

Abstract
Previous study suggested that cyclosporine A (CsA) could partially reduce ischaemia/reperfusion-induced injury in isolated heart, but the mechanism was still unclear. In this study, the possible mechanisms of cyclosporine A in regulating oxidative stress-induced cardiomyocyte apoptosis were examined.
Morphological (cell shrinkage, apoptotic body formation, and DNA fragmentation) and biochemical (annexin-V staining for exposed phosphatidylserine residues) evidences showed that both hydrogen peroxide (H2O2) and hypoxia/reoxygenation could induce apoptotic change in the embryonal rat heart myoblast-derived cells (H9c2). These effects were inhibited by pre-treatment with CsA at concentration of 0.01–1.0 μM for 24 h, but were increased with 10.0 μM CsA.
While examining the mechanisms of CsA in protecting cardiomyocyte apoptosis, we found that the collapse of mitochondria membrane potential (ΔΨm) induced by oxidative stress was partially reversed by CsA (0.01–1.0 μM).
Compared to the control, CSA at the concentration of 0.1 and 10.0 μM significantly increased the level of intracellular reactive oxygen species (ROS) to 117.2±12.4% and 234.4±9.3%, respectively. Co-incubating with the antioxidant, ascorbic acid (10.0 μM), could partially reduce the protective effect of CsA (0.01–1.0 μM) and the toxic effect of 10.0 μM CsA.
Pre-treatment with CsA at concentration of 0.01–1.0 μM for 24 h produced up-regulation of heat shock protein 70 (Hsp 70), inducible nitric oxide synthase (iNOS) and also induced NO production, indicating that these factors might be associated with the cell protective effects of CsA.
These results suggest that CsA could protect the oxidative stress-induced cardiomyocyte apoptosis not only by preventing the loss of ΔΨm in mitochondria, but also through ROS generation, Hsp70, and iNOS up-regulation.
Keywords: Cardiomyocytes, cyclosporine A, free radicals, membrane potential, mitochondria
Introduction
Understanding and prevention of cardiac cell death associated with myocardial infarction (MI), myocardial ischaemia plus subsequent reperfusion, cardiac allograft rejection, and heart failure is a major area of clinical interest (Haunstetter & Izumo, 1998). It becomes increasingly apparent that proportions of cardiomyocytes might preferentially undergo apoptosis following hypoxia, ischaemia/reperfusion, and treatment with hydrogen peroxide or nitric oxide (Arstall et al., 1999; Webster et al., 1999). However, the mechanism(s) to regulate myocardial injury remain unclear. Some recent studies have reported that the reactive oxygen species (ROS), mitochondrial permeability transition pore (PTP), Bcl-2 family, caspases, and cytochrome c might be involved in regulating the apoptosis of cardiomyocytes (Cook et al., 1999; Pinsky et al., 1999).
A large volume of evidence indicates that mitochondria plays a central role in triggering apoptosis and the functional studies indicate that drug-enforced opening or closing of the mitochondrial permeability transition pore, PTP, can induce or prevent apoptosis (Kroemer et al., 1997; Green & Reed, 1998; O'rourke, 1999). Cyclosporine A (CsA) is widely used as an immunosuppressant in organ transplantation (Kahan, 1989; Schreiber & Carbtree, 1992). Additionally, an in vitro study indicated that the Ca2+-induced permeability transition in isolated mitochondria was inhibited by low concentrations (<1.0 μM) of CsA (Halestrap & Davidson, 1990). CsA (the mitochondrial PTP inhibitor) was shown to regulate cell apoptosis by regulating a cyclosporin A-sensitive pore modulated by cyclophilin D, which might play a major role in cell death through modulating ATP depletion, disruption of Ca2+ homeostasis, and release of specific mitochondrial proteins in muscle cells (Bernardi, 1999). CsA has also been shown to stabilize the mitochondria transmembrane potential (ΔΨm) and prevent the release of cytochrome c from mitochondria, and thereby inhibit apoptosis induced by different stimuli in human endothelial cells (Zamzami et al., 1996; Walter et al., 1998). Experimental and clinical studies suggested that CsA might have the cardioprotective effect in the ischemia/reperfusion-induced injury (Massoudy et al., 1997; Griffiths & Halestrap, 1993). In contrast, other experimental and clinical studies suggest that CsA generated free radicals, thus inducing cell death and producing the side effects (Wolf et al., 1997). The mechanism of CsA on apoptosis remains to be elucidated.
Ischaemic preconditioning increased resistance to myocardial ischaemic injury following short sub-lethal periods of ischaemia (Marber, 2000). Several previous studies had shown that some endogenous systems are involved in ischaemic preconditioning of cardiomyocytes. It is suggested that the mitochondria and ROS might regulate some protective protein phosphorylation, activation, or expression, and subsequently were contributed to both early (1–2 h) and delayed (24–72 h) protection conferred by ischaemic preconditioning (Cook et al., 1999; Rubino & Yellon, 2000).
Mitochondria produce and are affected by ROS, which have been shown to paradoxically initiate both apoptosis and cardioprotection (Baines et al., 1997; Cai & Jones, 1998). The ROS serve as second messengers in different cell systems to mediate short-term signalling events, including cell growth or ischaemic preconditioning protection. ROS (e.g. superoxide, hydrogen peroxide, and hydroxyl radicals) generated from brief ischaemia/reperfusion have also been recognized as possible ‘triggers' in the initiation of preconditioning (Vanden Hoek et al., 1998). It might also play an important pathophysiological role in cardiac diseases characterized by apoptotic cell death (von harsdorf et al., 1999). Long-term effects of these radicals induced cellular damage and was tied with atherosclerosis, ischaemia/reperfusion, and hypoxia/reoxygenation (Sundaresan et al., 1995; Ballinger et al., 2000). Numerous studies have shown that several intracellular signal transduction pathways, such as the mitogen-activated protein kinases, protein kinase C, c-Jun NH2-terminal kinase, NF-κB, p53, and Hsp27 were regulated via ROS, but the mechanisms were still unclear (Duranteau et al., 1998; von harsdorf et al., 1999).
Reperfusion and/or reoxygenation-induced free radicals generation is believed to play a key role in the damage of cardiomyocytes, and has been shown to induce cell apoptosis (Duranteau et al., 1998; Saikumar et al., 1998; Kang et al., 2000). Thus, the direct free radical donor, H2O2, and the hypoxia/reoxygenation model were used in this study to mimic the oxidative stress in vitro. The aim of the present study was to investigate the possible mechanisms of CsA on the regulation of oxidative stress-induced cardiomyocyte apoptosis.
Methods
Cell culture
The H9c2 embryonal rat heart-derived cell line was obtained from the American Type Culture Collection (CRL1446) and was cultured in DMEM supplemented with 10% heat-inactivated foetal calf serum, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Kimes & Brandt, 1976). H9c2 cells were passaged at a 1 : 3 ratio and subcultured in 100-mm diameter tissue culture dishes for 4 days.
Hypoxia/Reoxygenation
Hypoxia was achieved by using an anaerobic jar (AnaeroPack Series, Mitsubishi Gas Chemical Co, Inc) equipped with an AnaeroPack, disposable O2-absorbing and CO2-generating agent, and an indicator to monitor oxygen depletion as previous study (Seko et al., 1996). The AnaeroPack jar is capable of depleting the concentration of O2 down to less than 0.1% in 2 h and of providing a 21% CO2 atmosphere. By placing flasks, which contain serum-free medium, in a AnaeroPack jar overnight, the medium was balanced with the hypoxic atmosphere. Cultured cardiac myocytes were subjected to hypoxic conditions by immediate replacement of the medium with the hypoxic medium in the AnaeroPack jar. To keep hypoxic conditions, all the procedures were performed in an airtight glove bag filled with 95% N2/5% CO2. After incubating in hypoxic conditions for 6 h, the cells were reoxygenated by immediate replacement of the hypoxic medium with a normoxic serum-free medium for another 12 h.
The experimental protocols
Two oxidative stress-induced cell injury models were used to identify the cell protective effects of CsA in cardiomyocytes (Figure 1). The H2O2 model (protocol 1): CsA was incubated with H9c2 cells for 0 h (control), 2 h (short-term pre-treatment) or 24 h (long-term pre-treatment) before H2O2 treatment for another 24 h. Second, the hypoxia/reoxygenation model (protocol 2): CsA was incubated with H9c2 cells 0 h (co-incubation), 2 h (short-term pre-treatment) or 24 h (long-term pre-treatment) before 6 h of hypoxia followed by 12 h of reoxygenation treatment.
Figure 1.
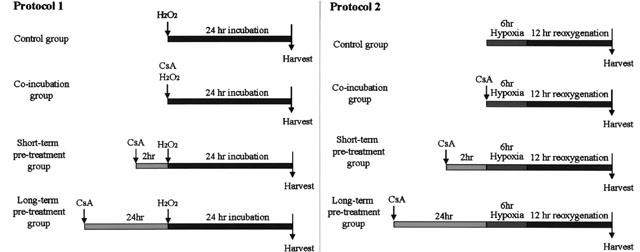
The experimental protocols.
Cell viability
In order to examine the cytotoxicity effects of H2O2 and hypoxia/reoxygenation, the H9c2 cells were exposed to H2O2 for 24 h or hypoxia/reoxygenation (12 h of reoxygenation after 6 h of hypoxia) with or without CsA-treatment. Cell viability was assessed by the Trypan blue exclusion and 3-(4,5-dimethyl-thiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT) uptake assay (Mosmann, 1983; Denizot & Lang, 1986).
Flow cytometry analysis for apoptosis
Apoptotic cells were detected by both PI and Annexin V labelling as described previously (Zamai et al., 1996; Chen et al., 2001). Double-labelling was performed at 37°C as follows: PI (50 μg ml−1) and Annexin V (2 μg ml−1) were added to the culture medium for 2 h, then the staining was immediately analysed on a FACScan (Becton Dickinson Heidelberg, Germany). Annexin V is a protein that binds to phosphatidylserine residues, which are exposed on the surface of apoptotic, but not normal, cells. In healthy cells, the distribution of the phosphatidylserine groups in the plasma membrane is asymmetrical with the groups being directed toward the interior of the cell; during apoptosis, this asymmetry is lost, and the phosphatidylserines are exposed to the exterior of the cell. Annexin V staining is therefore an established biochemical marker of apoptosis. The partial loss of membrane integrity or functionality is a useful criterion for distinguishing apoptotic from necrotic and living cells.
Determination of mitochondrial transmembranepotential (ΔΨm)
To evaluate ΔΨm, cells (5×105 cell ml−1) were incubated with chloromethyl Xarosamine (CMXRos) (Molecular Probe, Eugene, OR, U.S.A.; 30 nM for experiments without fixation, and 150 nM prior to fixation) or JC-1 (Molecular Probe, Eugene, OR, U.S.A.; 10.0 μg ml−1) for 15 min at 37°C. As a negative control, cells were simultaneously treated with the protonophase uncoupling agent carbonyl cyanide m-chlorophenylhydrazone (mCICCP, 50 μM, Sigma, St. Louis, MO, U.S.A.). The cells were then analysed using FACScan flow cytometer, as previously described (Mathony et al., 2000; Chen et al., 2001).
Fluorescent measurement of intracellular reactive oxygen species (ROS)
Intracellular oxidant stress was monitored by measuring the changes in fluorescence resulting from intracellular probe oxidation as previously (Boissy et al., 1989; Giardino et al., 1996). The probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA, 5.0 μM, Sigma, St. Louis, MO, U.S.A.) entered the cell and the acetate group on DCFH-DA was cleaved by cellular esterases, trapping the nonfluoresent DCFH inside. Subsequent oxidation by ROS, particularly H2O2 and hydroxyl radical, yielded the fluorescent product DCF. Thus, increases in DCFH oxidation to DCF (i.e. increases in DCF fluorescence) are suggestive of H2O2 or hydroxyl generation. Cells (106 per ml) were loaded with 10 μl DCFH-DA, and analysed by Becton Dickenson FACSCAN with excitation and emission settings of 495 and 525 nm, respectively.
Intracellular NO measurement
For direct intracellular NO measurements, cardiomyocytes at confluence were washed and maintained in DMEM without phenol red for the experiments. The NO-sensitive fluorescent probe 4,5-diaminofluorescein (DAF-2) (Calbiochem, San Diego, CA, U.S.A.) was pre-incubated at concentration of 10 μM for 1 h and maintained during the indicated treatment, as previous reports (Kojima et al., 1998; Navarro-Antolin & Lamas, 2001; Broillet et al., 2001). Following the incubation, cells were washed twice with phosphate buffered saline (PBS) and then analysed by Becton Dickenson FACSCAN with excitation and emission settings of 480 and 540 nM, respectively.
Western blotting
Cells were scraped into buffer A (mM) HEPES 10, mannitol 200, sucrose 70, pH 7.5), which contained protease and phosphatases inhibitors (1 μg ml−1 aprotinin, 1 μ ml−1 leupeptin, 1 μg ml−1 pepstatin A, 1 mM Na3VO4, 1 mM PMSF). Samples were centrifuged (500×g) to pellet nuclei, unbroken cells, and plasma membrane debris (nuclear fraction). The supernatants were re-centrifuged (10,000×g) to separate the mitochondrial fraction from the cytosolic fraction. The mitochondrial fraction was resuspended in buffer A containing 1% (v v−1) Triton X-100. The protein content of each fraction was determined by the BioRad Bradford assay. Samples were boiled with sample buffer and then for Western blotting to detect the Bax, Hsp27, Hsp70, or iNOS (1 : 100) with specific antibodies from Santa Cruz Biotechnol. Inc. (CA, U.S.A.) and then incubated with alkaline phosphatase-conjugated goat anti-mouse, goat anti-rabbit, or donkey anti-goat IgG antibody (1 : 1000). Membranes were developed using the substrate of alkaline phosphatase, nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (NBT/BCIT).
Statistical analysis
Data are presented as mean±s.e.mean. Paired Student's t-test and ANOVA were used to evaluate statistical significance of differences between paired observations. A value of P<0.05 was considered statistically significant.
Results
Hydrogen Peroxide or Hypoxia/Reoxygenation-induced cell apoptosis
Exposure of H9c2 to H2O2 (100 μM for 24 h) or hypoxia/reoxygenation (12 h of reoxygenation after 6 h of hypoxia) were resulted in apoptotic changes as compared to untreated control in morphology, including cells shrank and retracted from neighbouring cells (Figure 2). The annexin V procedure stains cell membrane that has lost the membrane integrity, a characteristic of cells in the early stages of apoptotic cell death. Using FITC-conjugated annexin-V (green-fluorescence) and propidium iodide (red-fluorescence for chromatin staining) staining, H9c2 cardiac myocytes exposure to H2O2 or hypoxia/reoxygenation were annexin-V positive (strong green fluorescence). The control group (untreated) only showed the background green fluorescence. The advanced apoptotic or necrotic cells showed the double positive staining for Annexin V and propidium iodide (green and red) indicating that the integrity of the cellular membrane was lost (arrowhead in Figure 2d).
Figure 2.
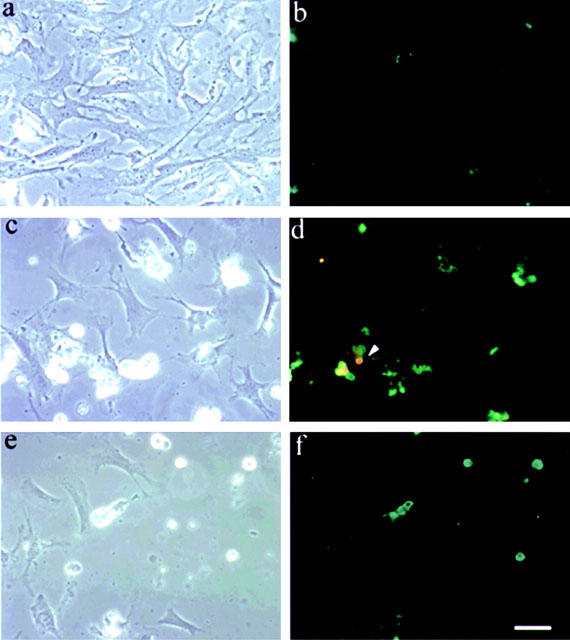
Oxidative stress-induced morphological change and apoptosis in cardiac myocytes. Shown are phase-contrast views (a, c and e) and fluorescence microscopic observation with annexin-V/propidium iodide staining (b, d and f). H9c2 cells both untreated (a and b) and treated with either 100 μM H2O2 (c and d) or hypoxia/reoxygenation (12 h of re-oxygenation after 6 h of hypoxia, e and f), were performed PI and Annexin V labelling as described in Materials and methods. The untreated cells (b) with no fluorescence (Annexin V−PI−) showed the cells were viable, which exclude PI and are negative for Annexin V binding. The green fluorescence in d and f (Annexin V++ PI−) represent the early apoptotic cells, Annexin V positive and PI negative, demonstrating cytoplasmic membrane integrity. Arrowhead in (d) showed advanced apoptotic cells or necrotic cells (Annexin V+ PI+), positive for Annexin V binding and for PI uptake which indicated that the integrity of cell membrane was lost. Similar results were obtained in four additional experiments using independent myocyte preparation, ×400. Bar = 30 μm.
Exposure of H9c2 cells to H2O2 or hypoxia/reoxygenation resulted in a 42.0±12.1% and 49.6±11.3% increase in cell apoptosis by the annexin-V/propidium iodide staining in the present study. These results indicated that the 100 μM H2O2 or hypoxia/reoxygenation-induced cell deaths were predominately apoptotic in both H9c2 cells and the freshly isolated adult rat ventricular myocytes, as demonstrated in previous study (Kang et al., 2000). Higher dose of the H2O2 (⩾500 μM) or hypoxia (⩾12 h) might induce cell necrosis in most of the cell (data not shown). According to these findings, the subsequent experiments were done with 100 μM H2O2 (24 h) or hypoxia/reoxygenation (12 h of reoxygenation after 6 h of hypoxia) to look for the protective effects of CsA on cardiomyocyte apoptosis.
The cell protective effects of cyclosporine A (CsA)
Treatment with CsA alone had no significant cytotoxicity at concentrations below 1.0 μM in H9c2 cells. The viability of cell was 98.7% at 1.0 μM as compared to untreated control. The threshold concentration of CsA to cause cell apoptosis was 10 μM in H9c2 cells (21.7±3.4% vs 6.3±4.1%, as compared to untreated control group, P<0.05, Figure 3). In order to examine the effects of CsA on oxidative stress-induced cell damage, CsA (0.01–10.0 μM) was co-incubated with H2O2 (100 μM) for 24 h. CsA (0.01–1.0 μM) showed no significant protective effect in H2O2-induced H9c2 cells apoptosis (Figure 3). However, co-incubation with CsA at the concentrations higher than 1.0 μM significantly increased the H2O2-induced cell apoptosis (67.6±4.3% and 80.3±3.9% when 1.0 or 10.0 μM CsA was co-incubated with 100 μM H2O2, compared to 100 μM H2O2 alone, 42.0±7.3%, P<0.05, Figure 3).
Figure 3.
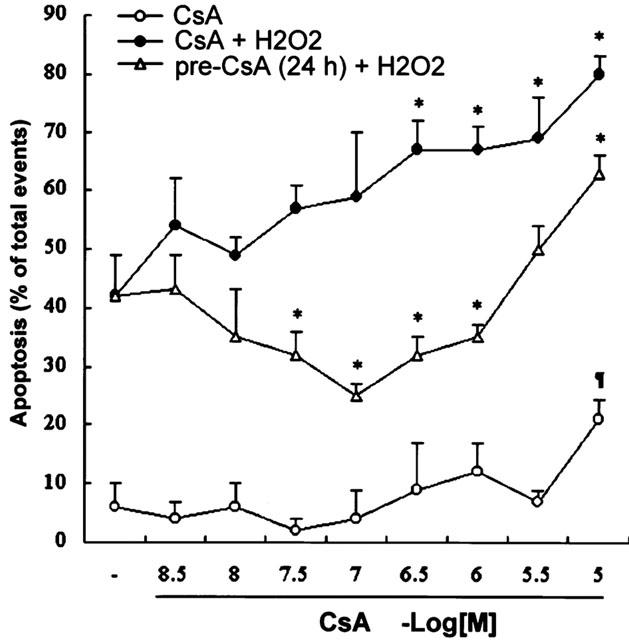
Flow cytometric analysis quantification of apoptosis in H9c2 cardiac myocytes exposed to different concentrations of CsA for 24 h, CsA combined with 100 μM H2O2 for 24 h, or pre-treated with CsA for 24 h before 100 μM H2O2 treatment for another 24 h. Cells were examined for cell apoptosis with the annexin-V/PI staining technique as described in Materials and methods. Data are expressed as mean±s.e.mean (n = 5). *P<0.05, as compared to the cells treated with 100 μM H2O2; ¶P<0.05, as compared to the untreated cells.
Contradictory, pretreatment with 0.01 to 1.0 μM CsA for 24 h showed protective effect on H2O2-induced cell apoptosis (Figure 3). CsA provided its maximal protective effects in H9c2 cells at the concentration of 0.1 μM. However, pretreated with 10.0 μM CsA has no significant effects on H2O2-induced cell apoptosis (36.7±6.1%, as compared to treatment with 100 μM H2O2 alone, P<0.05).
The present study also verified a similar result in hypoxia/reoxygenation injury. The fraction of apoptotic cardiomyocytes was increased from 15.4±6.2% to 50.2±11.0% (n=5, P<0.05). Pretreatment of cardiomyocytes with CsA (0.01–1.0 μM) for 24 h significantly, but not concentration-dependently, reduced the fraction of apoptotic cells from 50.2±11.0% to 35.5±3.6% (0.01 μM CsA, P<0.05), 25.0±3.4% (0.1 μM CsA, P<0.05), and 36.2±6.7% (1.0 μM CsA, P<0.05). Besides, CsA showed no significant protective effect for 2 h pre-treatment (46.8±6.3% and 44.5±4.7% at 0.1 and 0.01 μM, P>0.05). It implied that CsA-induced protective effect was with a time-dependent delay response in the concentration ranged from 0.01 to 1.0 μM.
CsA and mitochondrial membrane potential (ΔΨm)
Treatment of cardiomyocytes with H2O2 or hypoxia/reoxygenation resulted in an increase in cell apoptosis and significantly reduced the mitochondrial reductase activity, as assessed by the conversion of the tetrazolium dye MTT to its reduced form, a reaction indicates the integrity of mitochondrial function (Denizot & Lang, 1986). MTT reductase activity fell precipitously to 41.1±5.0% or 48.7±11.2% of control levels within 24 h of treatment with H2O2 (100 μM) or hypoxia/reoxygenation. It was surprisingly found that pretreated with 1.0 μM CsA for 24 h significantly prevented the loss of MTT reductase activity (Figure 4A). But, the effects of short-term pre-treatment with CsA for 2 h did not achieve a significant difference.
Figure 4.
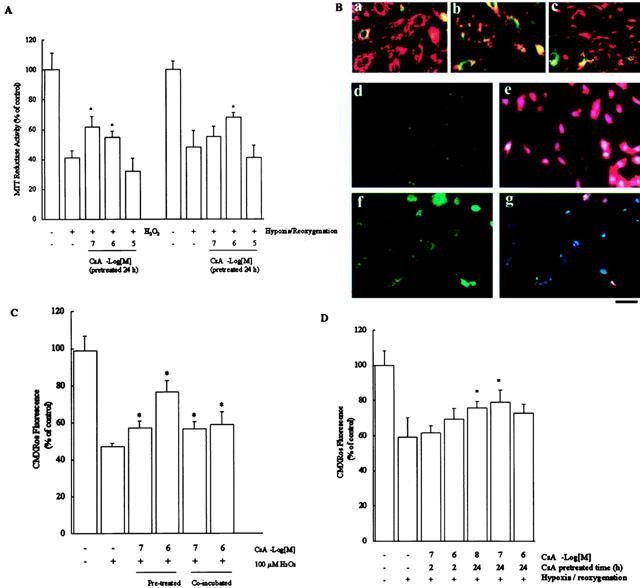
(A) Mitochondrial reductase activity was assayed by the reduction of MTT after the addition of 100 μmol l−1 H2O2 or hypoxia/reoxygenation (12 h of reoxygenation after 6 h of hypoxia. Data are expressed as mean±s.e.mean (n=5), and are reported as percentage of control. *P<0.05, as compared to the cells treated with 100 μmol l−1 H2O2 (left of the panel) or hypoxia/re-oxygenation (12 h of re-oxygenation after 6 h of hypoxia, right of the panel). (B) Mitochondrial membrane potential as assessed by JC-1 or CMXRos staining. H9c2 cells were cultured for 24 h in control medium (a) or medium containing 100 μmol l−1 H2O2(b). Note the loss of yellow-orange mitochondrial staining, representing JC-1 aggregates that accumulate at high membrane potential, in 100 μmol−1 H2O2-treated as compared to the control cells. This is an indication of loss of ΔΨm. Pretreatment with 0.1 μmol l−1 CSA (c) significantly prevented the loss of ΔΨm induced by H2O2. Correlation of apoptosis and mitochondrial membrane potential (ΔΨm) loss in hydrogen peroxide-treated cardiomyocytes. In H9c2 cells, untreated (d and e) and treated with 100 μmol l−1 H2O2 (f and g) were examined by fluorescence microscopy with annexin-V staining (d and f) and the loss of ΔΨm was analysed with CMXRos-staining (e and g). DAPI staining of DNA is shown for comparison (e and g). Similar results were obtained in three additional experiments using independent myocyte preparations, ×400. Bar=30 μm. The loss of ΔΨm induced by either 100 μmol l−1 H2O2 (C) or hypoxia/reoxygenation (12 h of reoxygenation after 6 h of hypoxia, D) was analysed (CMXRos, FL-2, FACS analysis) in H9c2 cells. Data are expressed as mean±s.e.mean (n=5) and are reported as percentage of control. *P<0.05, as compared to the cells treated with 100 μmol l−1 H2O2 (C) or hypoxia/reoxygenation (12 h of reoxygenation after 6 h of hypoxia, D).
In light of this change in mitochondrial function, we assessed whether there was any decrease in mitochondrial membrane potential (ΔΨm) in H2O2 or hypoxia/reoxygenation-induced cell apoptosis using potential-sensitive dye JC-1 (Figure 4B, a–c) and CMXRos (MitoTracker red, Figure 4B, e and g). The relationships between CsA in cardiac protection and the collapse of ΔΨm were further measured under diverse conditions in H9c2 cells. The membrane potential-dependent cellular localization of the positively charged lipophilic dye was visualized by fluorescence microscopy (Figure 4B) and measured by FACS analysis (Figure 4C). Cardiomyocytes exhibited a loss of ΔΨm after incubation in medium added with 100 μM of H2O2 for 12 h which led to a decrease of the transmembrane potential, and the loss of membrane potential persisted for 24 h (Figure 4b). The control cardiomyocytes showed the orange-red fluorescence in the cytosol granularly, indicating that the accumulation of JC-1 at high membrane potential mitochondria, and form the JC-1 aggregates (orange-red) also showed the integrity of mitochondria (Figure 4B, a). When the cells were treated with H2O2, they were turned to JC-1 monomer (green) (Figure 4B, b). Significantly, pre-treatment with 0.1 μM CsA was able to reverse the H2O2-induced the collapse of ΔΨm (Figure 4B, c).
To study the correlation between apoptosis and the ΔΨm loss, CMXRos co-staining with Annexin-V was used (Figure 4B, d–g). The control cardiomyocytes showed no annexin-V staining (Figure 4B, d) but exhibited strong staining with CMXRos (red fluorescence, Figure 4B, e). In Figure 4B, d and e, those cells that were positive for CMXRos showed no Annexin-V staining, indicating these were viable cells. In Figure 4B, f, the annexin-V positive, green fluorescence, cardiomyocytes indicated that H2O2-induced cell apoptosis and the red fluorescence (CMXRos) were reduced in H2O2-treated cells, indicating the collapse of mitochondria (Figure 4B, g).
Significantly, both co-incubated or pre-treated with 0.1 μM CsA reversed the H2O2-induced ΔΨm loss using CMXRos, which reduced the fluorescence intensity (FL-2) with the reduction of ΔΨm in FACS analysis (Figure 4c). Hypoxia/re-oxygenation (12 h of re-oxygenation after 6 h of hypoxia) also resulted in a 40.7±11.1% collapse of ΔΨm (Figure 4d), and pre-treatment with CsA for 24 h recovered the ΔΨm to 79.1±7.3% at the concentration of 0.01 μM (Figure 4d).
CsA and reactive oxygen species (ROS)
Because the generation of ROS is highly connected with mitochondrial function and could be a key component in activating the preconditioning protective effects and the apoptotic pathway in cardiovascular system (Semenza, 2000; Das et al., 1999a; Das et al., 1999b). We hypothesized that the ROS generation might play a role in the mechanisms of CsA in its cardioprotective effects and also in its cytotoxicity. Intracellular levels of ROS were measured by using the peroxide-sensitive fluorophore DCF. As demonstrated in Figure 5, levels of ROS was increased to 212.3±7.8% as compared with the control level when H2O2 was added for 4 h. Treatment of the cells with lethal concentration of CsA (10.0 μM), the ROS level was significantly increased to 234.4±9.3%. Sub-lethal concentrations of CsA (0.1–10.0 μM) also induced the generation of ROS, and this effect was increased when co-incubated with 100 μM H2O2 (from 117.2±12.4% to 231.6±20.8% at 0.1 μM CsA and from 143.5±6.7% to 272.3±9.1% at 1.0 μM CsA, P<0.05, Figure 5). Pre-treatment with CsA did not reduce H2O2 induced ROS generation, but they did increase ROS to levels higher than those of untreated cells (182.2±14.3% at 0.1 μM CsA, 173.8±15.8% at 1.0 μM CsA, and 212.3±7.8% H2O2 alone, as compared to untreated control, Figure 5).
Figure 5.
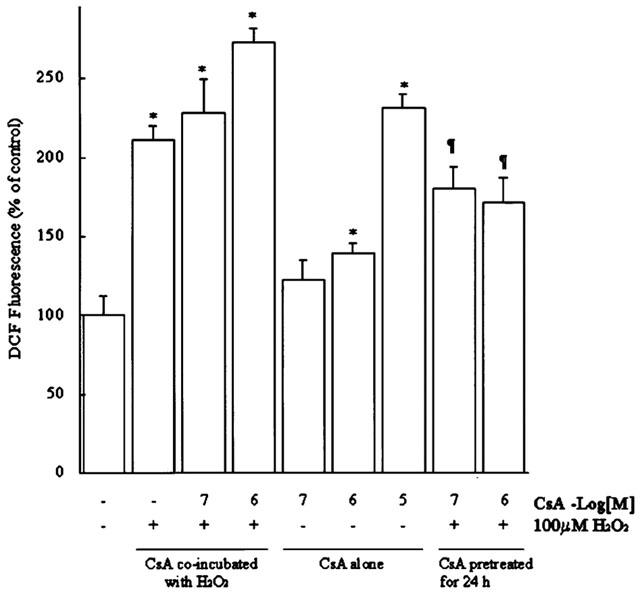
Pre-treatment with CsA lowered the H2O2-induced formation of DCF-detectable reactive oxygen species (H2O2 or hydroxyl radicals) but co-incubated with CsA enhanced the ROS generation in H9c2 cells. (A) Mean cellular DCF fluorescence is shown for 106 H9c2 myocytes loaded with DCFH-DA for 60 min in the absence or presence of H2O2 and CsA for the indicated conditions. DCF fluorescence was measured by flow cytometry of 10,000 cells and histograms of DCF fluorescence in all the cases yielded a single population. Mean DCF fluorescence is plotted. Mean DCF fluorescence increased when the myocytes were incubated with CsA or H2O2 alone or co-incubated with each other. Data are expressed as mean±s.e.mean (n=5) and are reported as percentage of control. *P<0.05, compared to the untreated cells; ¶P<0.05, compared to the cells coincubated with both H2O2 and CsA.
To understand whether the change in the intracellular ROS is an important mechanism in the cardioprotective effects of CsA, the myocytes was treated with the peroxide-scavenging cell-permeant antioxidant, N-acetylcysteine (NAC), and the other antioxidant (L-ascorbate) to scavenge the ROS. Treatment with NAC (10 μM) or L-ascorbate (10 μM) for 24 h during CsA pretreatment reduced the cardioprotective effects of CsA in H2O2-induced injury. The cell viability was reduced from 67.1±7.1% to 41.3±6.1 (NAC) and 49.1±3.2% (L-ascorbate) (n=6, P<0.05). Similarly, assessment of cellular ROS after NAC and L-ascorbate treatment might reduce the CsA-induced ROS generation (from 143.5±6.7% to 109.5±5.4% and 120.4±7.0%, as compared with control level). On the other hand, when L-ascorbate (10.0 μM) co-incubated with 10.0 μM CsA, the cytotoxicity of CsA was decreased (the apoptotic ratio was reduced from 21.7±3.4% to 9.7±1.8%). There are no significant effects in cell viability when H9c2 cells were treated with L-ascorbate (10.0 μM) or NAC (10.0 μM) in this study (102.4±6.4% and 98.1±7.9%, compared with untreated control).
Regulation of the preconditioning proteins by CsA
To regulate some cardiac protective genes is presumably an essential component of delayed protection achieved by ischaemic preconditioning. An increase in the expression of potentially protective proteins, such as heat shock proteins (Hsp) and the induced-nitric oxide synthase (iNOS), has been demonstrated during both classic and delayed preconditioning (Williams, 1997; Rubino & Yellon, 2000). The expressions of heat shock protein-70 (Hsp70, 53.4±12.1% increase as 0.1 μM CsA-treated, compare to control, P<0.05) and iNOS (73.5±5.7% increase as 1.0 μM CsA-treated, compare to control, P<0.05) were both increased after CsA (0.1–1.0 μM)-treatment in H9c2 cells (Figure 6). The free radical scavenger, ascorbic acid (10 μM), was found to prevent the up-regulation of these proteins when co-incubated with CsA (Figure 6). In contrast, CsA did not significantly influence Hsp27 expression. The expression of a pro-apoptotic protein, Bax, was also not achieved significance following CsA treatment (87.3±5.1% at 1.0 μM, P>0.05, Figure 6 and 103.2±12.8% at 10.0 μM, P>0.05, as compared with the control group).
Figure 6.
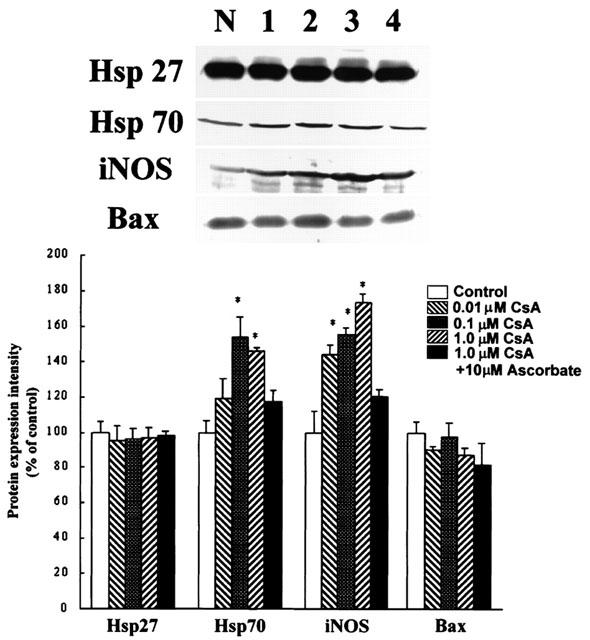
Effects of CsA on the levels of Hsp27, Hsp70, iNOS, and Bax protein expression were analysed after the treatment of 0.01, 0.1 and 1.0 μM CsA for 24 h by Western blot assay using the specific monoclonal antibodies as indicated in Materials and methods. Results are expressed in arbitrary units as compared to the protein expression level in the control group. The results are expressed as a percentage of the control level in the absence of CsA (% of control, n=5); the corresponding electrophoretic patterns of the proteins are shown in the left. Lane N, cells in the basal condition (%); lane 1–4, cells treated with 0.01 (%), 0.1 (%), 1.0 (%) μM CsA, and 1.0 μM CsA with 10 μM ascorbate (%). Each column represents the mean±s.e.mean (n=5) and are reported as percentage of control. *P<0.05, compared to the control.
Additionally, the cell-permeable fluorescent probe diaminofluorescein/diacetate (DAF-2/DA) was used with the flow cytometry technique to detect intracellular NO (Navarro-Antolin & Lamas, 2001). CsA-treated H9c2 cells showed an increased intracellular accumulation of diaminofluorescein triazol (DAF-2T) (oxidized and fluorescent form of the probe) indicated by the increase of intracellular NO production in H9c2 cells. A dose-dependent increase in the intracellular fluorescence of DAF-2 was detected in H9c2 cells treated with CsA (131.2±9.7% at 0.1 μM; 174.2±12.1% at 1.0 μM; 178.9±11.7% at 10.0 μM), which was similar to the results in endothelial cells treated with CsA (Navarro-Antolin et al., 2002). As a positive control, a significant increase in the intracellular fluorescence of DAF-2 was achieved in H9c2 cells treated with sodium nitroprusside (SNP, the NO donor, 171.4±14.3% at 0.1 μM).
Discussion
The present results suggested that pre-treatment with CsA could inhibit oxidative stress-induced cell apoptosis and co-incubation with CsA might augment hydrogen peroxide-induced cell apoptosis in cardiomyocyte. According to the results, we hypothesized that CsA, as a free radical generator, is a double-edged sword. When pre-treating cells with low dose of CsA (<1.0 μM) it exerts a ‘preconditioning like' effect by generating a small amount of ROS in cardiomyocytes (Rubino & Yellon, 2000). However, when cells were co-incubated with H2O2 and CsA or treated with higher dose of CsA (10 μM) alone, excessive free radicals were generated (e.g. peroxynitrite) (Navarro-Antolin et al., 2002), thus promoting cell death.
CsA, a potent inhibitor of mitochondrial respiration between cytochromes b and c, has been shown to stabilize the ΔΨm and inhibit cell apoptosis in endothelial cells (Halestrap & Davidson, 1990; Zamzami et al., 1996). Pre-treatment with CsA (0.75 μM) has been reported to reduce the infarct size from 28.7% to 10.0% in vivo, but the mechanism was still unclear (Griffiths & Halestrap, 1993; Massoudy et al., 1997; Weinbrenner et al., 1998). CsA was used in an attempt to elicit preconditioning-like cardioprotection in an ex vivo study. CsA pretreatment reduced infarct size (I/R ratio: 12.5±1.4%, P<0.001 vs control, 30.2±1.3%) similar to ischemic preconditioning (9.5±0.6%, P<0.001 vs control) in the Langendorff-perfused isolated rat hearts (Minners et al., 2000; Sanchez et al., 2001). In this study, pre-treatment with low dose of CsA (0.1–1.0 μM) could inhibit H2O2- or hypoxia/reoxygenation-induced ΔΨm loss and reduce the percentage of apoptosis in cardiomyocytes. To study the mechanisms involved in cell protective effect of CsA, we found that the protein levels of iNOS and Hsp 70 were up-regulated following CsA treatment. Heat shock proteins (Hsps) and iNOS have been demonstrated to play the role in ischaemic preconditioning effect (Kloner, 1998; Carroll & Yellon, 1998; Ping, 1999; Suzuki et al., 2000; Zhao et al., 2000). Previous evidence also indicated that CsA could induce eNOS expression through ROS generation (Lopez-Ongil et al., 1998; Rao et al., 1998; Esposito et al., 2000).
These results demonstrated that pre-treatment with CsA might evoke a protective response known as ‘ischaemic preconditioning effect'. Convincing evidence in the LLC-PK1 cells also showed that pre-treatment with sub-lethal concentration of CsA (50 μg ml−1) for 24 h following the exposure to a toxic concentration of CsA (200 μg ml−1) produced significant cytoprotection compared to untreated control (Yuan et al., 1996). Other evidences also indicated that low doses of CsA are capable of activating the heat shock transcription factors, HSF1 and HSF2 in the kidney BSC-1 cells and Hsp 70 in LLC-PK1 cells (Yuan et al., 1996; Paslaru et al., 2000). Drug induced Hsp70 elevation is a well known therapeutic approach for many applications in cardiovascular system (Vigh et al., 1997; Szigeti et al., 2000).
In contrast, other evidence indicates that CsA (10.0–1000.0 μM) may inhibit Ca2+ ATPase and NOS activities in the rat myocardium, and thus, may cause myocardial toxicity (Hutcheson et al., 1995). Some evidences have indicated that CsA enhances the generation of hydrogen peroxide and lipid peroxidation in vitro and in vivo (Wolf et al., 1997; Baliga et al., 1997). In transplanted patients, CsA-induced superoxide and free radical production, which might increase NO metabolism, could contribute to CsA-induced vasoconstriction and hypertension and predispose tp atherosclerosis (Calo et al., 2000). Furthermore, peroxynitrite formation and protein nitration are also considered a mediator of CsA-induced vascular damage, for example, to promote LDL oxidation during atherosclerotic lesion formation (Navarro-Antolin et al., 2001). The present study demonstrates that CsA alone could generate ROS in a concentration-dependent manner and induced cell apoptosis at 10.0 μM CsA or when co-incubating with H2O2. Antioxidants have been shown to be protective in cyclosporine A nephrotoxicity and reduced the side effects of CsA (Zhong et al., 1998; 1999; Buetler et al., 2000). It seems that CsA might enhance the oxidative stress in cells by generating excess ROS to increase cell apoptosis in cardiomyocytes which might be the major side-effects of CsA. Furthermore, CsA (10 μM) had no significant effect on the apoptotic regulator, Bax, which played an important role in mitochondria-dependent apoptotic cascade. These results indicated that CsA-regulated cell apoptosis might be through Bax-independent pathway.
The present study also showed that CsA could induce iNOS expression and NO production in H9c2 cells. The preponderance of the evidence indicated that endogenous NO is not required for the development of the early phase ofpre-conditioning effect. But, NO plays a dual role in the pathophysiology of the late phase of preconditioning, acting initially as the trigger and subsequently as the mediator of this adaptive response (Bolli, 2001). It seems that NO might play the role in the CsA-regulated cardioprotective effect, which is useful after 24 h pre-treatment (the late phase of preconditioning), not 2 h (the early phase of pre-conditioning effect). This pharmacologically-induced late preconditioning effect might have the potential for clinical use. However, whether the iNOS induction was of benefit to the cardiovascular system remains to be systematically investigated. Since NO-dependent vasodilatation might be good for myocardium, it was likely that elevation of serum NO2-/NO3- levels might cause myocardical toxicity by alerting cardiovascular NO pathway (Rao et al., 1998). Although the difference of CsA-induced NO production did not achieve significance between 1.0 μM and 10.0 μM, the ROS did significantly increase at 10.0 μM. Therefore, excess ROS (superoxide) might react with NO to produce peroxynitrite-induced cytotoxicity. These results demonstrated that CsA-induced NO and ROS production might regulate cardiomyocyte apoptosis in the myocardium, as previous study demonstrated (Navarro-Antolin et al., 2001).
These results suggested that the effects of CsA in cardioprotection or cytotoxicity might dependent on the dosage and correlate with ROS generation by CsA. Clinically therapeutic blood concentrations of CsA were in the range of 0.125 to 0.250 μM, which were able to generate NO (131.2±9.7% at 0.1 μM, compared with control). However, CsA had no significant effect in ROS generation at this concentration (117.2±12.4% at 0.1 μM CsA). It seemed that CsA might not produce enough ROS and peroxynitrite to damage cells, whereas the production of NO might be beneficial for cardiovascular system at the therapeutic range.
In summary, the present investigation demonstrated that pre-treatment with CsA inhibited oxidative stress-induced cell apoptosis in cardiomyocytes in vitro. Mechanisms underlying the protective effect of CsA appeared to be involved in stabilization of mitochondrial membrane potential, ROS generation, and some protective proteins (Hsp 70 and iNOS) elevation. To the best of our knowledge, this is the first study providing a direct link between CsA-induced ROS generation and induction of iNOS and Hsp70 that subsequently lead to ‘ischaemic preconditioning-like effect' in cardiomyocytes. These results suggested that CsA might protect cardiomyocytes against oxidative stress-induced cell apoptosis via a new mechanism, which might not be related to its immunosuppressive action. We concluded that CsA could mimic the protective response of ischaemic preconditioning and that might have the potential to improve the prognosis of myocardial infarction and also heart transplantation by modification of the timing and dosage of CsA-treatment.
Acknowledgments
This study was supported by grants from the National Science Council, the Department of Health and the National Taiwan University Hospital, Taiwan (89-2314-B-002-431, NTUH89A014 and NTUH89A023-10). Presented at 14th World Congress of Cardiology, Sidney, Australia, May 5–9, 2002.
Abbreviations
- ΔΨm
mitochondria membrane potential
- CsA
cyclosporine A
- H2O2
hydrogen peroxide
- Hsp70
heat shock protein 70
- iNOS
inducible nitric oxide synthase
- ROS
reactive oxygen species
References
- ARSTALL M.A., SAWYER D.B., FUKAZAWA R., KELLY R.A. Cytokine-mediated apoptosis in cardiac myocytes: the role of inducible nitric oxide synthase induction and peroxynitrite generation. Circ. Res. 1999;85:829–840. doi: 10.1161/01.res.85.9.829. [DOI] [PubMed] [Google Scholar]
- BAINES C.P., GOTO M., DOWNEY J.M. Oxygen radicals released during ischemia preconditioning contribute to cardioprotection in rabbit myocardium. J. Mol. Cell. Cardiol. 1997;29:207–216. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- BALIGA R., UEDA N., WALKER P.D., SHAH S.V. Oxidant mechanisms in toxic acute renal failure. Am. J. Kidney Disease. 1997;29:465–477. doi: 10.1016/s0272-6386(97)90212-2. [DOI] [PubMed] [Google Scholar]
- BALLINGER S.W., PATTERSON C., YAN C.N., DOAN R., BUROW D.L., YOUNG C.G., YAKES F.M., VAN HOUTEN B., BALLINGER C.A., FREEMAN B.A., RUNGE M.S. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ. Res. 2000;86:960–966. doi: 10.1161/01.res.86.9.960. [DOI] [PubMed] [Google Scholar]
- BERNARDI P. Mitochondria in muscle cell death. Ital. J. Neurol. Sci. 1999;20:395–400. doi: 10.1007/s100720050057. [DOI] [PubMed] [Google Scholar]
- BOISSY R.E., TRINKLE L.S., NORDLUND J.J. Separation of pigmented and albino melanocytes and concomitant evaluation of endogenous peroxide content using flow cytometry. Cytometry. 1989;10:779–787. doi: 10.1002/cyto.990100616. [DOI] [PubMed] [Google Scholar]
- BOLLI R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J. Mol. Cell. Cardiol. 2001;33:1897–1918. doi: 10.1006/jmcc.2001.1462. [DOI] [PubMed] [Google Scholar]
- BROILLET M., RANDIN O., CHATTON J. Related articles photoactivation and calcium sensitivity of the fluorescent NO indicator 4,5-diaminofluorescein (DAF-2): implications for cellular NO imaging. FEBS Lett. 2001;491:227–232. doi: 10.1016/s0014-5793(01)02206-2. [DOI] [PubMed] [Google Scholar]
- BUETLER T.M., COTTET-MAIRE F., KRAUSKOPF A., RUEGG U.T. Does cyclosporin A generate free radicals. Trend in Pharmacol. Sci. 2000;21:288–290. doi: 10.1016/s0165-6147(00)01508-x. [DOI] [PubMed] [Google Scholar]
- CAI J., JONES D.P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- CALO L., SEMPLICINI A., DAVIS P.A., BONVICINI P., CANTARO S., RIGOTTI P., D'ANGELO A., LIVI U., ANTONELLO A. Cyclosporin-induced endothelial dysfunction and hypertension: are nitric oxide system abnormality and oxidative stress involved. Transpl. Int. 2000;13:S413–S418. doi: 10.1007/s001470050374. [DOI] [PubMed] [Google Scholar]
- CARROLL R., YELLON D.M. Myocardial adaptation to ischaemia – the preconditioning phenomenon. Int. J. Cardiol. 1998;68:S93–S101. doi: 10.1016/s0167-5273(98)00297-6. [DOI] [PubMed] [Google Scholar]
- CHEN H.W., HSU M.J., CHIEN C.T., HUANG H.C. Effect of alisol B acetate, a plant triterpene, on apoptosis in vascular smooth muscle cells and lymphocytes. Eur. J. Pharmacol. 2001;419:127–138. doi: 10.1016/s0014-2999(01)00983-9. [DOI] [PubMed] [Google Scholar]
- COOK S.A., SUGDEN P.H., CLERK A. Regulation of Bcl-2 family proteins during development and in response to oxidative stress in cardiac myocytes: association with changes in mitochondrial membrane potential. Circ. Res. 1999;85:940–949. doi: 10.1161/01.res.85.10.940. [DOI] [PubMed] [Google Scholar]
- DAS D.K., ENGELMAN R.M., MAULIK N. Oxygen free radical signaling in ischemic preconditioning. Ann. New York Acad. Sci. 1999a;874:49–65. doi: 10.1111/j.1749-6632.1999.tb09224.x. [DOI] [PubMed] [Google Scholar]
- DAS D.K., MAULIK N., SATO M., RAY P.S. Reactive oxygen species function as second messenger during ischemic preconditioning of heart. Mol. Cell. Biochem. 1999b;196:59–67. [PubMed] [Google Scholar]
- DENIZOT F., LANG R. Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods. 1986;89:271–277. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- DURANTEAU J., CHANDEL N.S., KULISZ A., SHAO Z., SCHUMACKER P.T. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J. Biol. Chem. 1998;273:11619–11624. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- ESPOSITO C., FORNONI A., CORNACCHIA F., BELLOTTI N., FASOLI G., FOSCHI A., MAZZUCCHELLI I., MAZZULLO T., SEMERARO L., DAL CANTON A. Cyclosporine induces different responses in human epithelial, endothelial and fibroblast cell cultures. Kidney Int. 2000;58:123–130. doi: 10.1046/j.1523-1755.2000.00147.x. [DOI] [PubMed] [Google Scholar]
- GIARDINO I., EDELSTEIN D., BROWNLEE M. BCL-2 expression or antioxidants prevent hyperglycemia-induced formation of intracellular advanced glycation endproducts in bovine endothelial cells. J. Clin. Invest. 1996;97:1422–1428. doi: 10.1172/JCI118563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GREEN D.R., REED J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- GRIFFITHS E.J., HALESTRAP A.P. Protection by cyclosporine A of ischemia/reperfusion-induced damage in isolated rat hearts. J. Mol. Cell. Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- HALESTRAP A.P., DAVIDSON A.M. Inhibition of Ca2+-induced large amplitude swelling of liver and heart mitochondria by cyclosporine A is probably caused by the inhibitor binding to mitochondrial matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAUNSTETTER A., IZUMO S. Apoptosis: Basic mechanisms and implications for cardiovascular disease. Circ. Res. 1998;82:1111–1129. doi: 10.1161/01.res.82.11.1111. [DOI] [PubMed] [Google Scholar]
- HUTCHESON A.E., RAO M.R., OLINDE K.D., MARKOV A.K. Myocardial toxicity of cyclosporin A: inhibition of calcium ATPase and nitric oxide synthase activities and attenuation by fructose-1,6-diphosphate in vitro. Res. Commun. Mol. Pathol. Pharmacol. 1995;89:17–26. [PubMed] [Google Scholar]
- KAHAN B.D. Cyclosporine. N. Engl. J. Med. 1989;321:1725–1738. doi: 10.1056/NEJM198912213212507. [DOI] [PubMed] [Google Scholar]
- KANG P.M., HAUNSTETTER A., AOKI H., USHEVA A., IZUMO S. Morphological and molecular characterization of adult cardiomyocyte apoptosis during hypoxia and reoxygenation. Circ. Res. 2000;87:118–125. doi: 10.1161/01.res.87.2.118. [DOI] [PubMed] [Google Scholar]
- KIMES B.W., BRANDT B.L. Properties of a clonal muscle cell line from rat heart. Exp. Cell. Res. 1976;98:367–381. doi: 10.1016/0014-4827(76)90447-x. [DOI] [PubMed] [Google Scholar]
- KLONER R.A. Medical and cellular implication of stunning, hibernation and preconditioning. An NHLBI workshop. Circulation. 1998;97:1848–1867. doi: 10.1161/01.cir.97.18.1848. [DOI] [PubMed] [Google Scholar]
- KOJIMA H., NAKATSUBO N., KIKUCHI K., KAWAHARA S., KIRINO Y., NAGOSHI H., HIRATA Y., NAGANO T. Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal. Biochem. 1998;70:2446–2453. doi: 10.1021/ac9801723. [DOI] [PubMed] [Google Scholar]
- KROEMER G., ZAMZAMI N., SUSIN S.A. Mitochondrial control of apoptosis. Immun. Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- LOPEZ-ONGIL S., HERNANDEZ-PERERA O., NAVARRO-ANTOLIN J., PEREZ DE LEMA G., RODRIGUEZ-PUYOL M., LAMAS S., RODRIGUEZ-PUYOL D. Role of reactive oxygen species in the signalling cascade of cyclosporine A-mediated up-regulation of eNOS in vascular endothelial cells. Br. J. Pharmacol. 1998;124:447–454. doi: 10.1038/sj.bjp.0701847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARBER M.S. Ischemic preonditioning in isolated cells. Circ. Res. 2000;86:926–931. doi: 10.1161/01.res.86.9.926. [DOI] [PubMed] [Google Scholar]
- MASSOUDY P., ZAHLER S., KUPATT C., REDER E., MECKER B.F., GERLACH E. Cardioprotection by cyclosporine A in experimental ischemia and reperfusion-evidence for a nitric oxide-dependent mechanism mediated by endothelin. J. Mol. Cell. Cardiol. 1997;29:535–544. doi: 10.1006/jmcc.1996.0297. [DOI] [PubMed] [Google Scholar]
- MATHONY M., HONG Y., KEMP B.K., BARRIENTOS A.A., ERUSALIMSKY J.D. Evaluation of fluorescent dyes for the detection of mitochondrial membrane potential changes in cultured cardiomyocytes. Cardiovas. Res. 2000;46:126–138. doi: 10.1016/s0008-6363(00)00002-x. [DOI] [PubMed] [Google Scholar]
- MINNERS J., VAN DEN BOS E.J., YELLON DM., SCHWALB H., OPIE LH., SACK MN. Dinitrophenol, cyclosporin A, and trimetazidine modulate preconditioning in the isolated rat heart: support for a mitochondrial role in cardioprotection. Cardiovasc. Res. 2000;47:68–73. doi: 10.1016/s0008-6363(00)00069-9. [DOI] [PubMed] [Google Scholar]
- MOSMANN T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- NAVARRO-ANTOLIN J., LAMAS S. Nitrosative stress by cyclosporin A in the endothelium: studies with the NO-sensitive probe diaminofluorescein-2/diacetate using flow cytometry. Nephrol. Dial. Transplant. 2001;16:S6–S9. doi: 10.1093/ndt/16.suppl_1.6. [DOI] [PubMed] [Google Scholar]
- NAVARRO-ANTOLIN J., LOPEZ-MUNOZ M.J., SORIA J., LAMAS S. Superoxide limits cyclosporine-A-induced formation of peroxynitrite in endothelial cells. Free Radic. Biol. Med. 2002;32:702–711. doi: 10.1016/s0891-5849(02)00761-x. [DOI] [PubMed] [Google Scholar]
- O'ROURKE B. Apoptosis: rekindling the mitochondrial fire. Circ. Res. 1999;85:880–883. doi: 10.1161/01.res.85.10.880. [DOI] [PubMed] [Google Scholar]
- PASLARU L., RALLU M., MANUEL M., DAVIDSON S., MORANGE M. Cyclosporine A induces an atypical heat shock response. Biochem. Biophys. Res. Commun. 2000;269:464–469. doi: 10.1006/bbrc.2000.2295. [DOI] [PubMed] [Google Scholar]
- PING P. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rat: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ. Res. 1999;84:587–604. doi: 10.1161/01.res.84.5.587. [DOI] [PubMed] [Google Scholar]
- PINSKY D.J., AJI W., SZABOLCS M., ATHAN E.S., LIU Y., YANG Y.M., KLINE R.P., OLSON K.E., CANNON P.J. Nitric oxide triggers programmed cell death (apoptosis) of adult rat ventricular myocytes in culture. Am. J. Physiol. 1999;277:H1189–H1199. doi: 10.1152/ajpheart.1999.277.3.H1189. [DOI] [PubMed] [Google Scholar]
- RAO M.R., HUTCHESON A.E., MARKOV A.K. Changes in the cardiovascular nitric oxide pathway in cyclosporin-A treated rats. Drug & Chem. Toxicol. 1998;21:27–34. doi: 10.3109/01480549809017848. [DOI] [PubMed] [Google Scholar]
- RUBINO A., YELLON D.M. Ischaemic preconditioning of the vasculature: an overlooked phenomenon for protecting the heart. Trend in Pharmacol. Sci. 2000;21:225–230. doi: 10.1016/s0165-6147(00)01483-8. [DOI] [PubMed] [Google Scholar]
- SAIKUMAR P., DONG Z., WEINBERG J.M., VENKATACHALAM M.A. Mechanisms of cell death in hypoxia/reoxygenation injury. Oncogene. 1998;17:3341–3349. doi: 10.1038/sj.onc.1202579. [DOI] [PubMed] [Google Scholar]
- SANCHEZ H., ZOLL J., BIGARD X., VEKSLER V., METTAUER B., LAMPERT E., LONSDORFER J., VENTURA-CLAPIER R. Effect of cyclosporin A and its vehicle on cardiac and skeletal muscle mitochondria: relationship to efficacy of the respiratory chain. Br. J. Pharmacol. 2001;133:781–788. doi: 10.1038/sj.bjp.0704129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEKO Y., TOBE K., UEKI K., KADOWAKI T., YAZAKI Y. Hypoxia and hypoxia/reoxygenation activate Raf-1, mitogen-activated protein kinase kinase, mitogen-activated protein kinases, and S6 kinase in cultured rat cardiac myocytes. Circ. Res. 1996;78:82–90. doi: 10.1161/01.res.78.1.82. [DOI] [PubMed] [Google Scholar]
- SCHREIBER S.L., CARBTREE G.R. The mechanism of action of cyclosporine and FK506. Immunol. Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- SEMENZA G.L. Cellular and molecular dissection of reperfusion injury-ROS within and without. Circ. Res. 2000;86:117–118. doi: 10.1161/01.res.86.2.117. [DOI] [PubMed] [Google Scholar]
- SUNDARESAN M., YU Z.X., FERRANS V.J., IRANI K., FINKEL T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- SUZUKI K., SAWA Y., KAGISAKI K., TAKETANI S., ICHIKAWA H., KANEDA Y., MATSUDA H. Reduction in myocardial apoptosis associated with overexpression of heat shock protein 70. Basic Res. Cardiol. 2000;95:397–403. doi: 10.1007/s003950070039. [DOI] [PubMed] [Google Scholar]
- SZIGETI G., BANYASZ T., MAGYAR J., KORTVELY A., SZIGIGETI P., KOVACS L., JEDNAKOVITS A., NANASI P.P. Effects of bimoclomol, the novel heat shock protein coinducer, in dog ventricular myocardium. Life Sci. 2000;67:73–79. doi: 10.1016/s0024-3205(00)00604-4. [DOI] [PubMed] [Google Scholar]
- VANDENHOEK T.L., BECKER L.B., SHAO Z., LI C., SCHUMACKER P.T. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J. Biol. Chem. 1998;273:18092–18098. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- VIGH L., LITERATI P.N., HORVATH I., TOROK Z., BALOGH G., GLATZ A., KOVACS E., BOROS I., FERDINANDY P., FARKAS B., JASZLITS L., JEDNAKOVITS A., KORANYI L., MARESCA B. Biomoclomol: a nontoxic, hydroxylamine derive with stress protein-inducing activity and cytoprotective effects. Nat. Med. 1997;3:1150–1154. doi: 10.1038/nm1097-1150. [DOI] [PubMed] [Google Scholar]
- VON HARSDORF R., LI P.F., DIETZ R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation. 1999;99:2934–2941. doi: 10.1161/01.cir.99.22.2934. [DOI] [PubMed] [Google Scholar]
- WALTER D.H., HAENDELER J., GALLE J., ZEIHER A.M., DIMMELER S. Cyclosporine A inhibits apoptosis of human endothelial cells by preventing release of cytochrome C from mitochondria. Circulation. 1998;98:1153–1157. doi: 10.1161/01.cir.98.12.1153. [DOI] [PubMed] [Google Scholar]
- WEBSTER K.A., DISCHER D.J., KAISER S., HERNANDEZ O., SATO B., BISHOPRIC N.H. Hypoxia-activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53. J. Clin. Invest. 1999;104:239–252. doi: 10.1172/JCI5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEINBRENNER C., LIU G.S., DOWNEY J.M., COHEN M.V. Cyclosporine A limits myocardial infarct size even when administered after onset of ischemia. Cardiovasc. Res. 1998;38:678–684. doi: 10.1016/s0008-6363(98)00064-9. [DOI] [PubMed] [Google Scholar]
- WILLIAMS R.S. Heat shock proteins and ischemic injury to the myocardium. Circulation. 1997;96:4138–4140. [PubMed] [Google Scholar]
- WOLF A., TRENDELENBURG C.F., DIEZ-FERNANDEZ C., PRIETO P., HOUY S., TROMMER W.E., CORDIER A. Cyclosporine A-induced oxidative stress in rat hepatocytes. J. Pharmacol. Exp. Ther. 1997;280:1328–1334. [PubMed] [Google Scholar]
- YUAN C.M., BOHEN E.M., MUSIO F., CAROME M.A. Sublethal heat shock and cyclosporine exposure produce tolerance against subsequent cyclosporine toxicity. Am. J. Physiol. 1996;271:F571–F578. doi: 10.1152/ajprenal.1996.271.3.F571. [DOI] [PubMed] [Google Scholar]
- ZAMAI L., FALCIERI E., MARHEJKA G., VITALE M. Supravital exposure to propidium iodide identifies apoptotic cells in the absence of nucleosomal DNA fragmentation. Cytometry. 1996;23:303–311. doi: 10.1002/(SICI)1097-0320(19960401)23:4<303::AID-CYTO6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- ZAMZAMI N., MARCHETTI P., CASTEDO M., HIRSCH T., SUSIN S.A., MASSE B., KROEMER G. Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett. 1996;384:53–57. doi: 10.1016/0014-5793(96)00280-3. [DOI] [PubMed] [Google Scholar]
- ZHAO T., XI L., CHELLIAH J., LEVASSEUR J.E., KUKREJA R.C. Inducible nitric oxide synthase mediates delayed myocardial protection induced by activation of adenosine A1 receptors evidence from gene-knockout mice. Circulation. 2000;102:902–907. doi: 10.1161/01.cir.102.8.902. [DOI] [PubMed] [Google Scholar]
- ZHONG Z., ARTEEL G.E., CONNOR H.D., YIN M., FRANKENBERG M.V., STACHLEWITZ R.F., RALEIGH J.A., MASON R.P., THURMAN R.G. Cyclosporine A increases hypoxia and free radical production in rat kidneys: prevention by dietary glycine. Am. J. Physiol. 1998;275:F595–F604. doi: 10.1152/ajprenal.1998.275.4.F595. [DOI] [PubMed] [Google Scholar]
- ZHONG Z., CONNOR H.D., YIN M., MOSS N., MASON R.P., BUNZENDAHL H., FORMAN D.T., THURMAN R.G. Dietary glycine and renal denervation prevents cyclosporine A-induced hydroxyl radical production in rat kidney. Mol. Pharmacol. 1999;56:455–463. doi: 10.1124/mol.56.3.455. [DOI] [PubMed] [Google Scholar]