

Abstract
The extraneuronal monoamine transporter from rat (EMTr) was heterologously expressed by stable transfection in human embryonic kidney 293 cells and characterized in radiotracer experiments.
EMTr-mediated uptake of 1-methyl-4-phenylpyridinium (MPP+) was saturable, with a Km of 151 μmol l−1 and Vmax of 7.5 nmol min−1 mg protein−1.
Compared to the human orthologue EMTh (gene symbol SLC22A3), EMTr was about two orders of magnitude more resistant to most inhibitors, including disprocynium24 and corticosterone.
Strikingly, inhibitors and substrates at low concentration stimulated EMTr-mediated transport above control level with MPP+ and noradrenaline as substrate, but not with cimetidine. Results were confirmed with EMT from mouse.
With different IC50-values for different substrates, the standard method to calculate Ki-values is not applicable.
Our experiments suggest that activation is not caused by changes in membrane potential or trans-stimulation. Since the extent of activation depends markedly on the chemical structure of the monitored substrate, involvement of a receptor-mediated signalling pathway or recruitment of transporter reserve are implausible.
To explain activation, we present a kinetic model which assumes two binding sites for substrate or inhibitor per transporter entity, possibly resulting from the assembly of homodimers.
Activation explains previous reports about inhibitor-insensitive catecholamine transport in rat brain.
We speculate that activation may serve to keep the transporter working for specific substrates in the face of inhibitors.
Keywords: Extraneuronal monoamine transporter, transporter activation, 1-methyl-4-phenylpyridinium, organic cation transporter, catecholamine transport, uptake2, noradrenaline, cimetidine, trans-stimulation, Ki-value
Introduction
At the plasma membrane of neuronal and non-neuronal cells, specific transport proteins remove released monoamine transmitters, such as dopamine, noradrenaline, and 5-hydroxytryptamine, from the extracellular space and thus terminate and control signal transmission. Pharmacological blockade of these carriers serves to increase extracellular transmitter levels. The therapeutic utility of this principle is well acknowledged for neuronal transporters, which are molecular targets for e.g. clinically important antidepressants. We have a long-standing interest in the counterpart from non-neuronal cells, the extraneuronal monoamine transporter (EMT), originally discovered as a transport mechanism in rat heart and designated ‘uptake2' by Iversen and coworkers (Iversen, 1965). As part of both the central and peripheral aminergic nervous system (Eisenhofer et al., 1996; Russ et al., 1996), EMT could be involved in a number of clinically prominent human diseases, e.g. Parkinson's disease, schizophrenia, hypertension, and arrhythmia.
The functional characterization of EMT was pioneered with perfused organs and isolated tissues by the groups of Iversen and Trendelenburg (Grohmann & Trendelenburg, 1984; Iversen & Salt, 1970; Trendelenburg, 1988). Functional studies with neurotoxin 1-methyl-4-phenylpyridinium (MPP+) as excellent substrate on Caki-1 cells, a human kidney carcinoma cell line, have complemented the picture (Russ et al., 1992; Schömig et al., 1992; Schömig & Schönfeld, 1990). Based on this cell culture model, our group has developed highly potent and specific inhibitors for EMT, e.g., disprocynium24 and decynium22 (Russ et al., 1993). It is clear now that EMT differs immensely with respect to transport mechanism, drug sensitivity, substrate affinity, and substrate selectivity from the neuronal transporters e.g. for dopamine (DAT) and noradrenaline (NET) (Povlock & Amara, 1997).
Recently, we have succeeded in the molecular identification of human EMT (gene symbol SLC22A3) by cloning from a Caki-1 cDNA library and functional expression in 293 cells (Gründemann et al., 1998b). This assignment was thereafter confirmed for orthologuous cDNAs from rat (Kekuda et al., 1998; Wu et al., 1998) and mouse (Verhaagh et al., 1999; Wu et al., 2000). It has become evident by molecular cloning and by functional characterization based on heterologous expression that transporters in addition to EMT may participate in non-neuronal monoamine uptake, i.e. OCT1 (Breidert et al., 1998; Gründemann et al., 1994; Nagel et al., 1997), and OCT2 (Busch et al., 1998; Gründemann et al., 1997; Gründemann et al., 1998a; Okuda et al., 1996). All three transporters are members of the amphiphilic solute facilitator (ASF) family of transport proteins (Schömig et al., 1998) and accept catecholamines and MPP+ as substrates (Gründemann et al., 1999). EMT is expressed in many tissues, but OCT2 has been detected solely in kidney and brain, and OCT1 is confined to liver, kidney, and intestine. Collectively, we refer to these carriers as non-neuronal monoamine transporters.
The physiological and pathophysiological significance of non-neuronal monoamine transport is still fairly unclear. The first knock-outs in mouse emerge (Jonker et al., 2001; Zwart et al., 2001), but these may suffer from inherent methodological limitations. Important clues could come from the instant in vivo effects of potent and specific inhibitors in rat or mouse. Remarkably, in our experiments disprocynium24, the most efficacious known inhibitor of human EMT, turned out to be insufficiently potent on EMT from rat to be useful in vivo. We here report as likely cause a hitherto unknown activation mechanism for EMT from rat and mouse.
Methods
Construction of EMTr and EMTm expression vectors
For the cloning of EMTr, total RNA from Sprague–Dawley rat heart was prepared as described (Gründemann et al., 1997). A cDNA covering the open reading frame entirely was generated by RT–PCR with Pwo Polymerase (Roche, Mannheim, Germany) and with primers 5′-GAG AGA CAG GTA CCG CCA CCA TGC CCA CGT TCG ACC (forward primer, KpnI site underlined; this primer introduces a perfect Kozak sequence) and 5′-GAG AGA GAT CTA GAC GGC GCC ACC TAG AGG AGG TGA (reverse primer, XbaI site underlined). The amplicon (1.8 kb) was cloned, completely sequenced and eventually assembled into the KpnI and XbaI sites of pcDNA3 (Invitrogen, NV Leek, The Netherlands) to yield pcDNA3EMTr. Sequencing of four independent clones uniformly revealed a base disparity with the published cDNA sequence (accession number AF055286), 761G>A, which gives rise to amino acid substitution G124S. The variant base was confirmed by PCR on genomic DNA from rat liver and subsequent cloning and sequencing of the generated amplicon. For further corroboration, we used in PCR on genomic DNA two primers that differed only at the ultimate 3′-base, i.e. 5′-TCC ACA TAG CGC CAG TCG CC or …CG CT, as reverse primers in paired assays. With optimized conditions (forward primer as above, standard PCR (Schömig et al., 1998) plus 5% DMSO, 32 cycles of 30 s 94°C, 1 min 70°C, 500 ng EcoRI-digested genomic DNA from rat liver per assay) a product of expected size (0.40 kb) was generated solely with the primer ending in T.
A cDNA of EMTm (accession number AF078750) was kindly provided by Dr R. Zwart, Department of Molecular Genetics, The Netherlands Cancer Institute, Amsterdam, The Netherlands. The complete open reading frame was released from pCAGGS and inserted into pcDNA3 via EcoRI. The 5′-UTR was replaced by a perfect Kozak sequence. The junctions in the final construct, pcDNA3EMTm, were AAG CTTGCc acc atg ccc… (HindIII site of vector underlined; lower-case letters indicate original cDNA sequence) and …agg tgg cgA TAC CGT CGA GAA TCC (EcoRI site of vector underlined).
Cell culture and transfection
293 cells (ATCC CRL-1573), a transformed cell line derived from human embryonic kidney, were grown at 37°C in a humidified atmosphere (5% CO2) in plastic culture flasks (Falcon 3112, Becton Dickinson, Heidelberg, Germany). The medium was Dulbecco's Modified Eagle Medium (31885-023, Life Technologies, Eggenstein, Germany) supplemented with 10% foetal calf serum (Life Technologies). Medium was changed every 2–3 days and the culture was split every 7 days.
293 cells were transfected with supercoiled plasmid DNA by lipofection with the Tfx-50 reagent according to the protocol of the vendor (Promega, Mannheim, Germany). Stably transfected cells were then selected with G418 (Life Technologies) as described (Gründemann et al., 1997). Expression of EMTr and EMTm was verified by RT–PCR and functional characterization.
Transport assays
For measurement of uptake of radiolabeled solutes, cells were grown in surface culture on 60 mm polystyrol dishes (Nunclon 150288, Nunc, Wiesbaden, Germany) precoated with 0.1 g l−1 poly-L-ornithine in 0.15 mol l−1 boric acid-NaOH, pH 8.4. Cells were used for uptake experiments at a confluence of at least 70%.
Uptake was measured at 37°C. After preincubation for at least 20 min in 4 ml of Na+ buffer (125 mmol l−1 NaCl, 25 mmol l−1 HEPES-NaOH pH 7.4, 5.6 mmol l−1 (+)glucose, 4.8 mmol l −1 KCl, 1.2 mmol l−1 KH2PO4, 1.2 mmol l−1 CaCl2, 1.2 mmol l−1 MgSO4), the buffer was replaced with 3 ml of 3H-labelled substrate (at 100 nmol l−1 if not noted otherwise) and inhibitor as indicated in Na+ buffer. NaCl was iso-osmotically reduced if concentration of inhibitor exceeded 1 mmol l−1. In some experiments, K+ buffer was used instead of Na+ buffer K+ is identical to Na+ buffer except that NaCl and HEPES-NaOH were completely replaced by KCl and HEPES-KOH, respectively. Incubation was stopped (after 1 min if not noted otherwise) by rinsing the cells four times with each 4 ml ice-cold Na+ buffer. Subsequently, the cells were solubilized with 0.1% v v−1 Triton X-100 in 5 mmol l−1 TRIS-HCl pH 7.4, and radioactivity was determined by liquid scintillation counting. In experiments with noradrenaline, 1 mmol l−1 (+)ascorbic acid was present to prevent oxidation.
To avoid trans-stimulation effects with established or potential substrates, all inhibitors with a Ki larger than 1 μmol l−1 were absent during preincubation. By contrast, potent inhibitors (Ki smaller than 1 μmol l−1) were present during both the preincubation and uptake periods. This is necessary to achieve equilibrium binding with effective inhibitors – which are used at very low concentrations – and hence realize their full potency. These high-affinity inhibitors are at best transported very slowly and thus do not cause trans-stimulation.
Protein determination
Protein was measured by a modification of the Bradford Method (Zor & Selinger, 1996) with bovine serum albumin as standard.
Calculations and statistics
Analysis of the time course of substrate accumulation was based on a cone-compartment model as described earlier (Russ et al., 1992). Saturation curves were analysed as described (Schömig et al., 1993).
For EMTh, all Ki-values were calculated by non-linear regression fitting to the standard model (assuming a Hill coefficient of 1): vi/v0=1/(1+I/IC50) with vi: specific uptake velocity in the presence of inhibitor, v0: specific uptake velocity in the absence of inhibitor, and I: concentration of inhibitor. IC50-values are identical with Ki values, since non-saturating substrate concentrations were used (Cheng & Prusoff, 1973). For EMTr and EMTm, the general activation model according to Figure 9 can be factorized into vi/v0=(a1+a2 * I)/(a1+a3 * I+a4 * I2); parameters a1, a2, a3, and a4 are functions of dissociation constants, rate constants, and substrate concentration. Division by a1 yields vi/v0=(1+a * I)/(1+b * I+c * I2) with phenomenological constants a, b, and c. This equation was used for non-linear regression.
Figure 9.
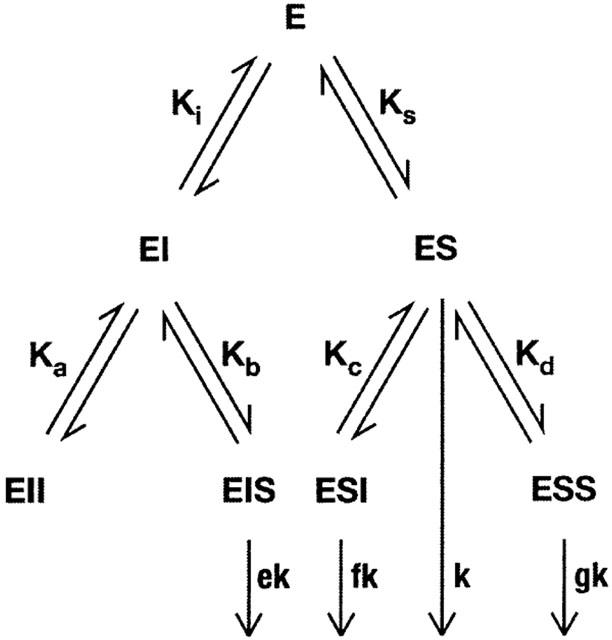
Kinetic model for the activation of EMTr and EMTm. The model assumes two binding sites for inhibitor (I) or radiolabeled substrate (S) per transporter entity (E). For a full mathematical description of complex formation and substrate turnover, 10 parameters are required, i.e. equilibrium dissociation constants (Ks, Ki, Ka, Kb, Kc, and Kd), a rate constant (k) and relative factors (e, f, and g).
Fitted parameters such as Km- and Ki-values are given as geometric mean with 95% confidence interval. Arithmetic means are given with s.e.mean. In the figures, error bars smaller than symbols are not shown.
Drugs
Radiotracers: 1-methyl-4-phenylpyridinium iodide (H-3, 2200 Bq pmol−1; ART-150, ARC, St. Louis, U.S.A.), (−)noradrenaline (norepinephrine) (H-3, 381 Bq pmol−1; NET-337, NEN, Zaventem, Belgium), and cimetidine (H-3, 455 Bq pmol−1; TK615, Amersham Pharmacia, Freiburg, Germany).
Unlabelled compounds: BU-224 and BU-226 (0725 and 1091, Tocris, Bristol, U.K.), cimetidine (28541-2, Aldrich, Steinheim, Germany), clobenpropit (0752, Tocris), corticosterone (C-2505, Sigma, Deisenhofen, Germany), decynium22 (1,1′-diethyl-2,2′-cyanine iodide, 32376-4, Aldrich), DMPBI (1,3-dimethyl-2-phenyl-3H-benzoimidazol-1-ium chloride, S136077, Sigma-Aldrich library of rare chemicals), histamine dihydrochloride (4370, Merck, Darmstadt, Germany), immepip (0932, Tocris), MPP+ (1-methyl-4-phenylpyridinium iodide, D-048, RBI, Natick, MA, U.S.A.), (−)noradrenaline hydrochloride (74480, Fluka, Buchs, Switzerland), papaverine (P-3510, Sigma), phentolamine hydrochloride (CIBA, Summit, NJ, U.S.A.), thioperamide (T-123, RBI), and trimazosine hydrochloride (Lappe Arzneimittel, Berg. Gladb., Germany). Disprocynium24 (1,1′-diisopropyl-2,4-cyanine iodide) was synthesized as described previously (Russ et al., 1993). All other chemicals were of analytical grade.
Results
Cloning and functional expression of EMTr in 293 cells
Based on the published cDNA sequence (Kekuda et al., 1998), we have cloned EMTr from rat heart by RT–PCR with specific primers. Sequencing and additional testing (see Methods for details) revealed a base disparity which gives rise to amino acid substitution G124S. Presumably, this amino acid exchange has no major functional consequence, since EMTm, the orthologue carrier from mouse, contains a glycine at the equivalent position and is functionally similar to our EMTr clone (see below). For functional expression, the cDNA of EMTr was inserted into the eukaryotic expression vector pcDNA3. The resulting plasmid, pcDNA3EMTr, was used to stably transfect 293 cells, our standard cell line for heterologous expression of non-neuronal monoamine transporters (Gründemann et al., 1999). Non-specific uptake was measured with control cells that were stably transfected with empty vector.
Functional expression of EMTr was verified in uptake experiments with adherent cells in plastic dishes. A detailed analysis of the time course of uptake of 3H-MPP+ into pcDNA3EMTr-transfected cells (Figure 1a) revealed rate constants for inwardly (kin) and outwardly (kout) directed MPP+ fluxes of 45±7 μl min−1 mg protein−1 and 0.6±0.1 min−1, respectively. The uptake at equilibrium (Amax) amounted to 7.9±0.3 pmol mg protein−1. Based on an intracellular water space of 6.7 μl mg protein−1 (Martel et al., 1996) and a conjectured transfection efficiency of 100%, at equilibrium a 12 fold accumulation of MPP+ relative to medium can be estimated. MPP+ uptake was largely, but not completely inhibited by 10 μmol l−1 disprocynium24. An uptake period of 1 min was chosen for subsequent experiments to approximate initial rates of transport.
Figure 1.
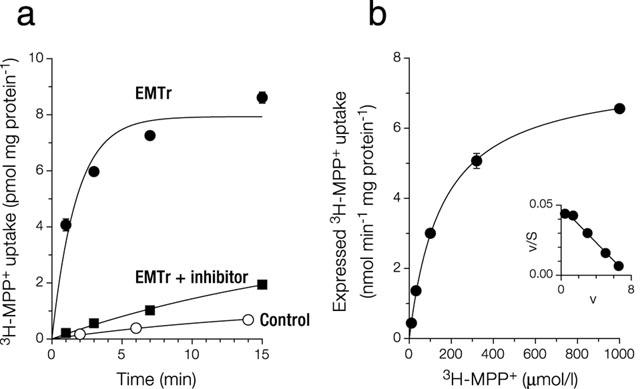
Characterization of 293 cells expressing EMTr in Na+ buffer. (a) Time course of MPP+ uptake. Cells grown in dishes were incubated at 37°C with 0.1 μmol l−1 MPP+. Shown is mean±s.e.mean (n=3). Shown are control cells (stably transfected with vector pcDNA3, kin=0.74±0.07 μl min−1 mg protein−1, kout=0.06±0.01 min−1), EmTr cells (see main text for kinetic constants) and EMTr cells in the presence of 10 μmol l−1 disprocynium24 (also during preincubation; kin=1.8±0.2 μl min−1 mg protein−1, kout=0.05±0.02 min−1). (b) Saturation of expressed uptake of MPP+. An uptake period of 1 min was chosen to approximate initial rates of transport. Shown is mean±s.e.mean (n=3). Expressed uptake was calculated by subtraction from total uptake of uptake into cells transfected with vector pcDNA3 alone. This control uptake increased linearly with MPP+ concentration, slope=0.66 μl min−1 mg protein−1. The graph shown was obtained by fitting v=Vmax * S / (Km+S) by non-linear regression, with v: uptake velocity, and S: substrate concentration. Inset: Eadie-Hofstee transformation. With our model of activation (see Figure 9), the corresponding equation is v=Vmax * S * (1+g * S/Kd ) / (Km+S * (1+S/Kd )). After fitting by non-linear regression, the graph coincides with the one shown.
Expressed uptake, i.e. total uptake minus total uptake into control cells, of MPP+ was saturable (Figure 1b), with an apparent Michaelis-Menten constant, Km, of 151 (95% confidence interval, 133–172) μmol l−1 and a maximal uptake rate, Vmax of 7.5±0.1 nmol min−1 mg protein−1. The Eadie-Hofstee plot is compatible with a single uptake mechanism.
Inhibitors of EMTh are less potent on EMTr
Inhibition of specific 3H-MPP+ uptake by disprocynium24, the most potent inhibitor available for the human EMT (Russ et al., 1993), was quantitatively examined for EMTr and EMTh, both expressed in stably transfected 293 cell lines (Figure 2a). Specific uptake was defined as that fraction of total uptake that was sensitive to 10 μmol l −1 disprocynium24. The affinity for disprocynium24 of EMTr (extrapolated IC50≈4 μmol l−1) appeared to be almost two orders of magnitude lower than that of EMTh (Ki=46 (42–51) nmol l−1). A large difference was also observed for corticosterone (Figure 2b), a well-established and relatively potent inhibitor of EMTh (Ki=0.35 (0.31–0.40) μmol l−1 in this work), but much less so for EMTr (IC50>10 μmol l−1). In additional experiments, estimated IC50-values of EMTr for decynium22 (6.4 (1.3-33) μmol l−1) and 3-O-methylisoprenaline (64 (29–144) μmol l−1) were higher than Ki-values of EMTh by a factor of 200 and 22, respectively.
Figure 2.
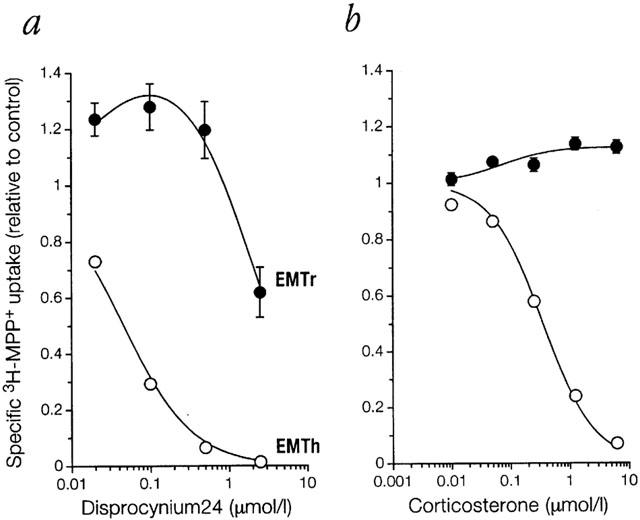
Inhibition by (a) disprocynium24 or (b) corticosterone of specific MPP+ uptake into 293 cells expressing EMTr or EMTh in Na+ buffer. Following preincubation for 20 min in the presence of inhibitor, uptake into 293 cells expressing EMTh or EMTr was measured for 1 min to approximate initial rates of transport. Shown as a function of inhibitor concentration is mean±s.e.mean (n=3) of the specific uptake of 3H-MPP+ (0.1 μmol l−1) relative to control (no inhibitor present). Specific uptake was defined as that fraction of total uptake that was sensitive to 10 μmol l−1 disprocynium24. The graphs shown result from fitting to different models for EMTh and EMTr as detailed in Methods.
In an effort to identify a powerful inhibitor for EMTr, we tested in parallel a range of compounds for inhibitory potency on EMTr and EMTh. Since histamine is an excellent substrate for EMTh (Gründemann et al., 1999), some compounds were histamine and imidazoline (Farsang & Kapocsi, 1999) receptor antagonists. Most compounds were by a factor of 10 to 100 more potent inhibitors on EMTh than on EMTr, and only very few such as e.g. BU-224 or BU-226 were about equally effective (Table 1). None of the tested inhibitors had a Ki below 100 nmol l−1, as would be desirable for an inhibitor to be effective in vivo (Schömig & Schönfeld, 1990). Surprisingly, for EMTr we noticed with some compounds, e.g. disprocynium24 (Figure 2a), clobenpropit, immepip, papaverine, thioperamide, and DMPBI (Figure 3a), a concentration-dependent stimulation of 3H-MPP+ uptake in the presence of inhibitor up to a factor of 1.5 relative to control. This activation was not seen with EMTh.
Table 1.
Inhibition by various compounds of specific 3H-MPP+ uptake into stably transfected 293 cells expressing either EMTh or EMTr
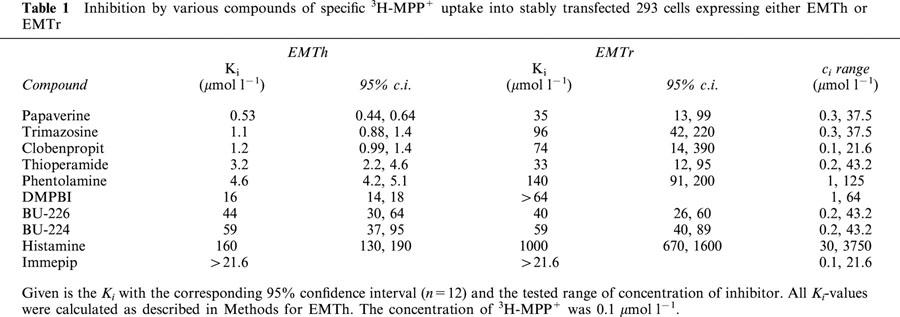
Figure 3.
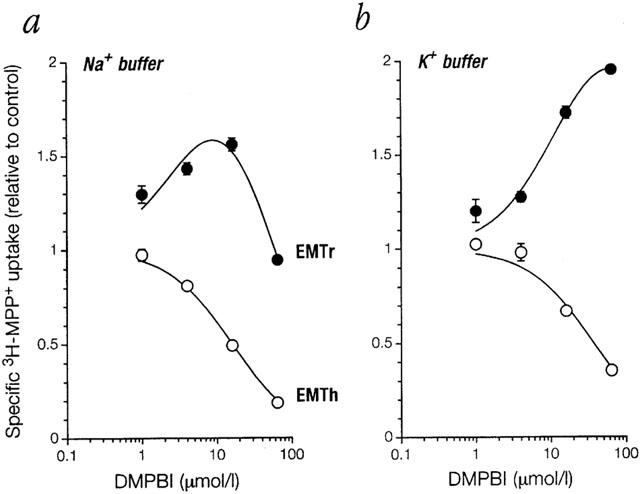
Inhibition by compound DMPBI of specific MPP+ uptake into 293 cells expressing EMTr or EMTh in (a) standard buffer or (b) high potassium buffer. In K+ buffer, all Na+ ions were replaced by K+ to depolarize and clamp membrane potential. For both conditions, cells were preincubated without inhibitor in the respective uptake buffers for 24 min. For EMTr, control uptake in K+ buffer was 44% of uptake in Na+ buffer, and non-specific uptake amounted to 5% of total uptake in Na+ buffer and 7% in K+ buffer. For further details see legend to Figure 2.
Mechanism of activation
The stimulation by compound DMPBI of EMTr-mediated 3H-MPP+ uptake was analysed in more detail. Transport of cationic substrates by non-neuronal monoamine transporters is markedly affected by membrane potential. In order to rule out activation of transport by hyperpolarization of plasma membrane, we measured uptake in a buffer with K+ in place of Na+ ions. This manoeuvre depolarizes the plasma membrane and clamps the potential essentially to 0 mV (Schömig et al., 1992). Under this condition the activation was not abolished but enhanced (Figure 3b). Therefore, hyperpolarization is not a plausible explanation for the observed activation. For most subsequent experiments K+ buffer was used to avoid any influence of membrane potential.
We next examined whether the activation was caused by trans-stimulation, i.e. acceleration of uptake by counter-transport of intracellular substrate. Cells expressing EMTr were preincubated for 24 min in K+ buffer with 1 mmol l−1 of unlabelled MPP+. Controls were incubated in K+ buffer alone. Following thorough washing, uptake of 3H-MPP+ was measured as usual. As expected, EMTr cells preloaded with MPP+ showed a marked increase in 3H-MPP+ uptake versus control cells (Figure 4). On the basis of specific uptake, this trans-stimulation amounts to a factor of 3.7±0.2. Nonetheless, in the presence of DMPBI the uptake was stimulated further (factor=1.5±0.1), similar to cells not preloaded with MPP+ (factor=1.8±0.1). If DMPBI was involved in trans-stimulation, it would have to compete intracellularly with a large excess of MPP+ and relative activation should decrease strongly. Thus, trans-stimulation and activation are probably independent mechanisms, and activation does not originate from trans-stimulation.
Figure 4.
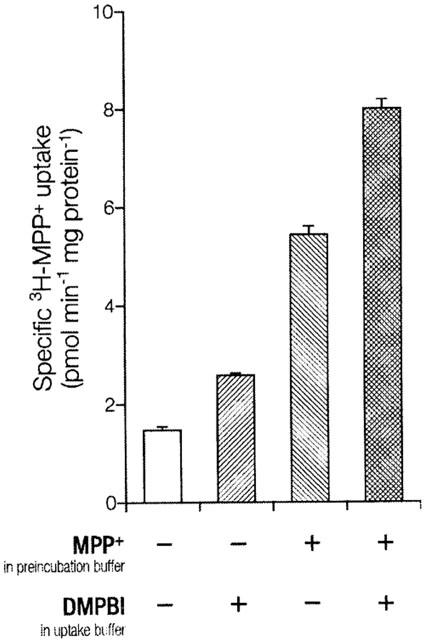
Trans-stimulation and activation of MPP+ uptake into 293 cells expressing EMTr in K+ buffer. Shown is mean±s.e.mean (n=3) of specific uptake. Specific uptake is total uptake minus non-specific uptake. Non-specific uptake (0.58±0.01 pmol min−1 mg protein−1) was measured in parallel assays with 10 μmol l−1 disprocynium24 both in preincubation and uptake buffers to inhibit EMTr. Stably transfected cells grown in dishes were preincubated for 24 min in K+ buffer (control), supplemented where indicated with 1 mmol l−1 of unlabelled MPP+ for loading. After washing three times with each 4 ml ice-cold buffer to remove substrate from extracellular space, uptake of 3H-MPP+ (0.1 μmol l−1, 1 min) in K+ buffer was measured. Parallel assays were performed with the addition of 50 μmol l−1 unlabelled DMPBI solely to uptake buffer.
3H-MPP+ uptake was also higher than control when cimetidine was used as inhibitor (Figure 5a). However, when 3H-cimetidine was used as substrate, unlabelled cimetidine did not stimulate uptake above control level. Hence, activation strikingly depends on the structure of the monitored substrate. In accordance with this concept, MPP+ as inhibitor did not stimulate uptake of 3H-cimetidine (Figure 5b), but markedly activated uptake of 3H-MPP+ (Figure 6).
Figure 5.
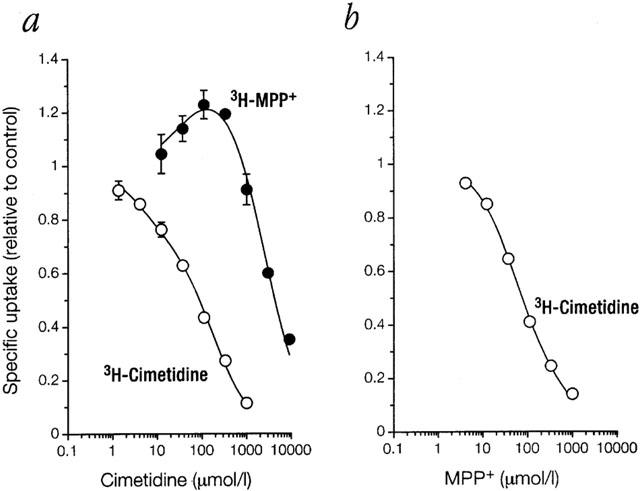
Inhibition by (a) cimetidine or (b) MPP+ of uptake into 293 cells expressing EMTr in K+ buffer. Shown as a function of inhibitor concentration is mean±s.e.mean (n=3) of specific uptake relative to control of 3H-MPP+ or 3H-cimetidine (each at 0.1 μmol l−1) as indicated. Cells were preincubated without inhibitor for 24 min. For further details see legend to Figure 2. With the special case of substrate equal to inhibitor and with the (unproved) assumption of complete symmetry (i.e. Ki=Ks, Ka=Kb=Kc=Kd, and e=f=g; see Figure 9), the general model of activation simplifies to vi/vo=(Ks+S+S2/Kd) * (Kd+g * S+2 * g * I) / ((Kd+g * S) * (Ks+S+I+1 / Kd * (S+I)2)). The parameters extracted for cimetidine by non-linear regression are Ks=4.0 (1.4–12) μmol l−1, Kd=180 (120–260) μmol l−1 and g=16 (6.2–40).
Figure 6.
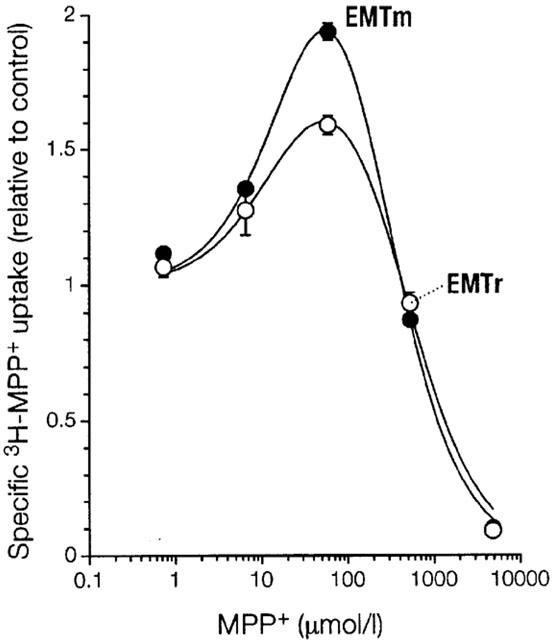
Comparison of EMT from rat and mouse. Shown as a function of inhibitor concentration is mean±s.e.mean (n=3) of specific uptake relative to control of 3H-MPP+ (0.1 μmol l−1) in K+ buffer into 293 cells expressing EMTr or EMTm as indicated. Cells were preincubated without inhibitor for 24 min. For further details see legend to Figure 2. Parameters fitted as described in legend to Figure 5 are Ks=26 (18–37) (EMTm) and 17 (6.5–42) μmol l−1 (EMTr), Kd =230 (190–280) and 440 (300–660) μmol l−1, and g=13 (8.3–20) and 27 (9.0–79).
The differential activation seen for uptake of 3H-cimetidine and 3H-MPP+ could relate to the use of inhibitors that are transported themselves, i.e. cimetidine and MPP+. We therefore tested activation of uptake by a non-transported inhibitor, disprocynium24 (Figure 7). Again, uptake of 3H-MPP+ was clearly stimulated above control level, while uptake of 3H-cimetidine was not. Stimulation of uptake of 3H-noradrenaline was intermediate. Note that IC50-values (disprocynium24 concentration at uptake relative to control=0.5) are considerably dissimilar for different substrates.
Figure 7.
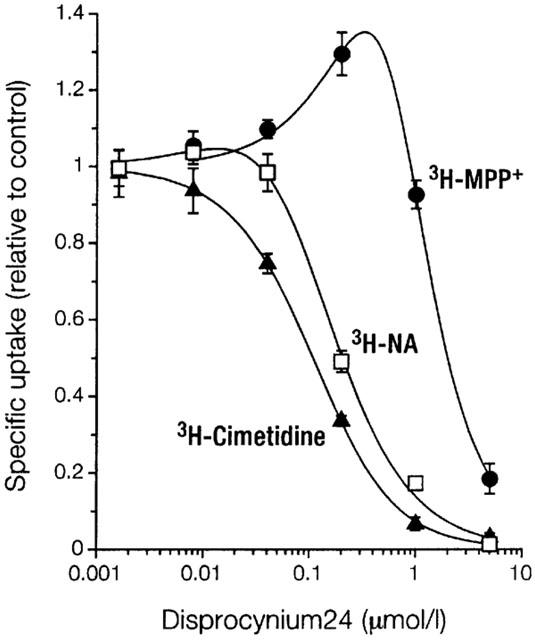
Inhibition by disprocynium24 of uptake of cimetidine, noradrenaline (NA), or MPP+ into 293 cells expressing EMTr in K+ buffer. Shown as a function of inhibitor concentration is mean±s.e.mean (n=3) of specific uptake relative to control of labelled substrate (each at 0.1 μmol l−1) as indicated. Cells were preincubated with inhibitor for 24 min. For further details see legend to Figure 2.
To confirm that the mechanism of activation also operates under physiological conditions, we measured stimulation of uptake of 3H-MPP+ by unlabelled MPP+ in Na+ buffer (Figure 8). Compared to uptake in K+ buffer the effect is somewhat smaller, but openly evident. This agrees well with the data presented in Figure 3.
Figure 8.
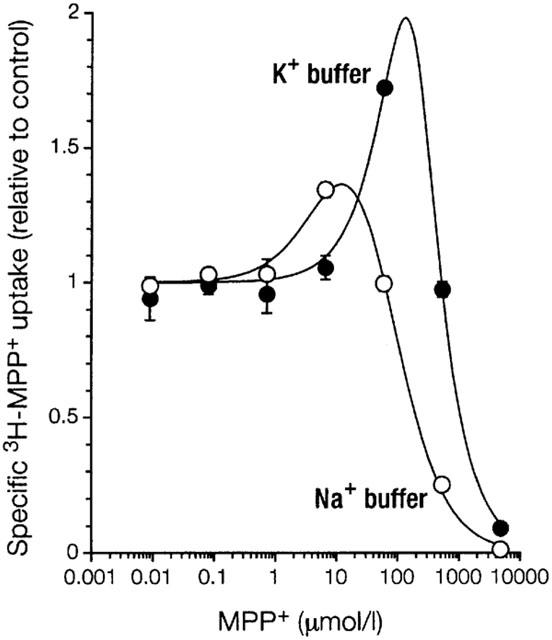
Comparison of activation of MPP+ uptake in standard buffer (Na+ buffer) or K+ buffer. Shown as a function of inhibitor concentration is mean±s.e.mean (n=3) of specific uptake of 3H-MPP+ (1 nmol l−1) relative to control into 293 cells expressing EMTr. For both conditions, cells were preincubated in the respective buffers for 24 min without inhibitor. For further details see legend to Figure 2.
For verification of results, we obtained the cDNA of the EMT orthologue from mouse, which is highly similar to EMTr (98.7% amino acid sequence identity). Functional expression of the EMTm cDNA in 293 cells was performed as described above. In excellent agreement with the results for EMTr, 3H-MPP+ uptake mediated by EMTm was strongly activated by unlabelled MPP+ (Figure 6). Also, the Ki-values of EMTm for disprocynium24 (0.29 (0.18–0.45) μmol l−1) and corticosterone (11 (4.4–26) μmol l−1) were similar to the above values for EMTr.
Discussion
The first hint that EMT from rat is markedly different in kinetic terms from other non-neuronal monoamine transporters and even from the human orthologue EMTh came from the Km-values for MPP+. Whereas OCT2 and OCT1 both from human and rat and EMT from human display very similar affinities (10–20 μmol l−1) (Gründemann et al., 1999; manuscript in preparation), EMT from rat was reported with a Km of 90–140 μmol l−1 in HRPE cells (Wu et al., 1998). Our present results confirm this difference (Figure 1b). We also report here that known highly potent EMTh inhibitors such as disprocynium24, decynium22, and corticosterone are much less potent on EMT from rat (Figure 2) and mouse. At the same time, there is a pressing need for potent inhibitors in rat or mouse in order to promote understanding of physiological function and pharmacology of EMT in these principal mammalian model organisms. Thus far, our efforts to identify a truly potent inhibitor for EMTr have not been successful. However, an unexpected finding may explain the peculiar characteristics of EMTr. In this work, we report that inhibitor may stimulate EMTr-mediated transport above control level, an unusual and hitherto unrecognized phenomenon which we refer to as activation.
In order to verify this observation, we have sought for rat cell lines that natively express EMTr. With RT–PCR, none of the tested lines (A10, C6, RINm5F, RGE, NRK-52E, and PC12) was positive (not shown), which is quite in contrast to EMTh which has been detected in many cell lines. However, the almost identical orthologue from mouse, EMTm, showed features very similar to EMTr, providing ample corroboration of our results. Note that whereas the concentration of unlabelled MPP+ giving maximum stimulation in a given buffer was equal for both transporters (Figure 6) and invariable between experiments, the actual factor of maximum stimulation varied (not shown).
Our experiments demonstrate that activation is affected, but not caused by changes in membrane potential (Figure 3). The stronger activation (relative to control) in K+ buffer versus Na+ buffer may originate from attenuated binding of inhibitor due to membrane potential depolarization. In other words, in K+ buffer, activation is more pronounced since the opposing inhibition of uptake is shifted to higher concentrations of inhibitor. Activation by inadvertent trans-stimulation (also known as accelerated exchange) has been reported (Deves & Krupka, 1987), but this mechanism depends on rapid passive diffusion of a substrate across the plasma membrane. In our case, disprocynium24 clearly evokes activation (Figures 2a and 7), but it should not be transported by EMTr at all. This and the experiment shown in Figure 4 suggest that activation is not based on trans-stimulation. Also, the experiments shown in Figures 5a and 7 effectively exclude the involvement of a receptor-mediated signalling pathway, since any receptor ligand would affect the transporter indirectly, irrespective of the substrate (compare MPP+ and cimetidine). For the same reason and since activation is immediate, i.e. without any preincubation of cells in the presence of inhibitor, an inhibitor-induced recruitment of a transporter reserve (Zhang & Ismail-Beigi, 1998) is implausible. An apparent increase in uptake activity could also result from protection of the labelled substrate against some extracellular inactivation with increasing concentrations of cold substrate or competitor. However, this explanation does not apply here, since (1) EMTh should be affected to the same extent as EMTr, and (2) e.g. MPP+ is highly resistant to metabolization.
Instead, for an explanation of activation of EMTr and EMTm, we propose a model that assumes the interaction of two binding sites for substrate or inhibitor (Figure 9). Note that the familiar cooperativity-based Hill model for multisite inhibition (Segel, 1975) is unsuitable, since uptake in the presence of inhibitor will never exceed control uptake. Compared to two sites that operate independently, our model of activation suggests that inhibitor bound to the first site actually diminishes binding, but accelerates turnover of substrate at the second site (not shown). Thus, with MPP+ at moderate inhibitor concentrations (Figure 6), the gain in speed dominates over loss of affinity and activation becomes apparent. At high inhibitor concentrations, uptake eventually declines because two molecules of inhibitor bind to the transporter. By contrast, with cimetidine as substrate (Figure 5), no activation is apparent. This is intriguing, since cimetidine (1) is rather a good substrate for EMTh, with a transport efficiency of 10% relative to MPP+ (Gründemann et al., 1999), and (2) has an affinity for EMTh similar to MPP+. Apparently, the structure of cimetidine does not sufficiently promote stimulation of turnover.
Since our model would require the fitting of 10 dissociation or rate constants, it is not possible, in general, to extract with confidence these parameters from the experiments reported in this article. However, the model achieves a good fit of relative uptake as a function of inhibitor concentration with three aggregate parameters (see Methods for equation). With the assumption of two kinetically indistinguishable interacting sites, the model simplifies considerably if substrate and inhibitor are identical (except for the radiolabel on the substrate). Thus, we have been able to calculate intrinsic Ki values for MPP+ and cimetidine (see legend to Figure 5). For MPP+, the fitted values of EMTr (17 μmol l−1) and EMTm (26 μmol l−1) agree very well with the corresponding value of EMTh (24 μmol l−1 (Russ et al., 1992)). For cimetidine, the extracted Ki of EMTr (4.0 μmol l−1) is somewhat lower than expected from experiments with EMTh (38 μmol l−1 (Gründemann et al., 1998b)).
The extent of activation may depend on substrate affinity. Substrates of EMT with high affinity but slow turnover like MPP+ (Gründemann et al., 1999) could strongly benefit from stimulation of turnover (structure permitting, see cimetidine), whereas substrates with low affinity but high Vmax such as noradrenaline are less likely to show this effect (c.f. Figure 7). It is evident from our study that no conclusions can be drawn from the observation of activation whether an inhibitor is actually transported, since both transported (e.g. MPP+, cimetidine) and non-transported inhibitors (disprocynium24, corticosterone) elicited activation.
It is a generally accepted pharmacological principle that a transporter protein has a definite affinity (Ki) for an inhibitor irrespective of substrate. However, with EMTr we demonstrate that inhibition potency if determined as usual from the IC50-values depends very much on substrate (Figures 5 and 7). Clearly, the useful pharmacological concept to compare and identify carrier proteins based on IC50 values may fail with EMTr.
In rat brain a transport mechanism for catecholamines has been described that matches the extraneuronal monoamine transporter (uptake2) in all aspects except for the inhibition profile (Hendley et al., 1970; Wilson et al., 1988). IC50-values (1) are relatively high for e.g., corticosterone and 3-O-methylisoprenaline and (2) do not correlate closely with values from heart, a reference organ for uptake2. In experiments with rat heart, with isoprenaline or noradrenaline as substrates, there are no obvious signs of activation (Grohmann & Trendelenburg, 1984). We may now suggest that the protein behind the unidentified mechanism in rat brain is EMTr, and that activation causes the unusual inhibition pattern, since (1) activation increases IC50-values and (2) the extent of activation (and hence the shift of IC50-values) will not be uniform for different inhibitors. Further work is necessary to clarify why activation is apparently inoperative in rat heart. Perhaps activation is proportional to local transporter density or is blocked by other proteins (see below).
What would be the physiological purpose of activation? In contrast to cooperative proteins such as haemoglobin or aspartate transcarbamoylase, the saturation curve of EMTr is not plainly sigmoidal (Figure 1b), i.e. the fit to the hyperbolic Michaelis-Menten equation even at low substrate concentrations is as good as the fit to an equation based on our more complicated model (see legend to Figure 1b). Thus, we feel that activation of EMTr does not serve regulation of activity in response to substrate concentration. However, a suitable endogenous compound could conceivably evoke stimulation of transporter activity. Alternatively, we speculate that in rodents activation may keep the transporter working as it encounters inhibitors. Much higher concentrations of blocking substances are necessary to shut the transporter down. This relates to the notion that rodents tolerate higher levels of xenobiotics than man.
The primary structures of EMTr and EMTh are very similar (87.1% amino acid identity), yet only EMTr shows activation. It will be interesting to determine the underlying molecular details. So far, it is not possible to tell whether the two postulated binding sites reside on a single transporter molecule (intramolecular activation) or whether a dimer of single-site transporters is in operation (intermolecular activation). The later case would open the possibility for the formation of heterodimers with novel functional characteristics within the ASF family, an aspect neglected so far in heterologous expression of cloned transporters.
In conclusion, it should be difficult to evaluate the role of the extraneuronal monoamine transporter in the rat or mouse model by the use of pharmacological inhibitors, since EMTr and EMTm may be much more resistant than the human orthologue. We have uncovered a mechanism of activation to explain this difference. The extent of activation critically depends on the chemical structure of the monitored substrate. Kinetic modelling suggests that activation is based on the interaction of two substrate binding sites per transporter entity.
Acknowledgments
Supported by Deutsche Forschungsgemeinschaft (SCHO 373/4-1 and FOR 302/2 B2). We thank Katarzyna Baran and Anke Ripperger for skillful technical assistance, and R. Zwart for providing the cDNA of EMTm.
Abbreviations
- EMT
extraneuronal monoamine transporter
- kb
kilobase(s)
- MPP+
1-methyl-4-phenylpyridinium
- OCT
organic cation transporter
- PCR
polymerase chain reaction
- RT
reverse transcriptase
- h r, and m
attached to a protein name designate species as being human, rat, or mouse, respectively
References
- BREIDERT T., SPITZENBERGER F., GRÜNDEMANN D., SCHÖMIG E. Catecholamine transport by the organic cation transporter type 1 (OCT1) Br. J. Pharmacol. 1998;125:218–224. doi: 10.1038/sj.bjp.0702065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUSCH A.E., KARBACH U., MISKA D., GORBOULEV V., AKHOUNDOVA A., VOLK C., ARNDT P., ULZHEIMER J.C., SONDERS M.S., BAUMANN C., WALDEGGER S., LANG F., KOEPSELL H. Human neurones express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol. Pharmacol. 1998;54:342–352. doi: 10.1124/mol.54.2.342. [DOI] [PubMed] [Google Scholar]
- CHENG Y.-C., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- DEVES R., KRUPKA R.M. Effects on transport of rapidly penetrating, competing substrates: activation and inhibition of the choline carrier in erythrocytes by imidazole. J. Membr. Biol. 1987;99:13–23. doi: 10.1007/BF01870618. [DOI] [PubMed] [Google Scholar]
- EISENHOFER G., MCCARTY R., PACAK K., RUSS H., SCHÖMIG E. Disprocynium24, a novel inhibitor of the extraneuronal monoamine transporter, has potent effects on the inactivation of circulating noradrenaline and adrenaline in conscious rat. Naunyn-Schmiedeberg's Arch. Pharmacol. 1996;354:287–294. doi: 10.1007/BF00171059. [DOI] [PubMed] [Google Scholar]
- FARSANG C., KAPOCSI J. Imidazoline receptors: from discovery to antihypertensive therapy (facts and doubts) Brain Res. Bull. 1999;49:317–331. doi: 10.1016/s0361-9230(99)00057-x. [DOI] [PubMed] [Google Scholar]
- GROHMANN M., TRENDELENBURG U. The substrate specificity of uptake2 in the rat heart. Naunyn-Schmiedeberg's Arch. Pharmacol. 1984;328:164–173. doi: 10.1007/BF00512067. [DOI] [PubMed] [Google Scholar]
- GRÜNDEMANN D., BABIN-EBELL J., MARTEL F., ÖRDING N., SCHMIDT A., SCHÖMIG E. Primary structure and functional expression of the apical organic cation transporter from kidney epithelial LLC-PK1 cells. J. Biol. Chem. 1997;272:10408–10413. doi: 10.1074/jbc.272.16.10408. [DOI] [PubMed] [Google Scholar]
- GRÜNDEMANN D., GORBOULEV V., GAMBARYAN S., VEYHL M., KOEPSELL H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature. 1994;372:549–552. doi: 10.1038/372549a0. [DOI] [PubMed] [Google Scholar]
- GRÜNDEMANN D., KÖSTER S., KIEFER N., BREIDERT T., ENGELHARDT M., SPITZENBERGER F., OBERMÜLLER N., SCHÖMIG E. Transport of monoamine transmitters by the organic cation transporter type 2 (OCT2) J. Biol. Chem. 1998a;273:30915–30920. doi: 10.1074/jbc.273.47.30915. [DOI] [PubMed] [Google Scholar]
- GRÜNDEMANN D., LIEBICH G., KIEFER N., KÖSTER S., SCHÖMIG E. Selective substrates for non-neuronal monoamine transporters. Mol. Pharmacol. 1999;56:1–10. doi: 10.1124/mol.56.1.1. [DOI] [PubMed] [Google Scholar]
- GRÜNDEMANN D., SCHECHINGER B., RAPPOLD G.A., SCHÖMIG E. Molecular identification of the corticosterone-sensitive extraneuronal monoamine transporter. Nature Neurosci. 1998b;1:349–351. doi: 10.1038/1557. [DOI] [PubMed] [Google Scholar]
- HENDLEY E.D., TAYLOR K.M., SNYDER S.H. 3H-normetanephrine uptake in rat brain slices. Relationship to extraneuronal accumulation of norepinephrine. Eur. J. Pharmacol. 1970;12:167–179. doi: 10.1016/0014-2999(70)90062-2. [DOI] [PubMed] [Google Scholar]
- IVERSEN L.L. The uptake of catecholamines at high perfusion concentrations in the rat isolated heart: a novel catecholamine uptake process. Br. J. Pharmacol. 1965;25:18–33. doi: 10.1111/j.1476-5381.1965.tb01753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IVERSEN L.L., SALT P.J. Inhibition of catecholamine uptake by steroids in the isolated rat heart. Br. J. Pharmacol. 1970;40:528–530. doi: 10.1111/j.1476-5381.1970.tb10637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONKER J.W., WAGENAAR E., MOL C.A., BUITELAAR M., KOEPSELL H., SMIT J.W., SCHINKEL A.H. Reduced hepatic uptake and intestinal excretion of organic cations in mice with a targeted disruption of the organic cation transporter 1 (oct1) Mol. Cell Biol. 2001;21:5471–5477. doi: 10.1128/MCB.21.16.5471-5477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEKUDA R., PRASAD P.D., WU X., WANG H., FEI Y.-J., LEIBACH F.H., GANAPATHY V. Cloning and functional characterization of a potential-sensitive, polyspecific organic cation transporter (OCT3) most abundantly expressed in placenta. J. Biol. Chem. 1998;273:15971–15979. doi: 10.1074/jbc.273.26.15971. [DOI] [PubMed] [Google Scholar]
- MARTEL F., VETTER T., RUSS H., GRÜNDEMANN D., AZEVEDO I., KOEPSELL H., SCHÖMIG E. Transport of small organic cations in the rat-liver - the role of the organic cation transporter OCT1. Naunyn-Schmiedeberg's Arch. Pharmacol. 1996;354:320–326. doi: 10.1007/BF00171063. [DOI] [PubMed] [Google Scholar]
- NAGEL G., VOLK C., FRIEDRICH T., ULZHEIMER J.C., BAMBERG E., KOEPSELL H. A reevaluation of substrate specificity of the rat cation transporter rOCT1. J. Biol. Chem. 1997;272:31953–31956. doi: 10.1074/jbc.272.51.31953. [DOI] [PubMed] [Google Scholar]
- OKUDA M., SAITO H., URAKAMI Y., TAKANO M., INUI K. cDNA cloning and functional expression of a novel rat kidney organic cation transporter, OCT2. Biochem. Biophys. Res. Commun. 1996;224:500–507. doi: 10.1006/bbrc.1996.1056. [DOI] [PubMed] [Google Scholar]
- POVLOCK S.L., AMARA S.G.The structure and function of norepinephrine, dopamine, and serotonin transporters Neurotransmitter transporters. Structure, function, and regulation 1997Totowa: Humana Press; 1–28.ed. Reith M.E.A. pp [Google Scholar]
- RUSS H., ENGEL W., SCHÖMIG E. Isocyanines and pseudoisocyanines as a novel class of potent noradrenalin transport inhibitors: synthesis, detection, and biological activity. J. Medicinal Chem. 1993;36:4208–4213. doi: 10.1021/jm00078a010. [DOI] [PubMed] [Google Scholar]
- RUSS H., GLIESE M., SONNA J., SCHÖMIG E. The extraneuronal transport mechanism for noradrenalin (uptake2) avidly transports 1-methyl-4-phenylpyridinium (MPP+) Naunyn-Schmiedeberg's Arch. Pharmacol. 1992;346:158–165. doi: 10.1007/BF00165297. [DOI] [PubMed] [Google Scholar]
- RUSS H., STAUDT K., MARTEL F., GLIESE M., SCHÖMIG E. The extraneuronal transporter for monoamine transmitters exists in cells derived from human central nervous system glia. Eur. J. Neurosci. 1996;8:1256–1264. doi: 10.1111/j.1460-9568.1996.tb01294.x. [DOI] [PubMed] [Google Scholar]
- SCHÖMIG E., BABIN-EBELL J., RUSS H. 1,1′-Diethyl-2,2′-cyanine (decynium22) potently inhibits the renal transport of organic cations. Naunyn-Schmiedeberg's Arch. Pharmacol. 1993;347:379–383. doi: 10.1007/BF00165387. [DOI] [PubMed] [Google Scholar]
- SCHÖMIG E., BABIN-EBELL J., RUSS H., TRENDELENBURG U. The force driving the extraneuronal transport mechanism for catecholamines (uptake2) Naunyn-Schmiedeberg's Arch. Pharmacol. 1992;345:437–443. doi: 10.1007/BF00176622. [DOI] [PubMed] [Google Scholar]
- SCHÖMIG E., SCHÖNFELD C.L. Extraneuronal noradrenaline transport (uptake2) in a human cell line (Caki-1 cells) Naunyn-Schmiedeberg's Arch. Pharmacol. 1990;341:404–410. doi: 10.1007/BF00176331. [DOI] [PubMed] [Google Scholar]
- SCHÖMIG E., SPITZENBERGER F., ENGELHARDT M., MARTEL F., ÖRDING N., GRÜNDEMANN D. Molecular cloning and characterization of two novel transport proteins from rat kidney. FEBS Lett. 1998;425:79–86. doi: 10.1016/s0014-5793(98)00203-8. [DOI] [PubMed] [Google Scholar]
- SEGEL I.H. Enzyme kinetics. New York: John Wiley & Sons; 1975. [Google Scholar]
- TRENDELENBURG U.The extraneuronal uptake and metabolism of catecholamines Catecholamines I 1988Berlin: Springer-Verlag; 279–319.eds. Trendelenburg, U. & Weiner, N. pp [Google Scholar]
- VERHAAGH S., SCHWEIFER N., BARLOW D.P., ZWART R. Cloning of the mouse and human solute carrier 22a2 (Slc22a3/SLC22A3) identifies a conserved cluster of three organic cation transporters on mouse chromosome 17 and human 6q26-q27. Genomics. 1999;55:209–218. doi: 10.1006/geno.1998.5639. [DOI] [PubMed] [Google Scholar]
- WILSON V.G., GROHMANN M., TRENDELENBURG U. The uptake and O-methylation of 3H-(+/−)-isoprenaline in rat cerebral cortex slices. Naunyn-Schmiedeberg's Arch. Pharmacol. 1988;337:397–405. doi: 10.1007/BF00169530. [DOI] [PubMed] [Google Scholar]
- WU X., HUANG W., GANAPATHY M.E., WANG H., KEKUDA R., CONWAY S.J., LEIBACH F.H., GANAPATHY V. Structure, function, and regional distribution of the organic cation transporter OCT3 in the kidney. Am. J. Physiol. 2000;279:F449–F458. doi: 10.1152/ajprenal.2000.279.3.F449. [DOI] [PubMed] [Google Scholar]
- WU X., KEKUDA R., HUANG W., FEI Y.-J., LEIBACH F.H., CHEN J., CONWAY S.J., GANAPATHY V. Identity of the organic cation transporter OCT3 as the extraneuronal monoamine transporter (uptake2) and evidence for the expression of the transporter in the brain. J. Biol. Chem. 1998;273:32776–32786. doi: 10.1074/jbc.273.49.32776. [DOI] [PubMed] [Google Scholar]
- ZHANG J.Z., ISMAIL-BEIGI F. Activation of Glut1 glucose transporter in human erythrocytes. Arch. Biochem. Biophys. 1998;356:86–92. doi: 10.1006/abbi.1998.0760. [DOI] [PubMed] [Google Scholar]
- ZOR T., SELINGER Z. Linearization of the Bradford protein assay increases its sensitivity: theoretical and experimental studies. Anal. Biochem. 1996;236:302–308. doi: 10.1006/abio.1996.0171. [DOI] [PubMed] [Google Scholar]
- ZWART R., VERHAAGH S., BUITELAAR M., POPP-SNIJDERS C., BARLOW D.P. Impaired activity of the extraneuronal monoamine transporter system known as uptake-2 in Orct3/Slc22a3-deficient mice. Mol. Cell Biol. 2001;21:4188–4196.. doi: 10.1128/MCB.21.13.4188-4196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]