

Abstract
The present study was performed to determine the characteristics of the endothelium-derived hyperpolarizing factor (EDHF) that mediates the nitric oxide (NO)- and prostacyclin (PGI2)-independent hyperpolarization and relaxation of porcine renal interlobar arteries.
Bradykinin-induced changes in isometric force or smooth muscle membrane potential were assessed in rings of porcine renal interlobar artery preconstricted with the thromboxane analogue U46619 in the continuous presence of Nω-nitro-L-arginine and diclofenac to inhibit NO synthases and cyclo-oxygenases.
Inhibition of NO- and PGI2-production induced a rightward shift in the concentration-relaxation curve to bradykinin without affecting maximal relaxation. EDHF-mediated relaxation was abolished by a depolarizing concentration of KCl (40 mM) as well as by a combination of charybdotoxin and apamin (each 100 nM), two inhibitors of calcium-dependent K+ (K+Ca) channels. Charybdotoxin and apamin also reduced the bradykinin-induced, EDHF-mediated hyperpolarization of smooth muscle cells from 13.7±1.3 mV to 5.7±1.2 mV.
In addition to the ubiquitous α1 subunit of the Na-K-ATPase, the interlobar artery expressed the γ subunit as well as the ouabain-sensitive α2, α3 subunits. A low concentration of ouabain (100 nM) abolished the EDHF-mediated relaxation and reduced the bradykinin-induced hyperpolarization of smooth muscle cells (13.6±2.8 mV versus 5.20±1.39 mV in the absence and presence of ouabain).
Chelation of K+, using cryptate 2.2.2., inhibited EDHF-mediated relaxation, without affecting NO-mediated responses. Elevating extracellular KCl (from 4 to 14 mM) elicited a transient, ouabain-sensitive hyperpolarization and relaxation that was endothelium-independent and insensitive to charybdotoxin and apamin.
These results indicate that in the renal interlobar artery, EDHF-mediated responses display the pharmacological characteristics of K+ ions released from endothelial K+Ca channels. Smooth muscle cell hyperpolarization and relaxation appear to be dependent on the activation of highly ouabain-sensitive subunits of the Na-K-ATPase.
Keywords: EDHF, smooth muscle hyperpolarization, interlobar artery, Ca2+-dependent K+ channels, Na-K-ATPase, relaxation
Introduction
Endothelium-dependent, but nitric oxide (NO)- and prostacyclin (PGI2)-independent relaxation has been demonstrated in different arteries from various species and has been attributed to the existence of one or more endothelium-derived hyperpolarizing factor(s) (EDHF). The term EDHF most probably describes several factors as the pharmacological characteristics of the NO/PGI2-independent, endothelium-dependent relaxation (and hyperpolarization) in different arteries and species are clearly distinct (McGuire et al., 2001).
Current experimental evidence favours three explanations for the EDHF phenomenon (Busse et al., 2002). The first is an EDHF dependent upon the activation of a cytochrome P450 (CYP) epoxygenase in endothelial cells. We have previously demonstrated that altering the expression of a CYP epoxygenase homologous to CYP 2C8/9 in the endothelium of porcine coronary arteries, alters the magnitude of EDHF-mediated hyperpolarization and relaxation (Fisslthaler et al., 1999; 2000). However, CYP epoxygenases are not expressed in all endothelial cells and in other arteries, CYP enzymes do not play an important role in the regulation of vascular tone. In rat mesenteric and hepatic arteries, for example, CYP inhibitors do not affect NO/PGI2-independent relaxation (Edwards et al., 1998). In these arteries, EDHF is thought to be K+ ions which efflux from endothelial cells through Ca2+-activated K+ (K+Ca) channels, the open probability of which is enhanced by agonist stimulation. The accumulation of K+ in the myo-endothelial space has been proposed to elicit the hyperpolarization of smooth muscle cells by activating inwardly rectifying K+ channels and/or the Na-K-ATPase (Edwards et al., 1998). A third possible explanation for the EDHF phenomenon, is an as yet incompletely defined mechanism involving gap junctional communication, in particular myo-endothelial gap junctional communication and cyclic AMP (Taylor et al., 2001; Rozen & Skaletsky, 2000). At least in the microcirculation, endothelium-dependent hyperpolarization may be mediated by low electrical resistance coupling via myo-endothelial gap junctions (Coleman et al., 2001), although it cannot be excluded that a diffusible factor may transfer from one cell type to another through gap junctions (Fleming, 2000b).
The aim of the present investigation was to characterize the agonist-induced NO/PGI2-independent relaxation of porcine renal interlobar arteries and to determine which mechanism mediates this EDHF response.
Methods
Vessel preparation
Porcine kidneys were obtained from a local slaughterhouse, placed immediately into ice-cold modified Tyrode's solution and transported to the laboratory. Renal interlobar arteries were dissected, cleaned from adventitial adipose and connective tissue and cut into rings (3–4 mm) for the measurement of isometric force. The period between isolation and the start of experiments was less than 6 h.
Vascular reactivity studies
Renal interlobar arteries were mounted between force transducers (Hugo Sachs Elektronik-Harvard Apparatus, Germany) and a rigid support for measurement of isometric force and incubated in static organ baths containing Tyrode's solution of the following composition (mM): NaCl 132, KCl 4, CaCl2 1.6, MgCl2 0.98; NaHCO3, 23.8; NaH2PO4 0.36; glucose 10; Ca-Titriplex 0.05 and gassed with 20% O2, 5% CO2 and 75% N2 to give a pO2 of approximately 140 mmHg and pH 7.4 at 37°C.
Passive tension was adjusted over a 120 min period to 2 g; thereafter segments were repeatedly exposed to a modified Tyrode's solution rich in KCl (80 mM) until stable contractions were obtained. The mean level of tension developed by buffer containing 80 mM of KCl was 6.4±0.2 g.
After washing, arterial rings were exposed to U46619 (0.01–0.5 μM) to achieve a stable contraction which was approximately 80% of the maximal KCl (80 mM)-induced contraction. Thereafter, the integrity of the endothelium was assessed by the application of bradykinin (1 μM) and vessels exhibiting less than 80% relaxation were discarded.
Experiments were performed in the continuous presence of diclofenac (10 μM) and, with the exception of experiments in which NO-mediated responses were evaluated, Nω-nitro-L-arginine (L-NA, 300 μM). To determine whether or not increasing the concentration of extracellular K+ can relax the porcine renal interlobar artery we tested the effects of single and cumulative doses of KCl. In the latter experiments the extracellular concentration of KCl was increased in steps of 2 mM from 4 to 14 mM. In control experiments the concentration of NaCl was increased in similar steps.
Electrophysiological measurements
Renal interlobar artery rings (4–5 mm) were carefully slit along the longitudinal axis. The strips were pinned, luminal or adventitial side downwards, to the bottom of a chamber and continuously superfused (1 ml min−1) with Tyrode's solution containing diclofenac (10 μM), L-NA (300 μM) and U46619 (0.1 μM) to mimic conditions in the organ chamber. Smooth muscle membrane potentials were measured using conventional microelectrodes, pulled with a vertical pipette puller (Sutter Instruments, U.S.A.) from filamented borosilicate glass tubing (Science Products, Germany). Microelectrodes were filled with 3 M KCl. The electrical resistance of the electrodes, measured in modified Tyrode's solution was 50 to 80 MΩ. Impalements were performed from either the adventitial or the intimal side of the vessel. The potential measured was followed on an oscilloscope and traced with a pen recorder. Primary criteria for successful impalement were a sharp voltage deflection upon entering the cell, as judged visually on the oscilloscope, and a sharp return to the baseline value upon withdrawal of the pipette.
Reverse transcriptase PCR (RT–PCR)
For the design of the PCR primers the sequences for the human, rat and, when available, porcine α1, α2, α3 and γ subunits of the Na-K-ATPase from the NCBI database were compared with each other and highly homologous cDNA sequences identified. The sequences were analysed for suitability in PCR with the Primer 3 software provided by the Whitehead Institute for Biomedical Research (Rozen & Skaletsky, 2000) and subsequent structural analysis by the ‘mfold' software provided by the Rensselaer Polytechnic Institute (Santalucia, 1998). The primers used were: for the subunit α1: upstream: AGGGCAGTGTTTCAGGCTAAC, downstream: CCGTGGAGGAGGATAGA(A/G)CTG (fragment size 287 bp); α2: upstream: TCCTGTGGCTCAGTGAGGAA, downstream: GCATCTCCTTGTCAAG(A/C)GGGAT, (fragment size 213 bp); α3 upstream: CCTGTACTCAAGAGGGA(C/T)GTGG downstream: CGCCCTTCATCACTAACAGGTATC (fragment size 158 bp); γ upstream: GACTATGAAACCGT(C/T)CGCAA, downstream: CGTCACAG(C/T)TCATCTTCATTGA (fragment size 139 bp).
Total RNA was isolated with Tri Reagent (Sigma) from interlobar arteries ground to a powder in liquid nitrogen. One microgram total RNA was used for reverse transcription (Promega, AMV polymerase) with random hexamer primers in a total volume of 50 μl. An aliquot (2 μl) from each reaction was used for amplification with the oligonucleotides indicated above. The fragments were amplified using TAQ polymerase (Amersham/Pharmacia) using the following conditions: initial denaturation: 95°C, 10 min; 40 cycles of annealing (1.5 min), elongation (72°C, 1 min) and denaturation (95°C, 1 min) and final elongation at 72°C for 7 min. The annealing temperature for the α2, α3 and γ subunits was 58°C but 55°C for the α1 subunit. After the reactions the fragments were separated on a 1.5% agarose gel and stained by incubation with ethidium bromide. RNA prepared from porcine and rat brain which contain all of the Na-K-ATPase subunits served as a positive control.
Materials
Bradykinin and charybdotoxin were from Bachem (Heidelberg, Germany). U46619 was from Alexis (Grünberg, Germany), nifedipine was from Bayer (Wuppertal, Germany) and Ca-Titriplex was from Merck (Darmstadt, Germany). All other substances were obtained from Sigma (Deisenhofen, Germany).
Statistics
Data are expressed as the mean±s.e.mean and statistical evaluation was performed using Student's t-test for paired or unpaired data, one-way analysis of variance (ANOVA) followed by a Bonferroni t-test, or ANOVA for repeated measures where appropriate. Values of P<0.05 were considered statistically significant. Rmax represents the maximal relaxation recorded in response to the cumulative addition of a given agonist and pD2 (−log EC50) values were calculated by non-linear regression of the concentration-relaxation curves to bradykinin.
Results
NO/PGI2-independent relaxation
In renal interlobar artery rings precontracted with the thromboxane agonist U46619, bradykinin elicited concentration-dependent relaxations. The inclusion of L-NA (300 μM) in the organ bath did not affect the maximal bradykinin-induced relaxation but resulted in a significant rightward shift in the concentration-relaxation curve to bradykinin (pD2 values were 8.5±0.3 versus 7.5±0.3 in the absence and presence of L-NA, n=6, P<0.01; Figure 1a). The L-NA-insensitive relaxation to bradykinin was not affected by the addition of the soluble guanylyl cyclase inhibitor, NS2028 (not shown). A depolarizing concentration of KCl (40 mM) increased the basal tone of interlobar arteries (by 3.9±0.2 g in the absence and 6.1±0.1 g in the presence of L-NA) but prevented the bradykinin-induced, NO/PGI2-independent relaxation of arterial rings which were preconstricted to the same extent with either KCl or KCl and U46619 (Figure 1b).
Figure 1.
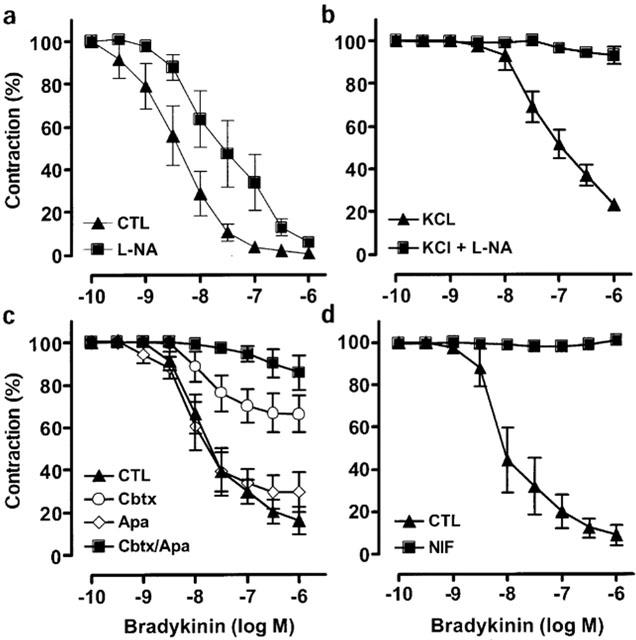
Pharmacological characterization of NO/PGI2-independent relaxation of the interlobar artery. (a) Arterial rings were preconstricted with U46619 (0.01–0.5 μM) in the continuous presence of diclofenac (10 μM) and concentration-relaxation curves to bradykinin (0.1 nM to 1 μM) were obtained in the absence (CTL) and presence of Nω-nitro-L-arginine (L-NA, 300 μM). (b) Arterial rings were preconstricted with a combination of KCl (40 mM) and U46619 and concentration-relaxation curves to bradykinin were obtained in the absence (KCl) and presence of L-NA (KCl+L-NA). (c) Arterial rings were preconstricted with U46619 in the continuous presence of diclofenac and L-NA and concentration-relaxation curves to bradykinin were obtained in the absence (CTL) and presence of charybdotoxin (Cbtx, 100 nM), apamin (Apa, 100 nM) or a combination of charybdotoxin and apamin. (d) Arterial rings were preconstricted with U46619 (0.01–0.6 μM) in the continuous presence of diclofenac, L-NA and concentration-relaxation curves to bradykinin (0.1 nM to 1 μM) were obtained. Experiments were performed in the absence (CTL) and presence of nifedipine (NIF, 1 μM). Presented are the mean±s.e.mean of five to six separate experiments.
Charybdotoxin (100 nM), a blocker of intermediate and large conductance K+Ca channels markedly inhibited NO/PGI2-independent relaxations (Figure 1c). The combination of charybdotoxin and the blocker of small-conductance K+Ca channels, apamin (100 nM), almost completely prevented bradykinin-induced, NO/PGI2-independent relaxation (Figure 1c). Apamin alone did not significantly affect NO/PGI2-independent relaxations (Figure 1c), whereas iberiotoxin (100 nM), which targets large conductance K+Ca channels significantly attenuated NO/PGI2-independent relaxations (Table 1). Since the sensitivity to charybdotoxin/apamin is a hallmark of EDHF-mediated responses, the NO/PGI2-independent relaxation of the interlobar artery is subsequently referred to as an EDHF-mediated relaxation.
Table 1.
Pharmacological characterization of the EDHF-mediated relaxation of porcine renal interlobar arteries to bradykinin
EDHF-mediated smooth muscle hyperpolarization is thought to induce relaxation by decreasing the open probability of voltage-dependent Ca2+ channels. We therefore determined the effects of the Ca2+ antagonist nifedipine (1 μM) on the bradykinin-induced EDHF-mediated relaxation of interlobar arteries. Significantly more U46619 was required to contract rings pre-treated with nifedipine to the reference level of 80% of the maximal KCl-induced contraction (0.60±0.04 versus 0.09±0.004 μM in nifedipine and solvent-treated rings respectively, P<0.001, n=6). However, in nifedipine pre-treated rings bradykinin failed to elicit an EDHF-mediated relaxation (Figure 1d). The inhibitory effect of nifedipine was selective for EDHF as the NO-mediated relaxation was potentiated by the Ca2+ antagonist (Table 2), while the endothelium-independent relaxations elicited by sodium nitroprusside were unaffected by nifedipine (Table 2).
Table 2.
Effect of nifedipine pretreatment on the NO-mediated relaxation of porcine interlobar arteries to bradykinin and sodium nitroprusside
Characterization of the EDHF-mediated relaxation
17-Octadecynoic acid (10 μM), a suicide inhibitor of some CYP enzymes, had no effect on EDHF-mediated relaxations (not shown). Sulphaphenazole, a specific inhibitor of CYP 2C9 (Sai et al., 2000) which inhibits the EDHF-mediated relaxation of large porcine coronary arteries (Fisslthaler et al., 1999), caused a small, but significant, rightward shift of the concentration-relaxation curve to bradykinin (Table 1).
The gap junction blocker 18α-glycyrrhetinic acid (30 μM), which inhibits the EDHF-mediated relaxation in the rabbit mesenteric artery (Chaytor et al., 2000) had no effect on the basal or U46619-induced tone of arterial rings and did not affect the EDHF-mediated relaxation of the interlobar artery (Table 1).
As combined blockade of the inwardly rectifying K+ channels by barium and the Na-K-ATPase by ouabain has been reported to abolish EDHF-mediated hyperpolarization (Edwards et al., 1998), we determined the effects of these inhibitors on the bradykinin-induced relaxation of interlobar artery rings. Barium (30 μM) did not affect the EDHF-mediated relaxation (data not shown). Moreover, concentrations of ouabain (10–100 μM) previously reported to inhibit EDHF-mediated relaxations (Wetzel & Sweadner, 2002; Beguin et al., 1997), elicited a contraction that was equivalent to that induced by 80 mM KCl. In rings of interlobar artery, markedly lower concentrations of ouabain, which did not affect basal tension, attenuated the bradykinin-induced EDHF-mediated relaxation. Rmax was reduced from 91.3±3.0% to 15.5±8.1% (P<0.01, n=6) in the presence of 10 nM ouabain while EDHF-mediated relaxation was abolished by 100 nM ouabain (Figure 2a).
Figure 2.
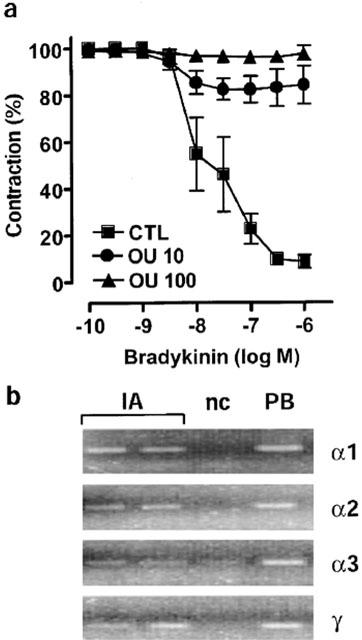
Effect of ouabain on EDHF-mediated relaxations and expression of the Na-K-ATPase in the interlobar artery. (a) Concentration-response curves to bradykinin (0.1 nM to 1 μM) were obtained in rings preconstricted with U46619 (0.01–0.5 μM) in the absence (CTL) and presence of 10 or 100 nM of ouabain (OU 10, OU 100, respectively). At the doses tested ouabain did not affect basal tension of preconstricted rings. Presented are the mean±s.e.mean of seven separate experiments. (b) Total RNA was isolated from samples of porcine interlobar artery (IA) and porcine brain (PB) and subjected to RT–PCR as described in the text. PCR products were visualized by staining the agarose gel with ethidium bromide. A negative control (n.c., no reverse transcriptase in the RT reaction) was included in each experiment. Identical results were obtained in three additional experiments and each sample was obtained from a different animal.
Comparison of EDHF-mediated and KCl-induced relaxation
The high sensitivity of EDHF-mediated responses in the interlobar artery to ouabain implied that this vessel expresses one or more of the ouabain-sensitive α2, α3 subunits in addition to the ubiquitous but relatively ouabain-insensitive α1 subunit. RT–PCR revealed that the porcine interlobar artery expresses the mRNA of the three known α-subunits, among them the ouabain-sensitive α2 and α3 subunits (Figure 2b). The interlobar artery also expresses mRNA encoding the γ subunit which is expressed almost exclusively in the kidney (Wetzel & Sweadner, 2002). The expression of the α1 and α2 subunits was confirmed by Western blotting (not shown).
Low concentrations of KCl (0.1 to 5 mM) are known to activate the Na-K-ATPase in vascular smooth muscle cells (Campbell et al., 1996), and increasing the extracellular concentration of KCl from 4 to 12 mM, elicited an endothelium-independent relaxation of interlobar artery rings (Figure 3a). The kinetic of the KCl-induced relaxation was similar to that of the EDHF-mediated relaxation (compare Figures 3a and 4a). Equivalent concentrations of NaCl were without effect (data not shown). The KCl-induced relaxation was converted to contraction in vessels pre-treated with ouabain (100 nM, Figure 3c), was unaffected by either barium (30 μM, data not shown), or the combination of charybdotoxin and apamin (each 100 nM; Figure 3c) and abolished by nifedipine (Figure 3d). The KCl-induced relaxation was comparable in arteries preconstricted with either U46619 or with phenylephrine (data not shown). The relaxant response to cromakalim (10 μM) was not affected by ouabain indicating that, under the experimental conditions used, the hyperpolarization of smooth muscle cells was still able to elicit relaxation (data not shown). In the presence of the K+ chelator cryptate 2.2.2 (2 mM), which was reported to inhibit the bradykinin-induced, EDHF-mediated relaxation of porcine coronary arteries (Beny & Schaad, 2000), the EDHF-mediated relaxation of the interlobar artery was significantly attenuated (Figure 4b). This effect was reversible and after 2 h wash-out, EDHF-mediated relaxations to bradykinin were restored (data not shown). Inclusion of cryptate 2.2.2 did not influence basal tone or the sodium nitroprusside-induced relaxation (data not shown), however less U46619 was required for preconstriction of cryptate-treated arterial rings (47±2.3 versus 87±3.9 nM U46619, n=5, P<0.05).
Figure 3.
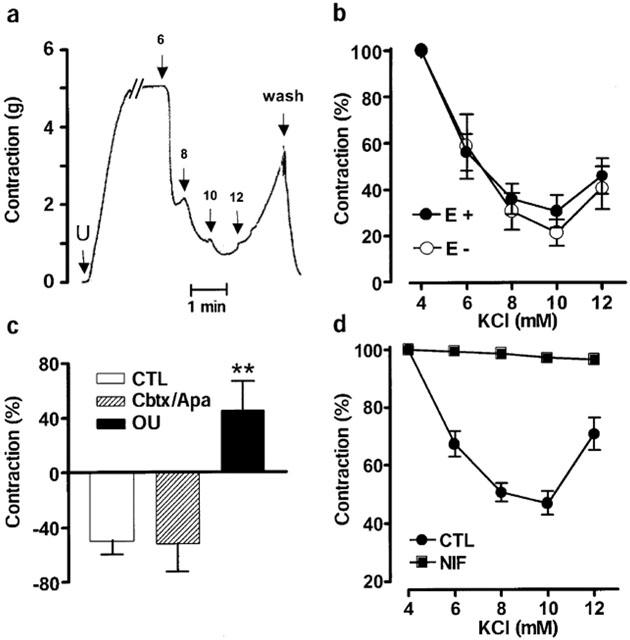
Pharmacological characterization of K+-induced relaxation. Original tracing in an endothelium-intact ring of interlobar artery (a) and statistical summary (b) showing the relaxant effect of the cumulative addition of KCl on the tone of arterial rings preconstricted with U46619 (U, 80 nM). Experiments were performed using endothelium-intact (E+) and endothelium-denuded (E-) rings. (c) Effect of the combination of charybdotoxin and apamin (Cbtx/Apa, each 100 nM) and ouabain (OU, 100 nM) on the KCl (10 mM; 14 mM final bath concentration)-induced relaxation of endothelium-denuded arterial rings, preconstricted with U46619 (80 nM). (d) Effect of nifedipine (1 μM) on the relaxation of U46619-contracted arterial rings induced by the cumulative addition of KCl. Presented are the mean±s.e.mean of six separate experiments.
Figure 4.
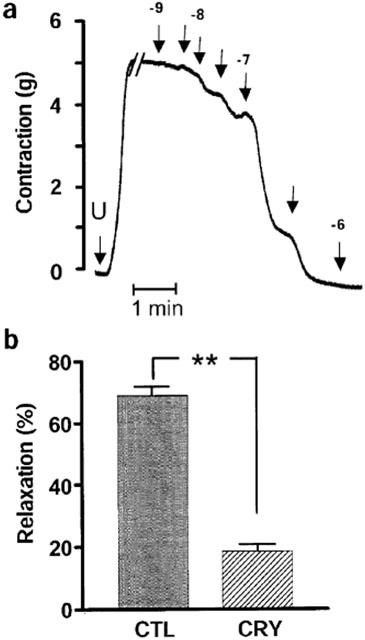
Effect of K+ chelation on EDHF-mediated relaxations. (a) Original tracing in an endothelium intact ring of interlobar artery showing the bradykinin (0.1 nM to 1 μM, arrows indicate application, interim concentrations 3 nM etc are unlabelled)-induced, EDHF-mediated relaxation of arterial rings preconstricted with U46619 (U, 80 nM). (b) Effect of the K+ chelator cryptate 2.2.2 (CRY, 2 mM added 10 min before preconstriction) on the bradykinin-induced (100 nM), EDHF-mediated relaxation of U46619-preconstricted rings of interlobar artery. Experiments were performed in the presence of L-NA (300 μM) and diclofenac (10 μM). Presented are the mean±s.e.mean of six separate experiments; **P<0.01.
Electrophysiological measurements
Electrophysiological experiments to record changes in smooth muscle membrane potential were performed in the presence of L-NA, diclofenac and U46619, to mimic the conditions in which tone was assessed. The inclusion of U46619 (100 nM, the concentration most routinely used in the reactivity studies) did not depolarize vascular smooth muscle cells (the resting membrane potential was −54.2±1.83 versus −54.0±2.63 mV in the absence and presence of U46619, n=7). However, increasing the concentration of U46619 to 600 nM (as in the experiments assessing the effect of nifedipine on NO/PGI2-independent relaxations) did elicit the depolarization of vascular smooth muscle cells (the resting membrane potential was −54.2±1.83 versus −45.6±3.02 mV in the absence and presence of U46619, P<0.05, n=7).
In the presence of L-NA, diclofenac and U46619 (100 nM), bradykinin elicited a significant, endothelium-dependent hyperpolarization of vascular smooth muscle cells (Figure 5a,b). The presence of charybdotoxin and apamin had no significant effect on the resting membrane potential (−47.9±1.1 versus −49.4±3.1 mV in the absence and presence of charybdotoxin/apamin, n=7), but significantly attenuated the bradykinin-induced, EDHF-mediated hyperpolarization of smooth muscle cells (Figure 5b). Ouabain (100 nM) induced a significant depolarization of smooth muscle cells (the resting membrane potential being −53.2±2.9 mV versus −44.4±2.2 in the absence and presence of ouabain, P<0.05, n=5) and markedly attenuated the bradykinin-induced hyperpolarization (Figure 5c). Increasing the extracellular KCl concentration from 4 to 14 mM resulted in the transient hyperpolarization of smooth muscle cells (Figure 6). Depolarization rather than hyperpolarization was observed when the same experiment was repeated in the presence of ouabain (Figure 6).
Figure 5.
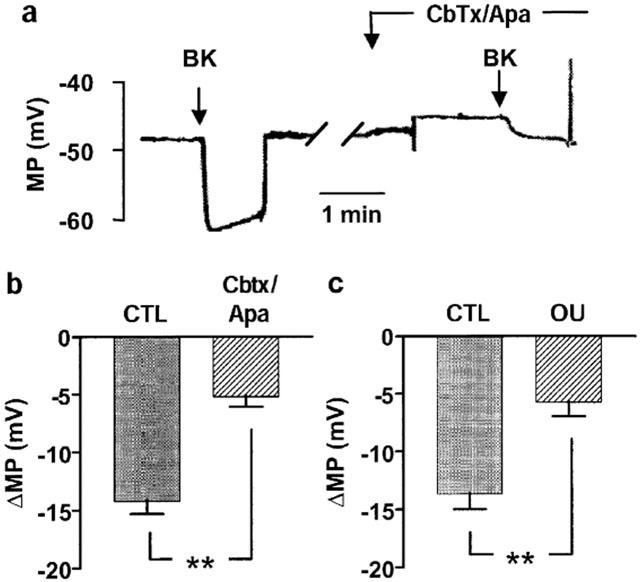
Pharmacological characterization of the bradykinin-induced, EDHF-mediated hyperpolarization of the interlobar artery. (a) Original tracings showing the effect of bradykinin (Bk, 100 nM) on the membrane potential (MP) of porcine interlobar smooth muscle cells (impaled from the intimal surface). Experiments were performed in the absence (CTL) and presence of charybdotoxin and apamin (Cbtx/Apa, each 100 nM). (b,c) Statistical summaries showing the effect of (b) charybdotoxin and apamin (Cbtx/Apa, each 100 nM) and (c) ouabain (OU, 100 nM) on the bradykinin-induced hyperpolarization (ΔMP) of porcine interlobar smooth muscle cells. All experiments were performed in the continuous presence of L-NA (300 μM) diclofenac (10 μM) and U46619 (100 nM). Presented are the mean±s.e.mean of seven separate experiments; **P<0.01.
Figure 6.
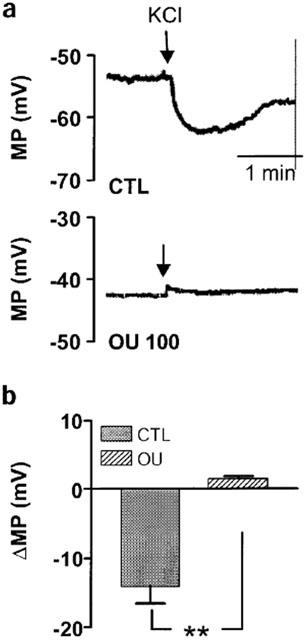
KCl-induced hyperpolarization of the interlobar artery. (a) Original showing the effect of KCl (10 mM final bath concentration) on the membrane potential (MP) of interlobar artery smooth muscle cells (impaled from the intimal surface). Experiments were performed in the absence and presence of ouabain (OU, 100 nM). (b) Statistical summary showing the effect of ouabain on the KCl-induced hyperpolarization (ΔMP) of the porcine interlobar artery. All experiments were performed in the continuous presence of L-NA (300 μM) diclofenac (10 μM) and U46619 (100 nM). Presented are the mean±s.e.mean of five separate experiments; **P<0.01.
In all experiments, identical results were obtained irrespective of whether smooth muscle cells were impaled from the intimal or adventitial side of the arteries. The resting membrane potential in the presence of L-NA, diclofenac and 100 nM U46619 was −53±1.83 mV when impaled from the intimal side and −51.5±3.89 mV when impaled from the adventitial side.
Discussion
The results of the present study demonstrate that a substantial portion of the bradykinin-induced relaxation of the porcine interlobar artery is mediated by a mechanism which does not involve the generation of either NO or PGI2. This NO/PGI2-independent relaxation was associated with the hyperpolarization of smooth muscle cells and could therefore be attributed to the generation and/or transfer of an endothelium-derived hyperpolarizing factor (EDHF). EDHF-mediated relaxation could be mimicked by increasing the extracellular concentration of KCl and was attenuated in the presence of a K+ chelator. The EDHF- as well as the K+-induced relaxation of interlobar arteries was exquisitely sensitive to low concentrations of the Na-K-ATPase inhibitor ouabain, supporting the concept that K+ released from endothelial cells elicits the hyperpolarization and relaxation of smooth muscle cells by activating the Na-K-ATPase.
Three types of NO/PGI2-independent relaxation can be characterized pharmacologically. One EDHF is dependent on the activation of CYP epoxygenases in endothelial cells and can be inhibited by blocking CYP activity (Fleming, 2000a). Another type of EDHF-mediated relaxation is sensitive to uncouplers of gap junctional communication (Taylor et al., 2001). This EDHF is thought to involve the transfer of an electrical or humoral signal from endothelial to smooth muscle cells via myo-endothelial gap junctions. The third EDHF is thought to be K+ ions which efflux from activated endothelial cells through K+Ca channels (Edwards et al., 1998).
The first two types of EDHF appear to play only a minor role in the EDHF-mediated hyperpolarization and relaxation of porcine interlobar arteries. A process involving gap junctional communication is unlikely to account for the EDHF-mediated relaxation observed, as the gap junction blocker 18α-glycyrrhetinic acid did not affect bradykinin-induced, EDHF-mediated responses. However, we did not investigate the effects of other agents that affect gap junctional communication. A CYP-dependent process plays only a limited role in the EDHF response inasmuch as sulphaphenazole, a selective inhibitor of CYP 2C9, and iberiotoxin, which inhibits the hyperpolarization induced by CYP products such as the epoxyeicosatrienoic acids (Busse et al., 2002), exhibited only modest inhibitory effects on EDHF-mediated relaxation. Rather, the EDHF-mediated responses in the porcine interlobar artery could be explained by the release of K+ ions from the agonist-stimulated endothelium and the subsequent activation of the Na-K-ATPase.
One characteristic of EDHF-mediated responses is their sensitivity to a combination of the K+Ca channel blockers, charybdotoxin and apamin (Chaytor et al., 1997). The fact that only the combination of the two toxins is effective implies that all three types of K+Ca channel are implicated in EDHF-mediated responses as apamin inhibits small conductance K+Ca channels, whereas charybdotoxin inhibits large and intermediate conductance K+Ca channels. The K+Ca channels activated in response to agonist stimulation are reported to be situated on endothelial cells rather than vascular smooth muscle cells (Baron et al., 1996), implying that the activation and hyperpolarization of endothelial cells is an absolute pre-requisite for the EDHF-mediated hyperpolarization and relaxation of smooth muscle cells. In the porcine renal interlobar artery, charybdotoxin and apamin attenuated EDHF-mediated hyperpolarization and abolished the associated relaxation. However, charybdotoxin alone markedly inhibited EDHF-mediated relaxation, perhaps indicating a greater dependence on intermediate and large conductance K+Ca channels in these arteries, a finding which fits with the observations made using iberiotoxin.
As an increase in extracellular KCl is reported to activate the inwardly rectifying K+ channel in rat coronary and intracerebral arteries (Knot et al., 1996), and dilation to K+ seems to be fully dependent on the Na-K-ATPase in rabbit arcuate arteries smooth muscle cells (Prior et al., 1998), we determined which effector mechanism was most likely to mediate the EDHF-induced relaxation of interlobar smooth muscle cells. Barium, which inhibits the inwardly-rectifying K+ channel, was without effect whereas low concentrations of the Na-K-ATPase inhibitor ouabain, attenuated both EDHF-mediated hyperpolarization and relaxation.
Ouabain was originally reported to inhibit the acetylcholine-induced hyperpolarization and relaxation of canine femoral and coronary arteries (Feletou & Vanhoutte, 1988). However, in these early studies such high concentrations of ouabain were employed that smooth muscle depolarization was pronounced and it was impossible to distinguish between inhibitory effects on the α1 subunit or the more ouabain-sensitive α2 and α3 subunits of the Na-K-ATPase. The fact that low concentrations of ouabain significantly inhibited the EDHF-mediated hyperpolarization and relaxation of the interlobar artery suggests that this response involves subunits of the Na-K-ATPase that are highly sensitive to ouabain (i.e., α2 or α3). Indeed, the mRNA of three Na-K-ATPase α subunits (α1, α2, α3) was detected in porcine interlobar smooth muscle cells. However, as a consequence of the lack of antibodies which recognise the porcine isoforms of the Na-K-ATPase it is currently not possible to speculate exactly which subunit is specifically targeted by ouabain in the interlobar artery. We also detected the expression of the renal specific γ subunit of the Na-K-ATPase (Arystarkhova et al., 2002), and although the regulation of pump activity by this subunit is not completely understood it has been proposed to regulate the sensitivity of the pump to K+ (Beguin et al., 1997). The finding that the porcine renal interlobar artery expresses the γ subunit of the Na-K-ATPase may at least partly explain the sensitivity of this artery to KCl.
Vascular smooth muscle cell hyperpolarization may elicit relaxation by a number of mechanisms, the most probable being the inhibition of voltage-dependent Ca2+ channels (Tomioka et al., 1999). In the porcine renal interlobar artery, the Ca2+ antagonist nifedipine abolished both the EDHF-mediated and K+-induced relaxations, a finding which implies that the closure of voltage-gated Ca2+ channels is involved in mediating hyperpolarization-induced relaxation.
The hypothesis that K+ can act as an EDHF has been questioned by several groups who were unable to show endothelium-independent relaxations in response to K+ in different arteries (Lacy et al., 2000; Doughty et al., 2000; Andersson et al., 2000). The failure to observe K+-induced relaxation in organ chamber experiments has been suggested to be an ‘artefact' associated with the use of a contractile agonist. For example, in rat isolated mesenteric arteries the relaxation to K+ was reported to depend on the extent of stimulation with the constrictor phenylephrine (Dora & Garland, 2001). Indeed, contractile agonists have been suggested to generate a ‘K+-cloud' which surrounds the contracting myocytes, rendering them insensitive to any further addition of extracellular K+ (Richards et al., 2001). Such a hypothesis implies that the more contracted an artery, the less likely it is that the EDHF-mediated relaxation can be attributed to the release of K+ from endothelial cells. However, in the interlobar artery whether precontracted with non-depolarizing concentrations of U46619 or with phenylephrine, the EDHF-mediated hyperpolarization and relaxation displayed characteristics consistent with the hypothesis that K+ released from endothelial cells activates the Na-K-ATPase in smooth muscle cells to induce hyperpolarization and elicit relaxation by reducing the open probability of voltage-dependent Ca2+ channels.
Acknowledgments
This study was supported by the Deutsche Gesellschaft für Kardiologie-Herz- und Kreislaufforschung (EB) and a research grant from the Institut de Recherches Internationales Servier. The authors are indebted to Tanja-Maria Mareczek for expert technical assistance.
Abbreviations
- ANOVA
analysis of variance
- Apa
Apamin
- Cbtx
Charybdotoxin
- CYP
cytochrome P450
- EDHF
endothelium-derived hyperpolarizing factor
- K+Ca channel
Ca2+-dependent K+ channel
- Na-K-ATPase
sodium potassium ATPase
- NO
nitric oxide
- NIF
nifedipine
- OU
Ouabain
- PGI2
prostacylin
References
- ANDERSSON D.A., ZYGMUNT P.M., MOVAHED P., ANDERSSON T.L., HOGESTATT E.D. Effects of inhibitors of small- and intermediate-conductance calcium-activated potassium channels, inwardly-rectifying potassium channels and Na+/K+ ATPase on EDHF relaxations in the rat hepatic artery. Br. J. Pharmacol. 2000;129:1490–1496. doi: 10.1038/sj.bjp.0703226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARYSTARKHOVA E., WETZEL R.K., SWEADNER K.J. Distribution and oligomeric association of splice forms of Na+-K+-ATPase regulatory γ-subunit in rat kidney. Am. J. Physiol. Renal Physiol. 2002;282:F393–F407. doi: 10.1152/ajprenal.00146.2001. [DOI] [PubMed] [Google Scholar]
- BARON A., FRIEDEN M., CHABAUD F., BENY J.L. Ca2+-dependent non-selective cation and potassium channels activated by bradykinin in pig coronary artery endothelial cells. J. Physiol. 1996;493:691–706. doi: 10.1113/jphysiol.1996.sp021415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEGUIN P., WANG X., FIRSOV D., PUOTI A., CLAEYS D., HORISBERGER J.D., GEERING K. The gamma subunit is a specific component of the Na,K-ATPase and modulates its transport function. EMBO J. 1997;16:4520–4560. doi: 10.1093/emboj/16.14.4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENY J.L., SCHAAD O. An evaluation of potassium ions as endothelium-derived hyperpolarizing factor in porcine coronary arteries. Br. J. Pharmacol. 2000;131:965–973. doi: 10.1038/sj.bjp.0703658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOULANGER C., VANHOUTTE P.M. Ouabain, Na+-free and K+-free solutions and relaxations to nitric oxide and nitrovasodilators. Gen. Pharmacol. 1991;22:337–340. doi: 10.1016/0306-3623(91)90460-n. [DOI] [PubMed] [Google Scholar]
- BUSSE R., EDWARDS G., FELETOU M., FLEMING I., VANHOUTTE P.M., WESTON A.H. EDHF: bringing the concepts together. Trends Pharmacol. Sci. 2002;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., GEBREMEDHIN D., PRATT P.F., HARDER D.R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. (Lond.) 1997;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAYTOR A.T., MARSH W.L., HUTCHESON I.R., GRIFFITH T.M. Comparison of glycyrrhetinic acid isoforms and carbenoxolone as inhibitors of EDHF-type relaxations mediated via gap junctions. Endothelium. 2000;7:265–278. doi: 10.3109/10623320009072213. [DOI] [PubMed] [Google Scholar]
- COLEMAN H.A., TARE M., PARKINGTON H.C. K+ currents underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig, rat and human blood vessels. J. Physiol. 2001;531:359–373. doi: 10.1111/j.1469-7793.2001.0359i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DORA K.A., GARLAND C.J. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H2424–H2429. doi: 10.1152/ajpheart.2001.280.6.H2424. [DOI] [PubMed] [Google Scholar]
- DOUGHTY J.M., BOYLE J.P., LANGTON P.D. Potassium does not mimic EDHF in rat mesenteric arteries. Br. J. Pharmacol. 2000;130:1174–1182. doi: 10.1038/sj.bjp.0703412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br. J. Pharmacol. 1988;93:515–524. doi: 10.1111/j.1476-5381.1988.tb10306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FISSLTHALER B., HINSCH N., CHATAIGNEAU T., POPP R., KISS L., BUSSE R., FLEMING I. Nifedipine increases cytochrome P4502C expression and endothelium-derived hyperpolarizing factor-mediated responses in coronary arteries. Hypertension. 2000;36:270–275. doi: 10.1161/01.hyp.36.2.270. [DOI] [PubMed] [Google Scholar]
- FISSLTHALER B., POPP R., KISS L., POTENTE M., HARDER D.R., FLEMING I., BUSSE R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- FLEMING I. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Trends Cardiovasc. Med. 2000a;10:166–170. doi: 10.1016/s1050-1738(00)00065-7. [DOI] [PubMed] [Google Scholar]
- FLEMING I. Myoendothelial gap junctions: the gap is there, but does EDHF go through it. Circ. Res. 2000b;86:249–250. doi: 10.1161/01.res.86.3.249. [DOI] [PubMed] [Google Scholar]
- KNOT H.J., ZIMMERMANN P.A., NELSON M.T. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J. Physiol. 1996;492:419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LACY P.S., PILKINGTON G., HANVESAKUL R., FISH H.J., BOYLE J.P., THURSTON H. Evidence against potassium as an endothelium-derived hyperpolarizing factor in rat mesenteric small arteries. Br. J. Pharmacol. 2000;129:605–611. doi: 10.1038/sj.bjp.0703076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGUIRE J.J., DING H., TRIGGLE C.R. Endothelium-derived relaxing factors: a focus on endothelium-derived hyperpolarizing factor(s) Can. J. Physiol. Pharmacol. 2001;79:443–470. [PubMed] [Google Scholar]
- PRIOR H.M., WEBSTER N., QUINN K., BEECH D.J., YATES M.S. K+-induced dilation of a small renal artery: no role for inward rectifier K+ channels. Cardiovasc. Res. 1998;37:780–790. doi: 10.1016/s0008-6363(97)00237-x. [DOI] [PubMed] [Google Scholar]
- RICHARDS G.R., WESTON A.H., BURNHAM M.P., FELETOU M., VANHOUTTE P.M., EDWARDS G. Suppression of K+-induced hyperpolarization by phenylephrine in rat mesenteric artery: relevance to studies of endothelium-derived hyperpolarizing factor. Br. J. Pharmacol. 2001;134:1–5. doi: 10.1038/sj.bjp.0704256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROZEN S., SKALETSKY H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- SAI Y., DAI R., YANG T.J., KRAUSZ K.W., GONZALEZ F.J., GELBOIN H.V., SHOU M. Assessment of specificity of eight chemical inhibitors using cDNA-expressed cytochromes P450. Xenobiotica. 2000;30:327–343. doi: 10.1080/004982500237541. [DOI] [PubMed] [Google Scholar]
- SANTALUCIA J., JR A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. U.S.A. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAYLOR H.J., CHAYTOR A.T., EDWARDS D.H., GRIFFITH T.M. Gap junction-dependent increases in smooth muscle cAMP underpin the EDHF phenomenon in rabbit arteries. Biochem. Biophys. Res. Commun. 2001;283:583–589. doi: 10.1006/bbrc.2001.4791. [DOI] [PubMed] [Google Scholar]
- TOMIOKA H., HATTORI Y., FUKAO M., SATO A., LIU M., SAKUMA I., KITABATAKE A., KANNO M. Relaxation in different-sized rat blood vessels mediated by endothelium-derived hyperpolarizing factor: importance of processes mediating precontractions. J. Vasc. Res. 1999;36:311–320. doi: 10.1159/000025659. [DOI] [PubMed] [Google Scholar]
- WETZEL R.K., SWEADNER K.J. Immunocytochemical localization of Na-K-ATPase alpha- and gamma-subunits in rat kidney. Am. J. Physiol. Renal Physiol. 2002;281:F531–F545. doi: 10.1152/ajprenal.2001.281.3.F531. [DOI] [PubMed] [Google Scholar]