

Abstract
Fibril formation of amyloid β peptide (Aβ) is considered to be responsible for the pathology of Alzheimer's disease (AD). The Aβ fibril is formed by a protein misfolding process in which intermolecular β-sheet interactions become stabilized abnormally. Thus, to develop potential anti-AD drugs, we screened an in-house library to find compounds which have a profile as a β-sheet breaker.
We searched for a β-sheet breaker profile in an in-house library of approximately 113,000 compounds. From among the screening hits, we focused on N,N′-bis(3-hydroxyphenyl)pyridazine-3,6-diamine (named RS-0406), which had been newly synthesized in our laboratory. This compound (10–100 μg ml−1) was found to be capable of significantly inhibiting 25 μM Aβ1–42 fibrillogenesis and, furthermore, disassembling preformed Aβ1–42 fibrils in vitro.
We then investigated the effect of RS-0406 on 111 nM Aβ1–42-induced cytotoxicity in primary hippocampal neurons, and found that 0.3–3 μg ml−1 RS-0406 ameliorates the cytotoxicity. Moreover, 3 μg ml−1 RS-0406 reversed 1 μM Aβ1–42-induced impairment of long-term potentiation in hippocampal slices.
In this study, we have succeeded in identifying RS-0406 which has potential to inhibit Aβ1–42 fibrillogenesis, and to protect neurons against Aβ1–42-induced biological toxicity in vitro. These results suggest that RS-0406 or one of the derivatives could become a therapeutic agent for AD patients.
Keywords: Amyloid β; Alzheimer's disease; fibrillogenesis; β-sheet breaker; 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; long-term potentiation; hippocampus
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by a slowly progressive cognitive decline. In the early stages, neuropathological hallmarks of AD include synaptic dysfunction, formation of neurofibrillary tangles and senile plaques, followed by loss of neurons (Anderton et al., 1998; Selkoe, 2001). Causal therapy has not yet been established, and AD is unfortunately still incurable. Available AD-specific therapy is currently limited to that based on the enhancement of cholinergic function, but this clinical effect is not sufficient in itself (Grundman & Thal, 2000). Therefore, substantial therapeutic intervention is required as soon as possible. Amyloid β protein (Aβ) is a 40–42 amino acid peptide that is the principal constituent of senile plaques. The potential role of Aβ as a neurotoxic agent has been demonstrated in vitro (Pike et al., 1993; Yankner et al., 1990) and in vivo (Emre et al., 1992; Moechars et al., 1999), supporting the hypothesis that Aβ is involved in the pathogenesis of AD.
The main targets for therapeutic intervention of Aβ cascade are classified as follows: (i) inhibition of Aβ production; (ii) inhibition of Aβ aggregation and fibril formation; and (iii) inhibition of the inflammatory response caused by Aβ deposition (plaques are surrounded by microglia which induce inflammation) (Grundman & Thal, 2000). The inhibition of fibril formation is of interest for the therapeutic treatment of AD (Findeis & Molineaux, 1999; Levine & Scholten, 1999) because the fibril form of Aβ becomes extremely toxic to neurons (Pike et al., 1993). The Aβ fibril is formed by a protein misfolding process in which intermolecular β-sheet interactions become stabilized abnormally. Therefore, much effort has been made to develop a therapeutic agent as a β-sheet breaker; however, no drugs have so far succeeded in clinical trials.
In this study, we screened an in-house library to find compounds which have a profile as a β-sheet breaker utilizing a thioflavin T binding assay. From among the screening hits, we focused on N,N′-bis(3-hydroxyphenyl)pyridazine-3,6-diamine (named RS-0406, Figure 1), and further investigated its pharmacological profile by using the following two experimental assays: Aβ1–42-induced inhibition of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction and impairment of long-term potentiation (LTP) by pretreatment with Aβ1–42. MTT reduction is widely used as an indicator of cytotoxicity in cells (Liu et al., 1997). LTP in the hippocampus is one form of synaptic plasticity and is thought to be a cellular mechanism underlying learning and memory (Malinow et al., 2000; Nicoll & Malenka, 1995), which brain functions are severely affected in AD patients.
Figure 1.
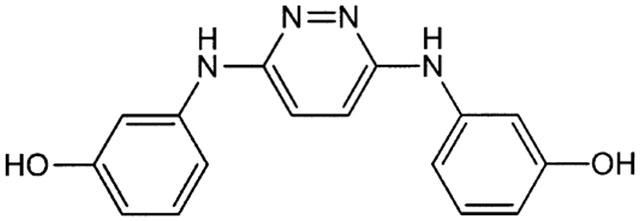
Chemical structure of RS-0406. C16H14N4O2, Mol. Wt.: 294.31.
Methods
Materials
Aβ1–42 (trifluoroacetic acid salt) was purchased from AnaSpec Inc. (San Jose, CA, U.S.A.) and Sigma (St. Louis, MO, U.S.A.), dissolved in 2 mM HCl to enhance the replacement of trifluoroacetic acid by Cl− as a counter ion, and then lyophilized before use (El-Agnaf et al., 1997). Aβ1–42 (HCl salt) was purchased from AnaSpec Inc. All of Aβ1–42 was dissolved at a concentration of 500 μM in phosphate-buffered saline (PBS), and stored in small aliquots at −20°C. RS-0406 was synthesized in our laboratory, and dissolved in dimethyl sulphoxide (DMSO) at a concentration of 30 mg ml−1 or in ethanol at a concentration of 5 mg ml−1. iAβ5 was purchased from Peptide Institute (Osaka, Japan), and dissolved at a concentration of 10 mM in distilled water. Thioflavin T was from Sigma, and MTT was from Wako (Osaka, Japan).
Thioflavin T binding assay
For Aβ1–42 fibrillogenesis, 25 μM Aβ1–42 (trifluoroacetic acid salt, Sigma, replaced by HCl before use) in PBS was incubated with 1–100 μg ml−1 RS-0406 or 2 mM iAβ5 at 37°C for 48 h. RS-0406 dissolved in ethanol was used in this assay, and the final concentration of ethanol was 10%. For dissolution of preformed fibrils, 25 μM Aβ1–42 alone was incubated for 72 h, and then further incubated for 48 h after addition of RS-0406 or iAβ5. Thioflavin T binding was evaluated as reported by Walsh et al. (1999). Briefly, 100 μl of sample was added to a 1 cm path length cuvette containing 800 μl of water and 1 ml of 100 mM glycine-NaOH, pH 8.5. The reaction was initiated by addition of 50 μl of 100 μM thioflavin T in the buffer. Fluorescence was measured for 1 min by using a spectrofluorometer with excitation and emission wavelengths of 435 and 490 nm, respectively. Data were collected from three independent experiments (n=3–9).
Neuronal culture
Primary hippocampal neurons were prepared from the hippocampi of 18-day-old embryos of Wistar rats (Japan SLC, Shizuoka, Japan) as described previously (Kaneko et al., 1995). The hippocampi were incubated in papain (10 U ml−1) for 30 min at 37°C, and dissociated by gentle pipetting. Cells were suspended in Dulbecco's modified Eagle's medium supplemented with 10% foetal bovine serum, 1 mM pyruvate and 20 mM HEPES. They were plated at a density of 18,000 cells per well on poly-L-lysine-coated 96-well tissue culture plates (Sumitomo Bakelite, Tokyo, Japan), and cultured in a humidified atmosphere (5% CO2–95% room air) at 37°C. One hour after plating, the culture medium was changed to a SUMILON Nerve Cell Culture System (SUMILON Medium; Sumitomo Bakelite). To generate fibrils, Aβ1–42 (trifluoroacetic acid, AnaSpec Inc., replaced by HCl before use) was diluted to 100 μM in water, NaCl and HEPES (final concentrations (in mM): NaCl 150, HCl 0.4 and HEPES 20), and then incubated at room temperature for 1–2 days before addition. On day 4, the preincubated 0.5 μg ml−1 (111 nM) Aβ1–42 was added, and 1 h after the addition of Aβ1–42, 0.3–3 μg ml−1 RS-0406 or vehicle (DMSO) was applied to the medium. The final concentration of DMSO was ⩽0.01%. The hippocampal neurons were further cultured for 4 days, and assayed for MTT reduction.
MTT assay
An MTT reduction assay was performed as described previously (Kaneko et al., 1995). Briefly, MTT was dissolved in PBS at 5 mg ml−1 (2.4 mM), and 5 μl solutions were added to 96-well plates containing 50 μl medium per well. After incubation for 3 h at 37°C, the absorbance at 570 nm (reference at 650 nm) of solubilized MTT formazan product was measured with a microplate reader. Data were collected from three independent experiments (n=4–13).
Electrophysiological recordings
To generate fibrils, Aβ1–42 (HCl salt, AnaSpec Inc.) was incubated for 1–2 days before electrophysiological recording in the same way as described in the Neuronal culture section. RS-0406 dissolved in ethanol was used in this experiment. Recording of field excitatory postsynaptic potentials (fEPSPs) in rat hippocampal slices was carried out as described in our previous report (Nakagami et al., 1997) with some modifications. Briefly, 3–4-week-old male Wistar rats (Japan SLC) were anaesthetized with ether, and rapidly decapitated. Transverse hippocampal slices, 400 μm thick, were prepared using a microslicer (Dosaka E.M., Kyoto, Japan). Slices were maintained in oxygenated (95% O2–5% CO2) artificial cerebrospinal fluid (ACSF) at 30°C for at least 1 h prior to pretreatment with drugs. ACSF had the following composition (in mM): NaCl 127, KCl 1.6, KH2PO4 1.24, MgSO4 1.3, CaCl2 2.4, NaHCO3 26 and glucose 10. After the recovery period, the slices were transferred to a small chamber filled with the same ACSF containing 0.1–1 μM Aβ1–42 and/or 3 μg ml−1 RS-0406, and then pretreated in the ACSF for 5 h. The compounds were vortexed, and diluted to desired final concentrations in ACSF immediately before pretreatment in each experiment (n=4–6). The final concentration of ethanol was ⩽0.006%. Control slices were also transferred to the same chamber filled with only ACSF.
After the pretreatment, the slices were transferred to a recording chamber in which they were continuously perfused with warmed (30°C) and oxygenated ACSF at a rate of 1.5–2.0 ml min−1. To thoroughly remove the drug-containing ACSF, the slices were perfused for at least 30 min before recording. The Schaffer collaterals were stimulated with a bipolar electrode, and the evoked fEPSPs were extracellularly recorded from the stratum radiatum of the CA1 region with a glass capillary microelectrode filled with 0.9% NaCl. A rectangular pulse of 50 μsec duration (20–40 μA) was delivered every 30 s with an intensity that evoked a fEPSP of 50–60% of the maximum fEPSP amplitude without a spike. High-frequency stimulation (HFS; 100 pulses at 100 Hz) was applied to induce LTP. For measuring basal synaptic response, fEPSPs were recorded for over 80 min without application of HFS. The degree of paired-pulse facilitation (PPF) was initially determined at an interpulse interval of 20, 50, 80, 140 and 250 msec, and data were expressed as the percentage increase in the slope of the second fEPSP compared to the first. All data were collected using a MacLab/2e system (ADInstruments, Australia), and analysed online using the program (Scope ver. 3.5).
High performance liquid chromatography (HPLC) procedure
In this measurement, RS-0406 dissolved in DMSO was used. RS-0406 was diluted at the concentration of 30 μg ml−1 in PBS, and then incubated at 37°C for 4 days. The final concentration of DMSO was 0.1%. To compare the stability, 30 μg ml−1 RS-0406 without incubation was also prepared before analysis by HPLC. These two samples (2.0 ml) were extracted with ethylacetate (1.0 ml×5 times). The collected organic layer was washed with distilled water and concentrated. The residue was dissolved in 200 μl of DMSO, and diluted with acetonitrile to 1.0 ml.
HP 1100 Binary Pump system (Cat. No. SE-1125; Hewlett-Packard, Avondale, PA, U.S.A.) was connected to an Imtakt Cadenza CD-C18 column (75×4.6 mm). The detector was an HP 1100 Diode Array Detector set up between 210 and 400 mm wavelengths, and connected to a PC for data acquisition. Separation of samples was obtained isocratically by using buffer of the following composition: 80% CH3CN, 20% H2O and 0.01% trifluoroacetic acid. The flow rate was 1.5 ml min−1 and the column temperature was constantly kept at 40°C. Chromatograms of RS-0406 were obtained at 280 nm wavelength.
Statistical analysis
All data in this study are expressed as means±s.e.mean. Significant differences of data were calculated by Tukey's test or Dunnett's test after analysis of variance. Probability values of P<0.05 were considered to represent significant differences.
Results
Effect of RS-0406 on Aβ1–42 fibrillogenesis
In the thioflavin T binding assay, 25 μM Aβ1–42 was incubated for 48 h at 37°C in PBS. This process produced a high proportion of Aβ1–42 fibrils, consistent with our previous report (Kaneko et al., 1995). Under the same conditions, Aβ1–42 was incubated in the presence of RS-0406. Two days after the incubation, RS-0406 significantly inhibited fibril formation of Aβ1–42 in a dose-dependent manner (Figure 2A). To examine the ability of RS-0406 to dissolve preformed fibrils, 25 μM Aβ1–42 was preincubated alone for 72 h at 37°C, and RS-0406 was added to the Aβ1–42 solution in the range of 1–100 μg ml−1. After further incubation for 48 h in the presence of RS-0406, thioflavin T binding was measured. The result showed that RS-0406 disassembled preformed fibrils (Figure 2B). iAβ5 (2 mM) affected neither fibril formation nor preformed fibrils.
Figure 2.
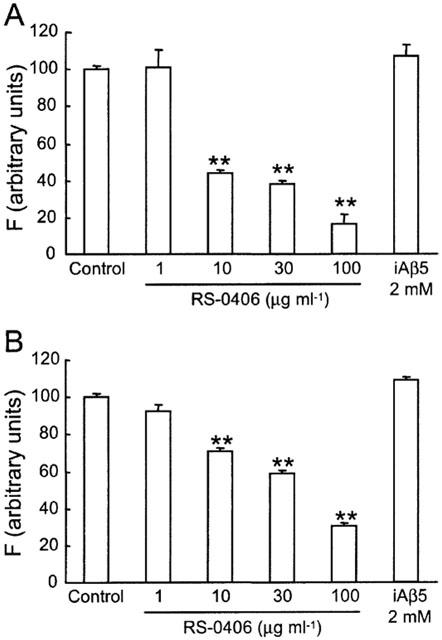
Inhibition of Aβ1–42 fibrillogenesis and dissolution of preformed fibrils by RS-0406. (A) Dose-dependent inhibition of 25 μM Aβ1–42 fibrillogenesis by RS-0406. Amyloid formation was measured by a thioflavin T fluorometric assay, and is expressed as a percentage of the value in the absence of RS-0406. (B) Concentration-dependent disassembly of preformed 25 μM Aβ1–42 fibrils. F: fluorescence intensity. **P<0.01 vs the value in the absence of RS-0406, Dunnett's test (n=3–9).
Effect of RS-0406 on Aβ1–42-induced inhibition of MTT reduction
We previously confirmed that a low concentration of Aβ (100–500 nM) significantly inhibited MTT reduction in primary hippocampal neurons (Kaneko et al., 1995). In this experiment, hippocampal neurons were treated with 0.5 μg ml−1 (approx. 111 nM) Aβ1–42 to detect Aβ1–42-induced cytotoxicity. As shown in Figure 3 , this treatment produced 45% inhibition in MTT reduction, which was almost the same as our previous report. The effect of RS-0406 alone was investigated in the range of 0.3–3 μg ml−1. Although 3 μg ml−1 RS-0406 showed a tendency to inhibit MTT reduction, the difference did not reach statistical significance. Finally, we investigated the effect of RS-0406 on Aβ1–42-induced inhibition of MTT reduction, and found that RS-0406 has potential to significantly ameliorate Aβ1–42-induced cytotoxicity.
Figure 3.
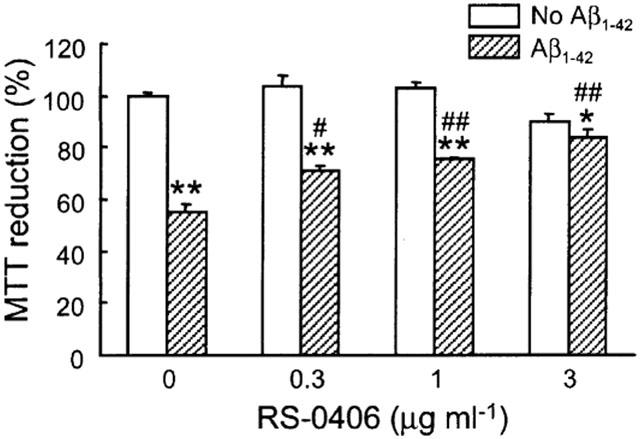
Effects of RS-0406 on Aβ1–42-induced inhibition of MTT reduction in hippocampal neurons. Cultures were treated with RS-0406 for 4 days in the absence (open columns) or presence (hatched columns) of 0.5 μg ml−1 (111 nM) Aβ1–42. *P<0.05, **P<0.01 vs no Aβ1–42 group without RS-0406, #P<0.05, ##P<0.01 vs Aβ1–42 group without RS-0406, Tukey's test (n=4–13 from three independent experiments).
Effect of RS-0406 on Aβ1–42-induced impairment of LTP
As RS-0406 was demonstrated to protect hippocampal neurons against Aβ1–42-induced cytotoxicity, we further investigated the profile of RS-0406 using the hippocampal slices, which more reflects physiological states than dispersed cells. In this study, we newly established an experimental protocol to detect Aβ1–42-induced impairment of LTP. PPF and basal synaptic response without HFS were investigated in control slices, and slices pretreated with 1 μM Aβ1–42 for 5 h. There was no apparent difference between the control slices and Aβ1–42-pretreated slices in PPF (Figure 4A), and the baseline response without HFS did not change over an 80 min period in the Aβ1–42-pretreated slices (Figure 4B). Furthermore, we checked the maximum fEPSP amplitude before the measurement of LTP in the following groups: control slices, slices pretreated with 1 μM Aβ1–42, slices pretreated with 3 μg ml−1 RS-0406, and slices pretreated with 1 μM Aβ1–42 and 3 μg ml−1 RS-0406. Although the maximum fEPSP amplitude in slices pretreated with Aβ1–42, and in slices pretreated with RS-0406, showed a tendency to decrease, it did not reach statistical significance (Figure 4C).
Figure 4.
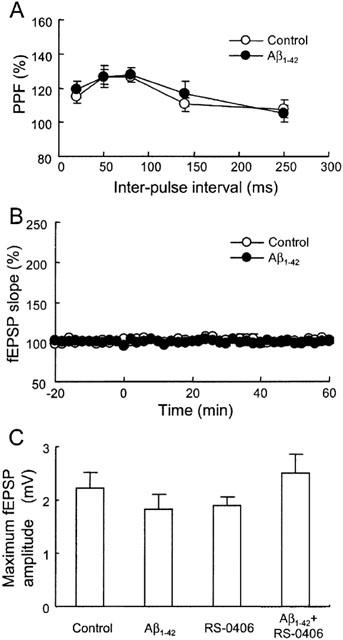
No effect of Aβ1–42 pretreatment on PPF, basal synaptic response and maximum fEPSP amplitude in hippocampal slices. (A) PPF in control slices (open circles; n=5), and slices pretreated with 1 μM Aβ1–42 for 5 h (closed circles; n=5). (B) The time course of basal synaptic response in control slices (open circles; n=4), and slices pretreated with Aβ1–42 (closed circles; n=4). HFS was not applied during the measurement. (C) The maximum fEPSP amplitude before the measurement of LTP. Control: control slices, Aβ1–42: slices pretreated with 1 μM Aβ1–42, RS-0406: slices pretreated with 3 μg ml−1 RS-0406, and Aβ1–42+RS-0406: slices pretreated with 1 μM Aβ1–42 and 3 μg ml−1 RS-0406 (n=5–6).
LTP was also studied in the above groups. In the control slices, application of HFS produced a robust potentiation of fEPSPs, which lasted for 60 min and reached up to a 160% level (Figure 5A,B, open circles). Subsequently, hippocampal slices were pretreated with 0.1–1 μM Aβ1–42 for 5 h. This concentration and application period did not induce any cell death in primary hippocampal neurons (our unpublished data). The same HFS was delivered at time 0, but fEPSPs gradually declined and returned to the baseline level in 1 μM Aβ1–42-pretreated slices (Figure 5A,B, closed circles). Utilizing this model, the effect of RS-0406 on the Aβ1–42-induced impairment of LTP was examined. Pretreatment with 3 μg ml−1 RS-0406 alone did not affect LTP, and RS-0406 blocked the impairment of LTP by pretreatment with Aβ1–42 (Figure 5A,B, open triangles). The absolute value of fEPSP slope at time 0 were as follows: −0.46±0.06 V s−1 in control slices, −0.38±0.03 V s−1 in slices pretreated with 1 μM Aβ1–42, −0.32±0.05 V s−1 in slices pretreated with 3 μg ml−1 RS-0406, and −0.36±0.05 V s−1 in slices pretreated with 1 μM Aβ1–42 and 3 μg ml−1 RS-0406, respectively, and there was no significant difference among the groups. However, comparisons of the slices showed that the magnitude of potentiation 0–60 min after HFS was significantly smaller in 1 μM Aβ1–42-pretreated slices than in control slices (Figure 5C). Furthermore, 3 μg ml−1 RS-0406 showed significant potential to reverse the impairment of LTP induced by pretreatment with 1 μM Aβ1–42.
Figure 5.
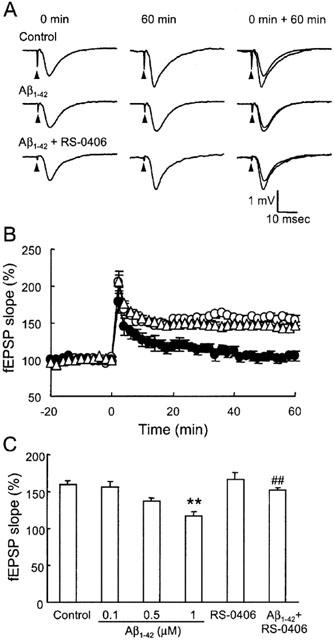
Effect of RS-0406 on Aβ1–42-induced impairment of LTP. (A) Typical fEPSPs recorded in the stratum radiatum of the CA1 area by stimulating the Schaffer collaterals. The fEPSPs immediately before and 60 min after HFS are imposed at the right. Test stimulation was delivered at the times indicated by the arrowheads. (B) The time course of LTP in control slices (open circles; n=5), slices pretreated with 1 μM Aβ1–42 for 5 h (closed circles; n=5), and slices pretreated with 1 μM Aβ1–42 and 3 μg ml−1 RS-0406 for 5 h (n=6). HFS was applied at time 0. LTP was plotted as a percentage of the baseline fEPSP slope. (C) Summary of the effect of pretreatment of Aβ1–42 and/or RS-0406 on LTP. The average percentage of the fEPSP slope 0–60 min after HFS was calculated for each slice. RS-0406: 3 μg ml−1 RS-0406, Aβ1–42+RS-0406: 1 μM Aβ1–42 and 3 μg ml−1 RS-0406, **P<0.01 vs control slices, ##P<0.01 vs 1 μM Aβ1–42-pretreated slices, Tukey's test (n=5–6).
Stability of RS-0406
Finally, we examined the stability of RS-0406 by using HPLC. The signal peak of RS-0406, which had been prepared immediately before the measurement, was detected at a retention time of 2.54 min, and the area under the curve (AUC) was 236.60 mAU/sec (Figure 6A). The two left signals at 0.84 and 0.97 min represented DMSO and ethylacetate, respectively. In the sample of RS-0406, which was incubated for 3 days at 37°C in PBS, the signal peak was detected at 2.54 min, and AUC was 232.81 mAU/sec (Figure 6B).
Figure 6.
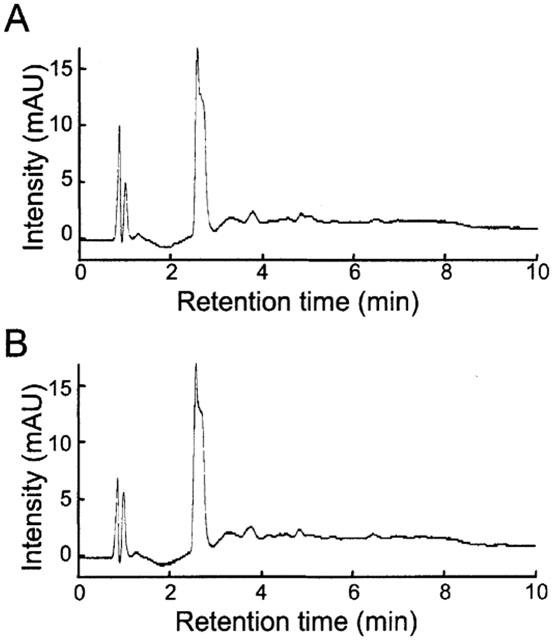
No effect of incubation on stability of RS-0406. (A) HPLC chromatogram of RS-0406, which was prepared immediately before measurement. (B) HPLC chromatogram of RS-0406, which was incubated at 37°C for 4 days. mAU: milli-arbitrary units.
Discussion
We have demonstrated for the first time that RS-0406, a newly synthesized compound, inhibits Aβ1–42 fibrillogenesis, ameliorates Aβ1–42-induced cytotoxicity, and reverses Aβ1–42-induced impairment of LTP. These results raise the possibility that RS-0406 or its derivatives have potential as a therapeutic agent for AD patients.
Fibril formation occurs either by the association of Aβ to existing plaques, or by micelles of Aβ developing into fibrils (Esler et al., 1999). There are several intermediates in the fibrillogenic pathway, and each of the following states is a potential therapeutic target: (i) Aβ monomers or oligomers; (ii) the nucleus of Aβ molecules; (iii) the process of Aβ fibril extension; and (iv) stacking of Aβ fibrils. A thioflavin T binding assay is not sufficient to elucidate how and in which state RS-0406 inhibits Aβ1–42 fibrillogenesis. Therefore, more detailed study is certainly needed to clarify the process at a molecular level, for example, by using atomic force microscopy. It is also important to understand the mechanisms by which Aβ fibril formation is enhanced, and preformed Aβ is disrupted. This information would be helpful to speed up development of drugs that retard formation of the fibrils.
To our knowledge, only the following two potent β-sheet breakers have been reported. A five-residue peptide (LPFFD), referred to as iAβ5, was demonstrated to have a profile that inhibits Aβ fibril formation and Aβ-induced neuronal death and, furthermore, significantly reduces Aβ deposition in vivo. It was also reported that 1.5 mg ml−1 (2.35 mM) iAβ5 was necessary to produce 72% inhibition of 0.5 mg ml−1 (111 μM) Aβ1–42 fibrillogenesis (Poduslo et al., 1999; Soto et al., 1998). We investigated the effect of iAβ5 on fibrillogenesis; however, this inhibitory effect was not apparent under our experimental conditions. As shown in Figure 2A, 30 μg ml−1 (102 μM) RS-0406 inhibited 25 μM Aβ1–42 fibril formation by 60%. Although experimental conditions such as incubation time, incubation buffer and Aβ source are different between those reports and our study, it is possible to assert that RS-0406 is also a strong β-sheet breaker, comparable to the previously reported iAβ5. The other β-sheet breaker previously reported is a natural plant extract, PTI-00703 (McCurley et al., 1998). This extract inhibits Aβ fibrillogenesis, and dissolves preformed Aβ fibrils from different sources, including AD brains and islet amyloid fibrils of type II diabetics. Moreover, the extract has anti-oxidant and anti-inflammatory activity, which may also prove useful for AD therapy. In the near future, an approach using these β-sheet breakers could be successful in developing anti-AD drugs which cure or halt the progression of AD neuropathological alterations.
RS-0406 significantly ameliorated Aβ1–42-induced inhibition of MTT reduction. Considering the profile of RS-0406 as a β-sheet breaker, it would be reasonable to assume that disassembly of Aβ1–42 by RS-0406 resulted in the protective effect. On the other hand, the possibility cannot be ruled out that the effect of RS-0406 is not mediated by disassembly of Aβ1–42, but by signals and/or receptors involved in Aβ1–42-induced cytotoxicity. This assumption might be supported by our preliminary experiments in which some RS-0406 derivatives showed a smaller inhibitory ratio of Aβ1–42 fibrils than that of RS-0406; however, they ameliorated Aβ1–42-induced inhibition of MTT reduction comparably (our unpublished data). In addition, it is unlikely that the above effect resulted from the resolved products of RS-0406, because no other signals except RS-0406 were detected after incubation for 4 days (Figure 6).
RS-0406 reversed the Aβ1–42-induced impairment of LTP in rat hippocampal slices. This finding is important from the point of view that the positive effect of RS-0406 is observed not only at the dispersed cell level but also at the slice level. Although the mechanisms by which RS-0406 reverses the Aβ1–42-induced impairment of LTP are not clear, the profile of RS-0406 as a β-sheet breaker is attributable to the positive effect. Furthermore, similar to the findings of the MTT assay, it remains to be solved how pretreatment with Aβ1–42 impairs LTP. There was no apparent difference between the control and Aβ1–42-pretreated slices in PPF or in basal synaptic response without HFS. Therefore, it is unlikely that pretreatment with Aβ1–42 attenuated the basal properties of presynaptic function or synaptic transmission. Lambert et al. (1998) reported that pretreatment with Aβ-derived diffusible ligands (ADDLs) inhibited hippocampal LTP in vitro. ADDLs comprise only oligomers in protofibril- and fibril-free states, and Fyn, a non-receptor protein kinase, is considered to be involved in ADDLs-induced toxicity (Lambert et al., 1998; Klein et al., 2001). Although there are few reports concerning the impairment of LTP by pretreatment with Aβ so far (Nakagami & Oda, 2002; Iwai & Oka, 2000), the model of pretreatment with Aβ is now available. This experimental protocol is important because a fast screening tool for Aβ inhibitors is indispensable for the development of new therapeutic drugs for AD (Findeis & Molineaux, 1999; Levine & Scholten, 1999).
In conclusion, we succeeded in obtaining a newly synthesized compound, RS-0406, which strongly inhibits Aβ1–42 fibrillogenesis. RS-0406 also has a pharmacological profile including inhibition of Aβ1–42-induced cytotoxicity and impairment of LTP. Further investigations are necessary to examine the mechanisms of RS-0406 and its effect in vivo. However, at present, our results raise the possibility that RS-0406 or one of its derivatives will be developed as a promising novel anti-AD drug in the future.
Abbreviations
- Aβ
amyloid β
- ACSF
artificial cerebrospinal fluid
- AD
Alzheimer's disease
- ADDLs
β-derived diffusible ligands
- AUC
area under the curve
- DMSO
dimethyl sulphoxide
- fEPSPs
field excitatory postsynaptic potentials
- HFS
high-frequency stimulation
- HPLC
high performance liquid chromatography
- LTP
long-term potentiation
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PBS
phosphate-buffered saline
- PPF
paired-pulse facilitation
- RS-0406
N,N′-bis(3-hydroxyphenyl)pyridazine-3,6-diamine
References
- ANDERTON B.H., CALLAHAN L., COLEMAN P., DAVIES P., FLOOD D., JICHA G.A., OHM T., WEAVER C. Dendritic changes in Alzheimer's disease and factors that may underlie these changes. Prog. Neurobiol. 1998;55:595–609. doi: 10.1016/s0301-0082(98)00022-7. [DOI] [PubMed] [Google Scholar]
- EL-AGNAF O.M., IRVINE G.B., GUTHRIE D.J. Conformations of β-amyloid in solution. J. Neurochem. 1997;68:437–439. [PubMed] [Google Scholar]
- EMRE M., GEULA C., RANSIL B.J., MESULAM M.M. The acute neurotoxicity and effects upon cholinergic axons of intracerebrally injected β-amyloid in the rat brain. Neurobiol. Aging. 1992;13:553–559. doi: 10.1016/0197-4580(92)90055-3. [DOI] [PubMed] [Google Scholar]
- ESLER W.P., STIMSON E.R., MANTYH P.W., MAGGIO J.E. Deposition of soluble amyloid-β onto amyloid templates: with application for the identification of amyloid fibril extension inhibitors. Methods Enzymol. 1999;309:350–374. doi: 10.1016/s0076-6879(99)09025-4. [DOI] [PubMed] [Google Scholar]
- FINDEIS M.A., MOLINEAUX S.M. Design and testing of inhibitors of fibril formation. Methods Enzymol. 1999;309:476–488. doi: 10.1016/s0076-6879(99)09032-1. [DOI] [PubMed] [Google Scholar]
- GRUNDMAN M., THAL L.J. Treatment of Alzheimer's disease: rationale and strategies. Neurol. Clin. 2000;18:807–828. doi: 10.1016/s0733-8619(05)70227-9. [DOI] [PubMed] [Google Scholar]
- IWAI T., OKA J. Blockade of LTP in the rat CA1 in vitro by β-amyloid protein (1-42): a role of endogenous GLP-1. Jpn. J. Pharmacol. 2000;82 Suppl 1:236. [Google Scholar]
- KANEKO I., YAMADA N., SAKURABA Y., KAMENOSONO M., TUTUMI S. Suppression of mitochondrial succinate dehydrogenase, a primary target of β-amyloid, and its derivative racemized at Ser residue. J. Neurochem. 1995;65:2585–2593. doi: 10.1046/j.1471-4159.1995.65062585.x. [DOI] [PubMed] [Google Scholar]
- KLEIN W.L., KRAFT G.A., FINCH C.E. Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum. Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- LAMBERT M.P., BARLOW A.K., CHROMY B.A., EDWARDS C., FREED R., LIOSATOS M., MORGAN T.E., ROZOVSKY I., TROMMER B., VIOLA K.L., WALS P., ZHANG C., FINCH C.E., KRAFT G.A., KLEIN W.L. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVINE H., SCHOLTEN J.D. Screening for pharmacologic inhibitors of amyloid fibril formation. Methods Enzymol. 1999;309:467–476. doi: 10.1016/s0076-6879(99)09031-x. [DOI] [PubMed] [Google Scholar]
- LIU Y., PETERSON D.A., KIMURA H., SCHUBERT D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J. Neurochem. 1997;69:581–593. doi: 10.1046/j.1471-4159.1997.69020581.x. [DOI] [PubMed] [Google Scholar]
- MALINOW R., MAINEN Z.F., HAYASHI Y. LTP mechanisms: from silence to four-lane traffic. Curr. Opin. Neurobiol. 2000;10:352–357. doi: 10.1016/s0959-4388(00)00099-4. [DOI] [PubMed] [Google Scholar]
- MCCURLEY D., CASTILLO G.M., SNOW A.D. Therapeutic implications of PTI-00703: a natural plant extract that is a potent dissolver of Alzheimer's disease amyloid. Neurobiol. Aging. 1998;19 Suppl 4:1070. [Google Scholar]
- MOECHARS D., DEWACHTER I., LORENT K., REVERSE D., BAEKELANDT V., NAIDU A., TESSEUR I., SPITTAELS K., HAUTE C.V., CHECLER F., GODAUX E., CORDELL B., VAN LEUVEN F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- NAKAGAMI Y., ODA T. Glutamate exacerbates amyloid β1-42-induced impairment of long-term potentiation in rat hippocampal slices. Jpn. J. Pharmacol. 2002;88:223–226. doi: 10.1254/jjp.88.223. [DOI] [PubMed] [Google Scholar]
- NAKAGAMI Y., SAITO H., MATSUKI N. Optical dye recording of trisynaptic pathway in rat hippocampal slices with a voltage-sensitive dye. Neuroscience. 1997;81:1–8. doi: 10.1016/s0306-4522(97)00161-9. [DOI] [PubMed] [Google Scholar]
- NICOLL R.A., MALENKA R.C. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- PIKE C.J., BURDICK D., WALENCEWICZ A.J., GLABE C.G., COTMAN C.W. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PODUSLO J.F., CURRAN G.L., KUMAR A., FRANGIONE B., SOTO C. β-sheet breaker peptide inhibitor of Alzheimer's amyloidogenesis with increased blood-brain barrier permeability and resistance to proteolytic degradation in plasma. J. Neurobiol. 1999;39:371–382. [PubMed] [Google Scholar]
- SELKOE D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- SOTO C., SIGURDSSON E.M., MORELLI L., KUMAR R.A., CASTANO E.M., FRANGIONE B. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy. Nat. Med. 1998;4:822–826. doi: 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- WALSH D.M., HARTLEY D.M., KUSUMOTO Y., FEZOUI Y., CONDRON M.M., LOMAKIN A., BENEDEK G.B., SELKOE D.J., TEPLOW D.B. Amyloid β-protein fibrillogenesis. J. Biol. Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- YANKNER B.A., DUFFY L.K., KIRSCHNER D.A. Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]