in vitro. Gastroenterology. 1992;103:800–806. doi: 10.1016/0016-5085(92)90009-n. [DOI] [PubMed] [Google Scholar]
- BOTTAZZI M.E., ZHU X., BOHMER R.M., ASSOIAN R.K. Regulation of p21(cip1) expression by growth factors and the extracellular matrix reveals a role for transient ERK activity in G1 phase. J. Cell. Biol. 1999;146:1255–1264. doi: 10.1083/jcb.146.6.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUNZ F., DUTRIAUX A., LENGAUER C., WALDMAN T., ZHOU S., BROWN J.P., SEDIVY J.M., KINZLER K.W., VOGELSTEIN B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- CARTER J.W., LANCASTER H., HARDMAN W.E., CAMERON I.L. Zinc deprivation promotes progression of 1,2-dimethylhydrazine-induced colon tumors but reduces malignant invasion in mice. Nutr. Cancer. 1997;27:217–221. doi: 10.1080/01635589709514529. [DOI] [PubMed] [Google Scholar]
- DHILLON A.S., MEIKLE S., YAZICI Z., EULITZ M., KOLCH W. Regulation of Raf-1 activation and signaling by dephosphorylation. EMBO J. 2002;21:64–71. doi: 10.1093/emboj/21.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELLWOOD K.C., ROEBUCK B.D., HATHCOCK J.N. Marginal zinc status does not exacerbate pancreatic carcinogenesis associated with dietary soybean trypsin inhibitor concentrate in rats. J. Nutr. 1994;124:894–900. doi: 10.1093/jn/124.6.894. [DOI] [PubMed] [Google Scholar]
- ENDRE L.F., BECK W.J., PRASAD A.S. The role of zinc in human health. J. Trace. Elem. Exp. Med. 1990;3:337–375. [Google Scholar]
- FONG L.Y., FARBER J.L., MAGEE P.N. Zinc replenishment reduces esophageal cell proliferation and N-nitrosomethylbenzylamine (NMBA)-induced esophageal tumor incidence in zinc-deficient rats. Carcinogenesis. 1998;19:1591–1596. doi: 10.1093/carcin/19.9.1591. [DOI] [PubMed] [Google Scholar]
- FONG L.Y., MAGEE P.N. Dietary zinc deficiency enhances esophageal cell proliferation and N-nitrosomethylbenzylamine (NMBA)-induced esophageal tumor incidence in C57BL/6 mouse. Cancer Lett. 1999;143:63–69. doi: 10.1016/s0304-3835(99)00191-3. [DOI] [PubMed] [Google Scholar]
- HALEVY O., NOVITCH B.G., SPICER D.B., SKAPEK S.X., RHEE J., HANNON G.J., BEACH D., LASSAR A.B. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- HANSSON A. Extracellular zinc ions induces mitogen-activated protein kinase activity and protein tyrosine phosphorylation in bombesin-sensitive Swiss 3T3 fibroblasts. Arch. Biochem. Biophy. 1996;328:233–238. doi: 10.1006/abbi.1996.0168. [DOI] [PubMed] [Google Scholar]
- LEE H.H., HILL G.M., SIKHA V.K., BREWER G.J., PRASAD A.S., OWYANG C. Pancreaticobiliary secretion of zinc and copper in normal persons and patients with Wilson's disease. J. Lab. Clin. Med. 1990;116:283–288. [PubMed] [Google Scholar]
- LEE H.H., PRASAD A.S., BREWER G.J., OWYANG C. Zinc absorption in human small intestine. Am. J. Physiol. 1989;256:G87–G91. doi: 10.1152/ajpgi.1989.256.1.G87. [DOI] [PubMed] [Google Scholar]
- LIANG J.Y., LIU Y.Y., ZHOU J., FRANKLIN R.B., COSTELLO L.C., FENG P. Inhibitory effect of zinc on human prostatic carcinoma cell growth. Prostate. 1999;40:200–207. doi: 10.1002/(sici)1097-0045(19990801)40:3<200::aid-pros8>3.0.co;2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU Y., MARTINDALE J.L., GOROSPE M., HOLBROOK N.J. Regulation of p21WAF/CIP expression through mitogen-activated protein kinase signaling pathway. Cancer Res. 1996;56:31–35. [PubMed] [Google Scholar]
- MARGALIOTH E.J., SCHENKER J.G., CHEVION M. Copper and zinc levels in normal and malignant tissues. Cancer. 1983;52:868–872. doi: 10.1002/1097-0142(19830901)52:5<868::aid-cncr2820520521>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- MARTIN MATEO M.C., MARTIN G. Influence of metallic carcinogenesis in lung and colorectal neoplasia. Metals in neoplastic processes. Clin. Physiol. Biochem. 1988;6:321–326. [PubMed] [Google Scholar]
- MELLOW M.H., LAYNE E.A., LIPMAN T.O., KAUSHIK M., HOSTETLER C., SMITH J.C., JR Plasma zinc and vitamin A in human squamous carcinoma of the esophagus. Cancer. 1983;51:1615–1620. doi: 10.1002/1097-0142(19830501)51:9<1615::aid-cncr2820510911>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- NAKANO K., MIZUNO T., SOWA Y., ORITA T., YOSHINO T., OKUYAMA Y., FUJITA T., OHTANI-FUJITA N., MATSUKAWA Y., TOKINO T., YAMAGISHI H., OKA T., NOMURA H., SAKAI T. Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53-negative human colon cancer cell line. J. Biol. Chem. 1997;272:22199–22206. doi: 10.1074/jbc.272.35.22199. [DOI] [PubMed] [Google Scholar]
- NISHI Y. Zinc and growth. J. Am. Coll. Nutr. 1996;15:340–344. doi: 10.1080/07315724.1996.10718608. [DOI] [PubMed] [Google Scholar]
- PAGES G., LENORMAND P., L'ALLEMAIN G., CHAMBARD J.C., MELOCHE S., POUYSSEGUR J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARK K-S., KIM N.G., KIM J.J., KIM H., AHN Y-H., CHOI K-Y. Differential regulation of MAP kinase cascade in human colorectal tumorigenesis. Br. J. Cancer. 1999;81:1116–1121. doi: 10.1038/sj.bjc.6690817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PENCE B.C., BUDDINGH F. Inhibition of dietary fat-promoted colon carcinogenesis in rats by supplemental calcium or vitamin D3. Carcinogenesis. 1988;9:187–190. doi: 10.1093/carcin/9.1.187. [DOI] [PubMed] [Google Scholar]
- PRASAD A.S. Clinical, biochemical, and pharmacological role of zinc. Ann. Rev. Pharmacol. Toxicol. 1979;19:393–426. doi: 10.1146/annurev.pa.19.040179.002141. [DOI] [PubMed] [Google Scholar]
- PRASAD A.S. Zinc: an overview. Nutrition. 1995;11:93–99. [PubMed] [Google Scholar]
- PUMIGLIA K.M., DECKER S.J. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. U.S.A. 1997;94:448–452. doi: 10.1073/pnas.94.2.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROJAS E., HERRERA L.A., POIRIER L.A., OSTROSKY-WEGMAN P. Are metals dietary carcinogens. Mutat. Res. 1999;443:157–181. doi: 10.1016/s1383-5742(99)00018-6. [DOI] [PubMed] [Google Scholar]
- ROZEN P., FIREMAN Z., FINE N., WAX Y., RON E. Oral calcium suppresses increased rectal epithelial proliferation of persons at risk of colorectal cancer. Gut. 1989;30:650–655. doi: 10.1136/gut.30.5.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAMET J.M., GRAVES L.M., QUAY J., DAILEY L.A., DEVLIN R.B., GHIO A.J., WU W., BROMBERG P.A., REED W. Activation of MAPKs in human bronchial epithelial cells exposed to metals. Am. J. Physiol. 1998;275:L551–L558. doi: 10.1152/ajplung.1998.275.3.L551. [DOI] [PubMed] [Google Scholar]
- SCHAEFFER H.J., WEBER M.J. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEWING A., WISEMAN B., LLOYD A.C., LAND H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol. Cell Biol. 1997;17:5588–5597. doi: 10.1128/mcb.17.9.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARON L.C., KHOSRAVI-FAR R., ROSSMAN K.L., CLARK G.J., DER C.J. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–1431. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- SONG M.K., HENG M.C., ROLANDELLI R., AMENT M.E., HENG M.K. Possible link between zinc intake and colon cancer. J. Natl. Cancer Inst. 1993;85:667–669. doi: 10.1093/jnci/85.8.667. [DOI] [PubMed] [Google Scholar]
- TOMBES R.M., AUER K.L., MIKKELSEN R., VALERIE K., WYMANN M.P., MARSHALL C.J., MCMAHON M., DENT P. The mitogen-activated protein (MAP) kinase cascade can either stimulate or inhibit DNA synthesis in primary cultures of rat hepatocytes depending upon whether its activation is acute/phasic or chronic. Biochem. J. 1998;330:1451–1460. doi: 10.1042/bj3301451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VALLEE B.L., FALCHUK K.H. The biochemical basis of zinc physiology. Physiol. Rev. 1993;73:79–118. doi: 10.1152/physrev.1993.73.1.79. [DOI] [PubMed] [Google Scholar]
- WOODS D., PARRY D., CHERWINSKI C., BOSCH E., LEES E., MCMAHON M. Raf-Induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip. Mol. Cell Biol. 1997;17:5598–5611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]