

Abstract
The bioreductive drug EO9 (3-hydroxy-5-aziridinyl-1-methyl-2[indole-4,7-dione]–prop-β-en-α-ol) has good pharmacodynamic properties in vitro, modest anti-tumour activity in experimental tumour models, but failed to show activity in clinical trials. Understanding the reasons for its poor efficacy in vivo is important in terms of progressing second generation analogues into the clinic.
In two human tumour xenografts, direct intra-tumoural injection resulted in improved anti-tumour activity compared with intravenous administration suggesting that drug delivery to tumours is suboptimal. Compared with Mitomycin C (MMC) and the experimental agent MeDZQ, EO9 was rapidly cleared from the systemic circulation (t1/2=1.8 min) whereas MMC and MeDZQ had significantly increased plasma t1/2 values (14 and 22 min respectively). These three compounds demonstrated similar pharmacodynamic properties in terms of potency towards the NQO1 (NAD(P)H:Quinone oxidoreductase) rich H460 cell line in vitro but differed significantly in their in vivo activity with growth delays of 17.7, 4.5 and 1.0 days for MMC, MeDZQ and EO9 respectively. EO9 was rapidly metabolized by red blood cells in vitro (t1/2=14.5 min) which must contribute to its rapid pharmacokinetic elimination in vivo whereas MMC and MeDZQ were metabolized at comparatively slower rates (t1/2>120 min and 77.0 min respectively).
In conclusion, the development of second generation EO9 analogues should address the issue of drug delivery and analysis of drug metabolism by murine whole blood in vitro could be utilized as a preliminary screen to identify lead compounds that are likely to have improved pharmacokinetic profiles in vivo.
Keywords: EO9, mitomycin C, NQO1, bioreductive drugs
Introduction
In the early to mid-1990's, the indolequinone compound EO9 (3-hydroxy-5-aziridinyl-1-methyl-2[indole-4,7-dione]–prop-β-en-α-ol) was the centre of considerable interest as a potential bioreductive agent. Its mechanism of action centres around the enzyme NQO1 (NAD(P)H : Quinone oxidoreductase EC 1.6.99.2) with the final outcome of therapy being determined by a complex interaction between enzyme activity and the oxygenation status of cells (Workman, 1994). EO9 is a good substrate for purified human NQO1 and is reduced to DNA damaging species (single-strand breaks and DNA cross-linking) in both cell free and cell-based assays (Beall et al., 1994; Walton et al., 1991; Bailey et al., 1997). Under aerobic conditions a good correlation between NQO1 activity and chemosensitivity in vitro has been reported by several groups (Fitzsimmons et al., 1996; Robertson et al., 1994; Collard et al., 1995; Plumb et al., 1994), whereas under hypoxic conditions, this relationship is lost. Good hypoxia selectivity is only seen in cell lines that have low levels or are devoid of NQO1 activity (Robertson et al., 1994; Plumb & Workman, 1994; Plumb et al., 1994). Under these conditions, one electron reductases such as cytochrome P450 reductase assume a more prominent role in drug activation (Saunders et al., 2000). Whilst the mechanistic basis for these different biological properties of EO9 are not fully understood (Workman, 1994), it is generally accepted that EO9 would eradicate the aerobic fraction of NQO1 rich tumours whereas in NQO1 deficient tumours, the hypoxic fraction would potentially be targeted.
EO9 was selected for clinical evaluation under the auspices of the European Organisation for the Research and Treatment of Cancer (EORTC) on the basis of its novel mechanism of action, good preclinical activity, and evidence that a number of human tumours possessed elevated levels of NQO1 activity (Hendriks et al., 1993; Malkinson et al., 1992). Despite three partial responses in phase I evaluation, no anti-tumour activity was reported in subsequent phase II studies against breast, gastric, non small cell lung cancer (NSCLC), pancreatic and colorectal cancers (Schellens et al., 1994; Dirix et al., 1996; Pavlidis et al., 1996). Several possible explanations have been put forward to account for EO9's lack of clinical efficacy, but more recent studies have focused on the issue of whether or not sufficient drug is delivered to the tumour to achieve a therapeutic response (Phillips et al., 1998). This proposal is based on the fact that EO9 has a very short half-life in plasma (t1/2 values ranging from 0.8 to 19 min in humans) in conjunction with EO9's relatively poor ability to penetrate through multicell layers in vitro (Phillips et al., 1998; Schellens et al., 1994). These findings suggest that EO9 will penetrate only a few microns from a blood vessel within its pharmacokinetic lifespan, and we have proposed that poor drug delivery to tumours is likely to be the main reason for EO9's lack of clinical efficacy (Phillips et al., 1998).
This view has, however, been challenged by Wilson & Hicks (1999), on the basis that poor drug penetration into tumours is not easy to reconcile with the fact that EO9 is active against several tumour models in mice (Walton et al., 1992; Hendriks et al., 1993; Roed et al., 1989; Adams et al., 1992). It is important to point out that its activity is relatively low, with specific growth delays following drug administration via either the intraperitoneal (i.p.) or intravenous (i.v.) route being typically <1.6 days (Hendriks et al., 1993; Roed et al., 1989). Whilst this level of activity might be expected from a classical bioreductive agent following single agent treatment, EO9 is active against NQO1 rich aerobic cells in vitro, suggesting that if drug delivery to tumours was efficient, much greater anti-tumour activity might be expected. The principle objective of this study was to evaluate two strategies designed to address the question of whether or not poor drug delivery to tumours is responsible for EO9's relative lack of in vivo efficacy. The first objective of this study was to examine the effect of routes of drug administration and drug scheduling on the activity of EO9 in two human tumour xenografts with different NQO1 activities. Two routes of administration were employed, intravenous (i.v.) and intratumoural (i.t.), the latter in an attempt to by-pass some of the pharmacological problems discussed. In addition, the question of drug pharmacokinetics and its potential impact of drug efficacy was determined using three compounds (Mitomycin C (MMC), MeDZQ (2,5-diaziridinyl-1,4-benzoquinone) and EO9, Figure 1) which have similar activity in vitro against H460 cells (Beall et al., 1995) but have marked differences in efficacy in vivo. These studies were conducted with the aim of defining pharmacokinetic parameters which result in good anti-tumour efficacy in vivo for use as a guide to new drug development. In this context, this study also describes a potential novel approach for identifying analogues of EO9 which may have improved pharmacokinetic profiles in vivo, based on previous observations that quinone based compounds are rapidly metabolized by murine whole blood in vitro (Loadman et al., 2000). An understanding of the pharmacological problems surrounding the issue of drug delivery should facilitate the identification of quinone based bioreductive drugs with greater potential for activity in vivo.
Figure 1.
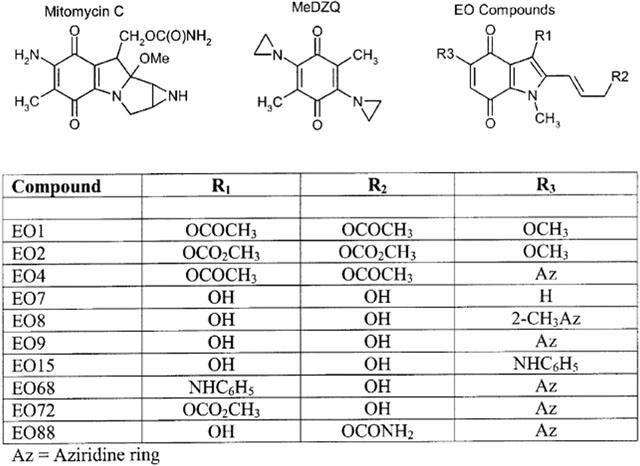
Chemical structures of Mitomycin C, MeDZQ, EO9 and its analogues.
Methods
Chemicals and reagents
EO9 was supplied via the Screening and Pharmacology Group of the EORTC. A series of indolequinone derivatives of EO9 have been extensively studied in both cell free and cellular based assays. These studies included substrate specificity for purified human NQO1, bioactivation to DNA damaging species, their selectivity for NQO1 rich cells in aerobic conditions and limited studies on hypoxia selectivity (Phillips, 1996; Jaffar et al., 1998). All compounds used in this study were initially synthesized by Oostveen & Speckamp (1987) and supplied via the Screening and Pharmacology Group of the EORTC. Compounds were selected on the basis of having a diverse range of properties with respect to NQO1 substrate specificity, selectivity for NQO1 rich cells etc. (Phillips, 1996). These compounds include EO1, EO2, EO4, EO7, EO8, EO9, EO15, EO68, EO72 and EO88, the chemical structures of which are presented in Figure 1. MMC and MeDZQ were used as reference compounds and were purchased from Sigma (Sigma Chemical Co. Ltd., Poole, U.K.) and gifted by Dr John Butler (Paterson Institute for Cancer Research, Manchester, U.K.) respectively. For in vitro experimentation all compounds were dissolved in dimethylsulphoxide (DMSO) to provide stock solutions of 20 mM. Stock solutions were stored at −20°C. High purity HPLC grade solvents (Fisher Scientific, Loughborough, U.K.), analytical grade chemicals (Sigma) and triple distilled water were used throughout.
Animals
NCR nude mice (NCR/nu) aged 6–8 weeks were obtained from the National Cancer Institute (NCI Bethesda, MD, U.S.A.). They were housed in isolation cabinets and fed a CRM pellet diet (CRM, Special Diets Service, Witham, Essex, U.K.) and water ad libitum. All animal experiments were carried out under a project licence approved by the Home Office, London, U.K., and U.K. CCCR guidelines (Workman et al., 1998) were followed throughout.
Tumour system
Human tumour xenografts were established in nude mice by subcutaneous (s.c.) inoculation of cell lines derived from the established cell cultures of the DLD-1 colon cancer cell line and H460 human NSCLC cells. NQO1 activity in the DLD-1 and H460 tumour cells was 27±4 nmol min mg−1 and 1139±120 nmol min mg−1 respectively (Collard et al., 1995; Phillips, 1996). Solid tumour xenografts were passaged from the established xenograft by the use of a trocar. Tumours were grown subcutaneously in the flank and treatment commenced when tumours could be reliably measured by calipers. In vivo chemotherapy studies were performed only on mice bearing tumours that had shown positive growth following a pre-treatment measurement.
Chemotherapy studies
The efficacy of EO9 was determined following administration of EO9 intravenously (i.v.) at 6 mg kg−1, a split schedule of 3 mg kg−1 (i.v.) every hour for 3 h and finally, an intra-tumoural (i.t.) administration of EO9 at 6 mg kg−1. EO9 was dissolved in saline for all (i.v.) administration experiments. For administration of EO9, a drug solution concentrated 4 fold was prepared in 10% dimethylsulphoxide/saline for (i.t.) injection of EO9 and 0.025 ml injected into the tumour per 10 g body weight. MMC and MeDZQ were dissolved in saline and 10% DMSO/saline respectively and administered at maximum tolerated doses (MTD). MMC was administered at 6 mg kg−1 (i.v.) and MeDZQ was administered intraperitoneally (i.p.) at 1 mg kg−1. Three analogues of EO9 were selected for in vivo studies on the basis of favourable properties in cell free and cellular based studies. These included EO4 (a substrate for NQO1 which is reduced to a DNA cross-linking agent, Phillips, 1996), EO72 (a poor substrate for NQO1 but its activity is significantly enhanced under acidic pH conditions, Phillips & Ward, 2001) and EO88 (a very good substrate for NQO1, unpublished data). EO4, EO72 and EO88 were dissolved in 10% DMSO/saline and administered (i.p.) as single injections at previously established MTD of 10, 6 and 10 mg kg−1 respectively. For all chemotherapy studies, groups of 5–10 tumour-bearing mice were treated, and therapeutic effects were assessed by twice weekly caliper measurements of the tumour. Tumour volumes were determined by the formula a2×b/2, where a is the smaller and b is the larger diameter of the tumour. Tumour volumes were normalized relative to their volume at the start of chemotherapy and graphs were plotted of relative tumour volume (RTV) against time. Therapeutic effects were expressed as tumour growth delay obtained by comparing the time taken by treated and control tumours to reach a RTV of two. Statistical comparisons between treated and control groups were made using the Mann–Whitney U-test.
Drug analysis
Chromatographic analysis of EO compounds was based on a previously published method (Phillips et al., 1992). A LiChrosorb RP-18 (25 cm×4.6 mm i.d.) column (Merck Ltd., U.K.) was used for the separation. A Waters 996 Photodiode Array Detector (λ=271) with Millenium Software (Waters Ltd., Watford, U.K.) was used for spectral analysis of the peaks of interest. The flow rate was set at 1.2 ml min−1 with Waters model 515 HPLC pump and samples introduced via Waters 717 plus autosampler. The mobile phase used was phosphate buffer (10 mM, pH 7.0)/methanol mixed at ratios of 44/56 (for EO1, EO2, EO4 and EO68), 50/50 (for EO7, EO8 and EO15) or 57/43 (for EO9, EO72 and EO88) buffer to methanol respectively. Chromatographic conditions for MeDZQ were as described using a mobile phase of methanol/ammonium acetate (0.048 M, pH 7.0) mixed at a ratio of 48 / 52 with detection at 327 nm. Conditions for the analysis of MMC have been reported previously (Phillips et al., 2000).
Pharmacokinetic studies
Pharmacokinetic analysis of plasma levels of EO9, MMC and MeDZQ following administration of drugs at maximum tolerated doses of 6 mg kg−1 (i.v.), 6 mg kg−1 (i.v.) and 1 mg kg−1 (i.p.) respectively were conducted in non-tumour bearing NCR-nu mice. At various time points after administration of drug, mice were anaesthetized and blood samples taken via cardiac puncture and collected in heparinized tubes. Three mice per time point were used for all pharmacokinetic studies. All samples were stored on ice prior to centrifugation at 3000 g for 5 min (at 4°C). Plasma was separated and stored at −20°C until required for analysis. Details of EO9 and MMC extraction from plasma have been described in detail elsewhere (Phillips et al., 1992; 2000). MeDZQ was extracted from plasma using acetonitrile (two volumes) to precipitate plasma proteins and samples centrifuged (3000 g for 5 min). The supernatant was analysed by HPLC. The pharmacokinetic parameters were estimated by standard non-compartmental methods. The terminal elimination rate (Kel) was calculated using linear regression analysis of the terminal log linear portion of the curve and the terminal t1/2 calculated from the equation t1/2=0.693/Kel. The area under the plasma concentration versus time curve (AUC) was calculated using the trapezoidal rule.
Drug metabolism by the blood
The ability of murine blood to metabolize EO9 (and its analogues), MMC and MeDZQ was determined using the chromatographic techniques outlined above. Murine blood samples were taken by cardiac puncture under ether anaesthesia from NMRI mice aged 6–8 weeks obtained from B and K Universal Ltd., (Hull, U.K.) and blood was placed into heparinized tubes on ice. Metabolism studies were conducted using whole blood and blood fractions which included red cells (complete cell or cytosolic and membrane fraction), white cells, plasma and platelet rich plasma. Blood fractions were obtained using Histopaque 1077 (Sigma, U.K.) using protocols specified by the manufacturer. The total blood volume that was separated into the various fractions was 1 ml. Isolated cells were resuspended in 1 ml phosphate buffered saline (PBS) in order to obtain cell concentrations that are equivalent to those which occur in the blood. Red cells were further treated by sonication to release cell contents and the membrane and cytosolic fractions were separated by centrifugation at 3000 g for 5 min (at 4°C). Plasma was separated from whole blood by centrifugation at 3000 g for 5 min. Platelet-rich plasma was obtained by centrifuging whole blood at 100 g for 5 min (at 4°C). Selected samples were heat inactivated (56°C for 20 min) to determine whether the loss of parent compound was due to biological activity. In addition, the stability of all compounds in aqueous solution (PBS) was assessed to obtain evidence of chemical breakdown.
Samples (195 μl) of whole blood or fractionated blood were pre-incubated at the appropriate temperature (37°C or 4°C) and then spiked with drug (20 μM final concentration). At specific time points, protein was precipitated in biological samples by the addition of ice-cold acetonitrile with a solvent/sample ratio of 2 : 1. Typically 30 μl of sample was mixed with 60 μl solvent and centrifuged at 7000 g for 5 min. The supernatant was then used for HPLC analysis, details of which are outlined above. Control (drug-free) samples were used both for calibration purposes and to calculate extraction efficiencies. Control samples were spiked with drug at the appropriate concentration and extracted as described above. Saline spiked with the appropriate concentration of drug was used as 100% control. Recovery from biological samples was then calculated as a percentage of the saline controls. Calibration curves were generated over the range of 0–20 μM. Concentrations of parent compound were monitored at intervals for 2 h and the half-life of the compounds were determined from least squares log linear regression analysis using the equation t1/2=0.693/Kd where Kd is the decay rate constant (i.e. Kd=the slope of the regression analysis×2.303).
Results
Anti-tumour activity and pharmacokinetics of EO9, MMC and RH1
The anti-tumour activity of EO9 against the NQO1 rich H460 tumour and the relatively NQO1 deficient DLD-1 tumour is shown in Table 1. Following a single (i.v.) administration of EO9, moderate activity was seen against the H460 tumour (growth delay of 2.2 days, P<0.05) whereas no significant activity was observed against DLD-1 tumours. Using a split schedule of 3 mg kg−1 administered (i.v.) at hourly intervals, growth delays of 1.0 and 0.9 days were obtained for H460 and DLD-1 tumours respectively. In contrast, direct intra-tumoural injection of EO9 induced significant growth delays of 7.0 and 2.1 days for H460 and DLD-1 human tumour xenografts respectively. MMC was highly effective against the H460 tumour following a single (i.v.) bolus injection (growth delay of 17.7 days). MeDZQ produced a significant growth delay of 4.5 days (P<0.01) in the H460 tumour xenograft following a single (i.p.) administration at 1 mg kg−1.
Table 1.
Response of H460 and DLD-1 human tumour xenografts to EO9, MMC, MeDZQ and EO compounds
Pharmacokinetic parameters following the administration of EO9, MMC and MeDZQ are presented in Table 2 and Figure 2. EO9 was rapidly cleared from the blood plasma with a t1/2 of 1.8 min, whereas MeDZQ and MMC had significantly greater t1/2 values of 22 and 14 min respectively. In terms of overall drug exposure, AUC values for MMC (4.54 μM.h) were significantly greater than MeDZQ (0.509 μM.h) with EO9 having the lowest AUC (0.184 μM.h).
Table 2.
Summary of pharmacokinetic parameters following the administration of MeDZQ, MMC and EO9

Figure 2.
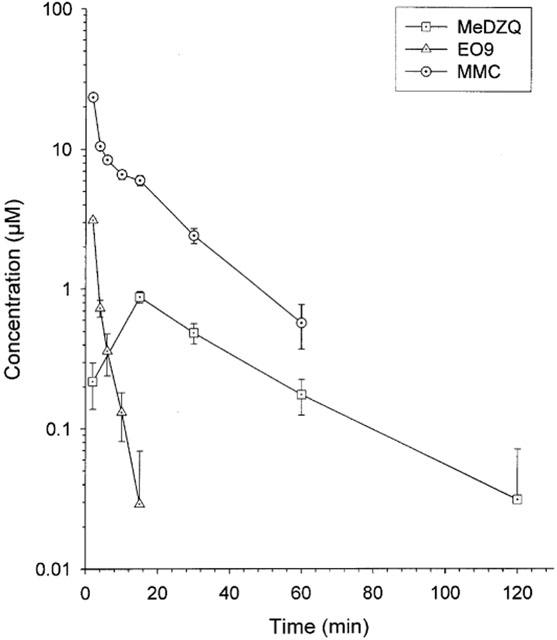
Plasma levels of EO9, MMC and MeDZQ. Each value represents the mean±standard deviation obtained from five mice per time point.
Metabolism of MMC, EO9 and MeDZQ by blood
The chromatographic systems described here produced reproducible data with extraction efficiencies consistently greater than 70% and enabled the stability of EO9 (and its analogues), MMC and MeDZQ to be investigated.
The metabolic breakdown of MMC, EO9 and MeDZQ by murine blood cells is presented in Table 3 and Figure 3. EO9 was stable in plasma but was rapidly metabolized by murine whole blood (t1/2=15.6±2.0 min). Heat inactivation of murine whole blood or cooling to 4°C significantly increased t1/2 values (>120 min) suggesting metabolism of EO9 by blood cells is responsible for its rapid half-life in murine whole blood at 37°C. No metabolites of EO9 were detected on any of the chromatograms (data not presented). Upon fractionation of murine blood, EO9 was not metabolized by white blood cells, plasma or platelet enriched plasma with t1/2 values >120 min (Figure 3). Red blood cells rapidly metabolize EO9 (t1/2=14.5±6.3 min) and further fractionation of the red cell population into membrane and cytosolic fractions demonstrates that EO9 is metabolized predominantly in the cytosol (t1/2=43.0±12.5 min). Both MMC and MeDZQ were found to be stable in murine plasma(t1/2=120 min) with MMC being stable in murine whole blood (t1/2>120 min) and MeDZQ having a calculated t1/2 of 77.0±12.2 min.
Table 3.
Metabolism of quinone based bioreductive drugs by whole and cellular fractions of murine or human blood
Figure 3.
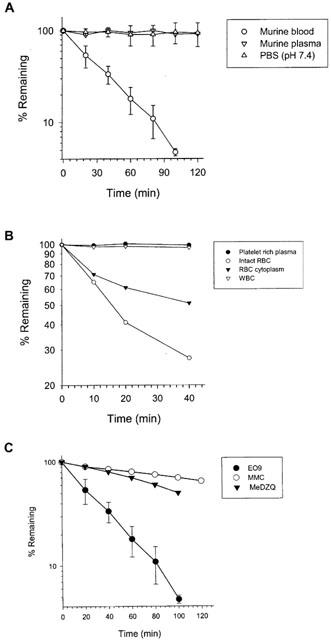
(A) Metabolism of EO9 by murine whole blood, heat inactivated blood, plasma and stability of EO9 in PBS (pH 7.4). (B) Metabolism of EO9 by cellular components of murine whole blood (including the cytosolic fraction of red blood cells). (C) Metabolism of EO9, MMC and MeDZQ by murine whole blood.
EO compounds
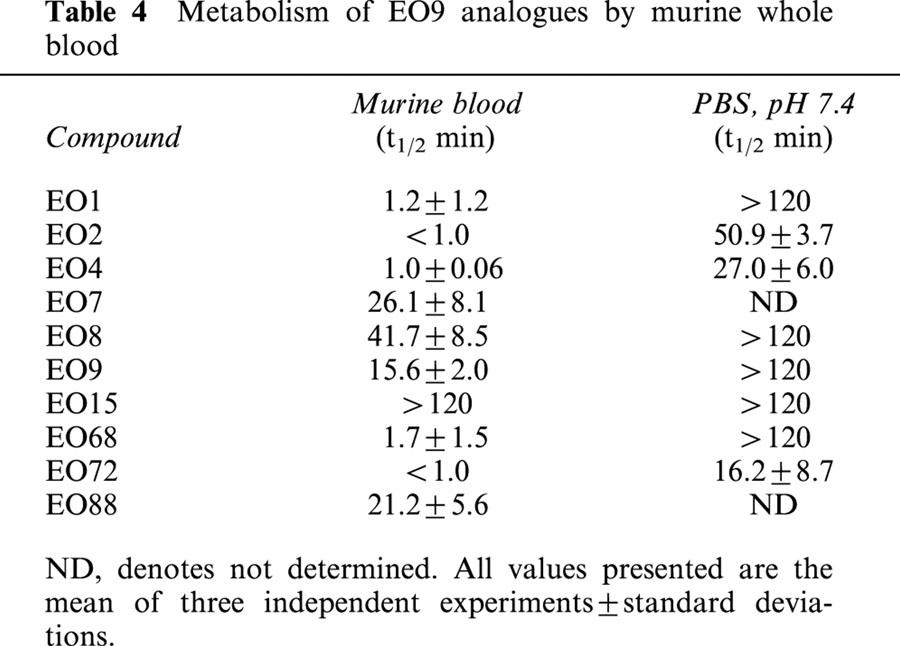
Metabolism of EO9 and its analogues by murine blood is presented in Table 4. A broad spectrum of metabolic rates in murine blood (expressed in terms of t1/2 values) was obtained ranging from no metabolism (EO15 t1/2>120 min) to very rapid metabolism (t1/2 values for EO2 and EO72 of <1 min). Chemical stability in aqueous solution (PBS, pH 7.4) of EO2, EO4 and EO72 were variable with t1/2 values of 50.9±3.7, 27.0±6.0 and 16.2±8.7 min respectively. In comparison with EO9, rates of metabolism by murine blood were similar for EO7, EO8 and EO88, whereas EO15 was not metabolized by murine blood. In comparison with MMC and MeDZQ (Table 3), all compounds with the exception of EO15 were metabolized at a significantly greater rate. The anti-tumour activity of EO4, EO72 and EO88 (selected on the basis of previously published cell free and cellular based screening procedures) against the NQO1 rich H460 human tumour xenograft is presented in Table 4. All three compounds were inactive against the H460 xenograft.
Table 4.
Metabolism of EO9 analogues by murine whole blood
Discussion
Translating activity in vitro into efficacy in vivo is recognized as a major hurdle in drug development and numerous factors associated with tumour biology and drug pharmacology are known to influence the outcome of chemotherapy in vivo (Phillips et al., 1990). These can broadly be separated into pharmacokinetic and pharmacodynamic factors. In the case of EO9, pharmacodynamic studies have clearly demonstrated that EO9 has potent and selective activity against well oxygenated NQO1 rich cells in vitro as well as selective activity against NQO1 deficient cells under hypoxic conditions (Plumb & Workman, 1994; Robertson et al., 1994; Workman, 1994). In addition, the potency of EO9 is significantly enhanced by mild acidic conditions in vitro and the drug is preferentially active against non-proliferating plateau phase cultures compared with cells in exponential growth (Phillips et al., 1992; Choudry et al., 2001; Phillips & Clayton, 1997). These studies suggest that EO9 would not be adversely affected by micro-environmental conditions typically found within poorly vascularized regions of solid tumours. In the in vivo setting, however, EO9's activity is disappointing with relatively small tumour growth delays produced following administration at the maximum tolerated dose (Hendriks et al., 1993; Roed et al., 1989; Collard et al., 1995). Similarly in this study, growth delays of 2.2 and 0 days were obtained following treatment of H460 and DLD-1 human tumour xenografts respectively with EO9 at 6 mg kg−1 administered (i.v.) (Table 1). Understanding the reasons why EO9's good pharmacodynamic properties in vitro do not translate into significant anti-tumour activity in vivo is important, particularly in view of significant efforts being made by several laboratories involved in the development of second generation analogues of EO9 and other quinones (Phillips, 1996; Beall et al., 1998; Jaffar et al., 1998; Naylor et al., 1997; 1998; Skibo et al., 1997).
Previous studies have raised questions over the issue of whether or not drug delivery to tumours is problematical in view of the fact that EO9 is rapidly cleared from the systemic circulation in conjunction with poor penetration across multi-cell layers in vitro (Schellens et al., 1994; Phillips et al., 1998). Whereas split scheduling of EO9 did not result in a significant increase in response, direct intra-tumoural injection of EO9 resulted in enhanced anti-tumour activity in both the H460 and DLD-1 tumour models (Table 1). Similar findings have been reported by Cummings et al. (1998) with significant anti-tumour responses produced following (i.t.) administration of EO9 in a panel of tumours displaying a broad range of NQO1 activity. It is intriguing to note, however, that in this study, the magnitude of the anti-tumour effect following (i.t.) administration remains relatively poor although drug distribution within the tumour after such administration may not be optimal as the drug may have to diffuse longer distances compared with delivery via a vascular route. This view is supported by the fact that multi-cell spheroids are significantly less responsive to EO9 than monolayer cultures suggesting that drug penetration barriers exist for EO9 (Bibby et al., 1993; Choudry et al., 2001). Nevertheless, the results of this and other studies (Cummings et al., 1998) suggest that if EO9 can be efficiently delivered to tumours, significant anti-tumour effects would be achieved.
As previously stated, there is considerable interest in the development of quinone based bioreductive drugs, and numerous compounds have emerged which have similar or improved properties compared with EO9 in terms of mechanisms of action and cellular response in vitro. The central issue that needs to be addressed in the preclinical development of these compounds is whether or not activity in vitro can be translated into efficacy in vivo. In the case of quinone based compounds such as EO9, it is clear that the pharmacological properties of novel compounds (particularly their pharmacokinetic profiles) need to be improved if enhanced efficacy in vivo is to be obtained. In this context, it would be extremely beneficial if pharmacokinetic parameters associated with good anti-tumour efficacy could be identified as these could be employed as yardsticks against which the relative merits of novel compounds can be judged. In this study, the response of H460 xenografts to MMC, MeDZQ and EO9 was determined and marked differences in response were obtained with growth delays of 17.7, 4.5 and 2.2 days respectively (Table 1). In terms of the pharmacokinetic characteristics of each compound, EO9 is rapidly cleared from the systemic circulation with a t1/2 of 1.8 min whereas MMC and MeDZQ have significantly greater plasma t1/2 values of 14 and 22 min respectively (Table 2). Similarly, AUC values for EO9 (0.184 μM.h) are lower than those for MeDZQ (0.509 μM.>h) and significantly lower than MMC (4.54 μM.h). Qualitatively therefore, a correlation between t1/2 and AUC values and growth delay in vivo exists although this conclusion is only valid if other properties (i.e. mechanism of action and cellular response in vitro) are similar for the three compounds evaluated. Extensive studies have demonstrated that all three compounds are substrates for purified NQO1 (although differences exist in substrate specificity with EO9 and MeDZQ being much better substrates than MMC) and are reduced to DNA damaging species (Beall et al., 1995; Bailey et al., 1997; Walton et al., 1991; Siegel et al., 1990). In terms of the response of H460 cells in vitro (Beall et al., 1995), IC50 values for MeDZQ and EO9 are comparable (IC50 values of 0.043 and 0.029 μM respectively) with MMC being relatively less potent (IC50 value of 0.21 μM). It is therefore, unlikely that the major differences in the in vivo anti-tumour activity of these compounds are due to differences in pharmacodynamic properties and that pharmacokinetic characteristics play a significant role in determining efficacy. For novel compounds with similar potencies in vitro to MMC, the results of this study suggest that good anti-tumour activity in vivo could be obtained provided the pharmacokinetic parameters (i.e. t1/2 and AUC) are equivalent to those of MMC. An additional consequence of greater plasma half-lives of compounds is that drug penetration into avascular tissue could be enhanced. Previous studies have demonstrated that the penetration of drugs across multicell layers in vitro is both concentration and time dependent (Phillips et al., 1998; Cowan et al., 1996). The differences in half-lives between EO9 and MMC or MeDZQ may therefore, be significant when the question of drug penetration from the vascular supply into avascular tissue is considered, although further studies are required to address this issue.
Pharmacokinetic analysis and efficacy studies in vivo on extensive numbers of compounds emerging from mechanistic and in vitro chemosensitivity studies is feasible but is compromised by cost and is in conflict with the ethical pressure to reduce animal experimentation. The results of this study offer a potential route by which only those compounds that are most likely to have efficacy in vivo are selected for evaluation. This proposal is based upon the fact that EO9 is rapidly metabolized by murine whole blood in vitro with a t1/2 of 15.6±2.0 min at 37°C (Table 3, Figure 3A,B). The exact metabolic route responsible for EO9's short half-life in murine blood in vitro is unclear, but metabolism is inferred by the fact that EO9 is stable in both heat inactivated blood and blood at 4°C (Table 3). No metabolites (or the major breakdown product EO5A, Phillips et al., 1992) of EO9 were detected on HPLC chromatograms, suggesting that the metabolic products of EO9 are reacting with cellular macromolecules via covalent binding (data not shown). Metabolism of EO9 is confined exclusively to the cytosolic fraction of red blood cells, with no evidence of metabolism by white cells, plasma, platelet enriched plasma or the membrane fraction of red cells. NQO1 activity is present in murine blood (16.7±1.9 nmol min mg−1) and it is conceivable that metabolism of EO9 is due to reduction by NQO1 (data not shown). Other enzymes must, however, play a role as MeDZQ is a good substrate for NQO1 (but is more stable in murine blood than EO9, Table 3) and compounds such as EO1, EO2 and EO72 which are poor substrates for NQO1 (Phillips, 1996) are rapidly metabolized by murine whole blood (Table 4). Whilst the mechanistic basis responsible for EO9 metabolism by murine whole blood is not known, this finding strongly suggests that metabolism by the cytosolic fraction of red blood cells is likely to play a significant role in the rapid pharmacokinetic elimination of EO9 in mice. Identification of compounds that are not metabolized by murine whole blood in vitro may therefore, provide a simple screen to select compounds that may have improved pharmacokinetic parameters in vivo. This is certainly the case with MMC (and to a lesser extent MeDZQ), where reduced rates of drug metabolism do translate into better pharmacokinetic profiles in vivo (Tables 2 and 3). Clearly other factors (particularly metabolism by the liver) will influence drug pharmacokinetics in vivo, but as a means of eliminating those compounds which are rapidly metabolized by murine whole blood in vitro, this procedure would select those compounds that have the potential for improved pharmacokinetic properties in vivo. To examine the potential value of such a screening process, the ability of murine whole blood to metabolize a series of indolequinones was evaluated. Within the panel of compounds evaluated, previous studies identified EO4, EO88 and EO72 as good candidates for in vivo evaluation, based on promising pharmacodynamic properties. EO4 is a good substrate for NQO1 and is reduced to a DNA cross-linking species in cell free assays (Phillips, 1996). Similarly, EO88 is also a good substrate for NQO1 and is selectively toxic to NQO1 rich cells in vitro (unpublished data). EO72 is not a good substrate for NQO1, but its potency in vitro is significantly enhanced under acidic conditions (Phillips & Ward, 2001) suggesting that EO72 could exploit regions of low extracellular pH which exist in solid tumours (Tannock & Rotin, 1989). EO1, EO2, EO4, EO68 and EO72 are extremely rapidly metabolized by murine whole blood with t1/2 values of <2 min whereas EO7, EO8 and EO88 have similar t1/2 values to EO9 (Table 4). EO15 is not metabolized by murine whole blood, but as this compound is a poor substrate for NQO1 and is inactive in vitro against cells with both high and low NQO1 activity, it would not have been considered for in vivo evaluation on this basis (Phillips, 1996). EO4, EO72 and EO88 were inactive against the H460 xenograft in vivo (Table 4). These findings demonstrate that compounds that are metabolized by murine whole blood at rates which are comparable or faster than EO9 are unlikely to have significant anti-tumour efficacy in vivo. In human blood, EO9 is also metabolized in vitro but at a reduced rate (t1/2=78.6±23.0 min) compared with murine blood (data not shown). It is important to stress however, that because of the pivotal role that experimental tumour models in mice play in the preclinical evaluation of novel compounds, the use of murine blood (as opposed to human blood) to identify quinone based compounds which potentially have improved pharmacokinetic properties is essential.
In conclusion, this study has provided further evidence to support the proposal that drug delivery to tumours is the major reason for EO9's disappointing efficacy against experimental tumour models. New drug development should address the issue of improving the pharmacokinetic profile of drugs, which is one factor that will have an obvious bearing on drug delivery. This study has demonstrated that a major reason for EO9's rapid elimination from murine blood is via metabolism by the blood itself. Within a limited panel of indolequinone derivatives of EO9, compounds which are metabolized by murine whole blood at rates that are faster or comparable to EO9 are unlikely to have good anti-tumour efficacy in vivo. Metabolism of novel compounds by murine whole blood in vitro could therefore be employed as part of a cascade to identify compounds which are likely to have better in vivo efficacy than EO9. Further studies to validate this proposal and to identify structural features of molecules that confer poor metabolism by murine whole blood are currently in progress.
Acknowledgments
This work was funded by Cancer Research U.K. (program grant number C459/A2579). The authors also thank C.M. Jarrett, S. Cowan and A.M. Matthew for assistance.
Abbreviations
- AUC
area under the curve
- DMSO
dimethylsulphoxide
- EO9
3-hydroxy-5-aziridinyl-1-methyl-2[indole-4,7-dione]–prop-β-en-α-ol
- EORTC
European Organisation for Research and Treatment of Cancer
- (i.p.)
intraperitoneal
- (i.t.)
intra-tumoural
- (i.v.)
intravenous
- MeDZQ
2,5-diaziridinyl-3,6-dimethyl-1,4-benzoquinone
- MMC
Mitomycin C
- NQO1
NAD(P)H Quinone oxidoreductase 1
- NSCLC
non small cell lung cancer
- PBS
phosphate buffered saline
- (s.c.)
subcutaneous
- RTV
relative tumour volume
References
- ADAMS G.E., STRATFORD I.J., EDWARDS H.S., BREMNER J.C.M., COLE S. Bioreductive drugs as post radiation sensitizers: comparison of dual function agents with SR 4233 and the mitomycin C analogue EO9. Int. J. Radiat. Oncol. Biol. Phys. 1992;22:717–720. doi: 10.1016/0360-3016(92)90510-o. [DOI] [PubMed] [Google Scholar]
- BAILEY S.M., WYATT M.D., FRIEDLOS F., HARTLEY J.A., KNOX R.J., LEWIS A.D., WORKMAN P. Involvement of DT-diaphorase (EC 1.6.99.2) in the DNA cross-linking and sequence selectivity of the bioreductive anti-tumour agent EO9. Br. J. Cancer. 1997;76:1596–1603. doi: 10.1038/bjc.1997.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEALL H.D., MULCHAHY T., SIEGEL D., TRAVER R.D., GIBSON N.W., ROSS D. Metabolism of bioreductive antitumour compounds by purified rat and human DT-diaphorase. Cancer Res. 1994;54:3196–3201. [PubMed] [Google Scholar]
- BEALL H.D., MURPHY A.M., SIEGEL D., HARGREAVES R.H.J., BUTLER J., ROSS D. Nicotinamide adenine dinucleotide (phosphate):quinone oxidoreductase (DT-diaphorase) as a target for bioreductive antitumour quinones: Quinone cytotoxicity and selectivity in human lung and breast cancer cell lines. Mol. Pharmacol. 1995;48:499–504. [PubMed] [Google Scholar]
- BEALL H.D., WINSKI S., SWANN E., HUDNOTT A.R., COTTERILL A.S., O'SULLIVAN N., GREEN S.J., BIEN R., SIEGEL D., ROSS D., MOODY C.J. Indolequinone antitumour agents: Correlation between quinone structure, rate of metabolism by recombinant human NQO1 and in vitro cytotoxicity. Bioorg. Med. Chem Lett. 1998;8:545–548. doi: 10.1016/s0960-894x(98)00069-9. [DOI] [PubMed] [Google Scholar]
- BIBBY M.C., CRONIN B.P., PHILLIPS R.M. Evaluation of the cytotoxicity of the indolequinine EO9 in a human colon adenocarcinoma model. Int. J. Oncology. 1993;3:661–666. doi: 10.3892/ijo.3.4.661. [DOI] [PubMed] [Google Scholar]
- CHOUDRY G.A., HAMILTON-STEWART P.A., DOUBLE J.A., KRUL M.R.L., NAYLOR B., FLANNIGAN G.M., SHAH T.K., BROWN J.E., PHILLIPS R.M. A novel strategy for NQO1 (NAD(P)H:quinone oxidoreductase, EC 1.6.99.2) mediated therapy of bladder cancer based on the pharmacological properties of EO9. Br. J. Cancer. 2001;85:1137–1146. doi: 10.1054/bjoc.2001.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLLARD J., MATTHEW A.M., DOUBLE J.A., BIBBY M.C. EO9: Relationship between DT-diaphorase levels and response in vitro and in vivo. Br. J. Cancer. 1995;71:1199–1203. doi: 10.1038/bjc.1995.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COWAN D.S.M., HICKS K.O., WILSON W.R. Multicellular membranes as an in vitro model for extravascular diffusion in tumours. Br. J. Cancer. 1996;74 Suppl XXVII:S28–S31. [PMC free article] [PubMed] [Google Scholar]
- CUMMINGS J., SPANSWICK V.J., GARDINER J., RITCHIE A., SMYTH J. Pharmacological and biochemical determinants of the antitumour activity of the indolequinone EO9. Biochem. Pharmacol. 1998;55:253–260. doi: 10.1016/s0006-2952(97)00265-7. [DOI] [PubMed] [Google Scholar]
- DIRIX L.Y., TONNESEN F., CASSIDY J., EPELBAUM R., TEN BOKKEL HUININK W.W., PAVLIDIS N., SORIO R., GAMUCCI T., WOLFF I., TE VELDE A., LAN J., VERWEIJ J. EO9 phase II study in advanced breast, gastric, pancreatic and colorectal carcinoma by the EORTC Early Clinical Studies Group. Eur. J. Cancer. 1996;32:A2019–A2022. doi: 10.1016/0959-8049(96)00226-2. [DOI] [PubMed] [Google Scholar]
- FITZSIMMONS S.A., WORKMAN P., GREVER M., PAULL K., CAMALIER R., LEWIS A.D. Reductase enzyme expression across the National Cancer Institute tumour cell line panel: correlation with sensitivity to mitomycin C and EO9. J. Natl. Cancer Inst. 1996;88:259–269. doi: 10.1093/jnci/88.5.259. [DOI] [PubMed] [Google Scholar]
- HENDRIKS H.R., PIZAO P.E., BERGER D.P., KOOISTRA K.L., BIBBY M.C., BOVEN E., DREEF-VAN DER MEULEN H.C., HENRAR R.E., FIEBIG H.H., DOUBLE JJ.A., HORNSTRA H.W., PINEDO H.M., WORKMAN P., SCHWARTSMANN G. EO9: a novel bioreductive alkylating indoloquinone with preferential solid tumour activity and lack of bone marrow toxicity in preclinical models. Euro. J. Cancer. 1993;29:A897–906. doi: 10.1016/s0959-8049(05)80434-4. [DOI] [PubMed] [Google Scholar]
- JAFFAR M., NAYLOR M.A., ROBERTSON N., LOCKYER S.D., PHILLIPS R.M., EVERETT S.A., ADAMS G.E., STRATFORD I.J. 5-Substituted analogues of 3-hydroxymethyl-5-aziridinyl-1-methyl-2-[1H-indole-4,7-dione]prop-2-en-1-ol (EO9, NSC 382459) and their regioisomers as hypoxia selective agents: Structure cytotoxicity in vitro. Anticancer Drug Des. 1998;12:105–123. [PubMed] [Google Scholar]
- LOADMAN P.M., PHILLIPS R.M., LIM L.E., BIBBY M.C. Pharmacological properties of a new aziridinylbenzoquinone, RH1 (2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone), in mice. Biochem. Pharmacol. 2000;59:831–837. doi: 10.1016/s0006-2952(99)00391-3. [DOI] [PubMed] [Google Scholar]
- MALKINSON A.M., SIEGEL D., FORREST G.L., GAZDAR A.F., OIE H.K., CHAN D.C., BUNN P.A., MABRY M., DYKES D.J., HARRISON S.D., ROSS D. Elevated DT-Diaphorase Activity and Messenger-RNA Content in Human Non-Small-Cell Lung-Carcinoma-Relationship to the Response of Lung-Tumour Xenografts to Mitomycin-C. Cancer Res. 1992;52:4752–4757. [PubMed] [Google Scholar]
- NAYLOR M.A., SWANN E., EVERETT S.A., JAFFAR M., NOLAN J., ROBERTSON N., LOCKYER S.D., PATEL K.B., DENNIS M.F., STRATFORD M.R.L., WARDMAN P., ADAMS G.E., MOODY C.J., STRATFORD I.J. Indolequinone antitumour agents: Reductive activation and elimination from (5-methoxy-1-methyl-4,7-dioxoindol-3-yl) methyl derivatives and hypoxia selective cytotoxicity in vitro. J. Med. Chem. 1998;41:2720–2731. doi: 10.1021/jm970744w. [DOI] [PubMed] [Google Scholar]
- OOSTVEEN E.A., SPECKAMP W.N. Mitomycin analogs. 1. Indoloquinones as (potential) bisalkylating agents. Tetrahedron. 1987;43:255–262. [Google Scholar]
- PAVLIDIS N., HANAUSKE A.R., GAMUCCI T., SMYTH J., LEHNERT M., TE VELDE A., LAN J., VERWEIJ J. A randomized phase II study with two schedules of the novel indoloquinone EO9 in non-small-cell lung cancer: a study of the EORTC Early Clinical Studies Group (ECSG) Ann. Oncol. 1996;7:529–531. doi: 10.1093/oxfordjournals.annonc.a010645. [DOI] [PubMed] [Google Scholar]
- PHILLIPS R.M. Bioreductive activation of a series of analogues of 5-aziridinyl-3-hydroxymethyl-1-methyl-2-[1H-indole-4,7-dione] prop-β-en-α-ol (EO9) by human DT-diaphorase. Biochem. Pharmacol. 1996;52:1711–1718. doi: 10.1016/s0006-2952(96)00521-7. [DOI] [PubMed] [Google Scholar]
- PHILLIPS R.M., BIBBY M.C., DOUBLE J.A. A critical appraisal of the predictive value of in vitro chemosensitivity assays. J. Natl. Cancer Inst. 1990;82:1457–1468. doi: 10.1093/jnci/82.18.1457. [DOI] [PubMed] [Google Scholar]
- PHILLIPS R.M., BURGER A.M., LOADMAN P.M., JARRETT C.M., SWAINE D.J., FIEBIG H.H. Predicting tumour response to mitomycin C on the basis of DT-diaphorase activity or drug metabolism by tumour homogenates: Implications for enzyme directed bioreductive drug development. Cancer Res. 2000;60:6384–6390. [PubMed] [Google Scholar]
- PHILLIPS R.M., CLAYTON M.R.K. Plateau phase cultures: An experimental model for identifying drugs which are bioactivated within the microenvironment of solid tumours. Br. J. Cancer. 1997;75:196–201. doi: 10.1038/bjc.1997.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PHILLIPS R.M., HULBERT P.B., BIBBY M.C., SLEIGH N.R., DOUBLE J.A. In vitro activity of the novel indoloquinone EO9 and the influence of pH on cytotoxicity. Br. J. Cancer. 1992;65:359–364. doi: 10.1038/bjc.1992.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PHILLIPS R.M., LOADMAN P.M., CRONIN B.P. Evaluation of a novel in vitro assay for assessing drug penetration into avascular regions of tumours. Br. J. Cancer. 1998;77:2112–2119. doi: 10.1038/bjc.1998.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PHILLIPS R.M., WARD T. Influence of extracellular pH on the cytotoxicity and DNA damage of a series of indolequinone compounds. Anticancer Res. 2001;21:1795–1802. [PubMed] [Google Scholar]
- PLUMB J.A., GERRITSEN M., WORKMAN P. DT-diaphorase protects cells from the hypoxic cytotoxicity of indoloquinone EO9. Br. J. Cancer. 1994;70:1136–1143. doi: 10.1038/bjc.1994.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLUMB J.A., WORKMAN P. Unusually marked hypoxic sensitisation to indoloquinone EO9 and Mitomycin C in a human colon cell line that lacks DT-diaphorase activity. Int. J. Cancer. 1994;56:134–139. doi: 10.1002/ijc.2910560124. [DOI] [PubMed] [Google Scholar]
- ROBERTSON N., HAIGH A., ADAMS G.E., STRATFORD I.J. Factors affecting sensitivity to EO9 in rodent and human tumour cells in vitro: DT-diaphorase activity and hypoxia. Eur. J. Cancer. 1994;30:A1013–A1019. doi: 10.1016/0959-8049(94)90134-1. [DOI] [PubMed] [Google Scholar]
- ROED H., AABO K., VINDELOV L., SPANG-THOMSEN M., CHRISTENESEN I.J., HANSEN H. In vitro and in vivo evaluation of the indolequinone EO-9 (NSC 382459) against human small cell carcinoma of the lung. Eur. J. Cancer Clin. Oncol. 1989;25:1197–1201. doi: 10.1016/0277-5379(89)90415-x. [DOI] [PubMed] [Google Scholar]
- SAUNDERS M., JAFFAR M., PATTERSON A.V., NOLAN J., NAYLOR M.A., PHILLIPS R.M., HARRIS A.L., STRATFORD I.J. The relative importance of NADPH:Cytochrome c (P450) reductase for determining the sensitivity of human tumour cells to the indolequinone EO-9 and related analogues lacking functionality at the C-2 and C-3 positions. Biochem. Pharmacol. 2000;59:993–996. doi: 10.1016/s0006-2952(99)00405-0. [DOI] [PubMed] [Google Scholar]
- SCHELLENS J.H.M., PLANTING A.S.T., VAN ACKER B.A.C., LOOS W.J., DE BOER-DENNERT M., VAN DER BURG M.E.L., KOIER I., KREDIET R.T., STOTER G., VERWEIJ J. Phase I and Pharmacological Study of the Novel Indoloquinone Bioreductive alkylating Cytotoxic Drug EO9. J. Natl. Cancer Inst. 1994;86:906–912. doi: 10.1093/jnci/86.12.906. [DOI] [PubMed] [Google Scholar]
- SIEGEL D., GIBSON N.W., PREUSCH P.C., ROSS D. Metabolism of MMC by DT-diaphorase: Role in MMC induced DNA damage and cytotoxicity in human colon carcinoma cells. Cancer Res. 1990;50:7483–7489. [PubMed] [Google Scholar]
- SKIBO E.B., GORDON S., BESS L., BORUAH R., HEILEMAN M.J. Studies of pyrrolo[1,2-alpha]benzimidazolequinone DT-diaphorase substrate activity, topoisomerase II inhibition activity, and DNA reductive alkylation. J. Med. Chem. 1997;40:1327–1339. doi: 10.1021/jm960546p. [DOI] [PubMed] [Google Scholar]
- TANNOCK I.F., ROTIN D. Acid pH in tumours and its potential for therapeutic exploitation. Cancer Res. 1989;49:4373–4384. [PubMed] [Google Scholar]
- WALTON M.I., BIBBY M.C., DOUBLE J.A., WORKMAN P. DT-diaphorase activity correlates with sensitivity to the indolequinone EO9 in mouse and human colon carcinomas. Eur. J. Cancer. 1992;28:A1597–A1600. doi: 10.1016/0959-8049(92)90049-8. [DOI] [PubMed] [Google Scholar]
- WALTON M.I., SMITH P.J., WORKMAN P. The role of NAD(P)H quinone reductase (EC 1.6.99.2, DT-diaphorase) in the reductive bioactivation of the novel indoloquinone antitumour agent EO9. Cancer Communications. 1991;3:199–206. doi: 10.3727/095535491820873164. [DOI] [PubMed] [Google Scholar]
- WILSON W.R., HICKS K.O. Measurement of extravascular drug diffusion in multicellular layers. Br. J. Cancer. 1999;79:1623–1626. [PubMed] [Google Scholar]
- WORKMAN P. Enzyme directed bioreductive drug development revisited: A commentary on recent progress and future prospects with emphasis on quinone anticancer agents and quinone metabolising enzymes, particularly DT-diaphorase. Oncol. Res. 1994;6:461–475. [PubMed] [Google Scholar]
- WORKMAN P., TWENTYMAN P., BALKWILL F., BALMAIN A., CHAPLIN D., DOUBLE J., EMBLETON J., NEWELL D., RAYMOND R., STABLES J., STEPHENS T., WALLACE J. United Kingdom co-ordinating committee on cancer research (UKCCCR), guidelines for the welfare of animals in experimental neoplasia Br. J. Cancer 1998771–10.2nd edn [DOI] [PMC free article] [PubMed]