

Abstract
Vascular endothelial growth factor (VEGF) plays an important role in the neovascularization of ischaemic retinal diseases such as proliferative diabetic retinopathy. We determined that bucillamine, an anti-rheumatic drug, inhibits the VEGF production induced by hypoxia in bovine retinal microcapillary endothelial cells (BREC). To further clarify the inhibitory mechanism, we investigated the possible mechanism by which bucillamine exerts this inhibitory effect.
Bucillamine (100 μM) decreased the hypoxia-induced increase of VEGF mRNA by 54.5% (P<0.001). Bucillamine (100 μM) reduced the hypoxia-induced VEGF content in culture media by 29.0% (P<0.001), while monosulfydryl drugs, N-acetylcysteine and D-penicillamine, did not.
Bucillamine (100 μM) did not affect VEGF mRNA half-life (hypoxia, 4.3 h; hypoxia+bucillamine, 3.9 h; normoxia, 2.7 h; normoxia+bucillamine, 2.7 h).
Reporter gene studies revealed that bucillamine reduced transcriptional activity in the 5′-flanking region of the VEGF gene by 74.0%. Hypoxia stimulated binding activity of BREC nuclear protein to a hypoxia responsive element (HRE), which was decreased by bucillamine.
Bucillamine inhibited hypoxic-induction of HIF-1α mRNA by 73.1% (P<0.001). Bucillamine also inhibited spontaneous VEGF mRNA expression by 26.6%. Furthermore, it inhibited activity of VEGF promoter and decreased binding activity to Sp1 and HRE, but did not alter AP1 and AP2 activity in normoxia.
These data suggest that bucillamine inhibits hypoxic induction of VEGF through inhibition of HIF-1 induction and binding activity in BREC. Bucillamine also inhibits the spontaneous expression of VEGF mRNA by its effect on Sp1 and HRE binding.
Keywords: Bucillamine, angiogenesis, retinal ischaemia, vascular endothelial growth factor
Introduction
Bucillamine (N-(mercapto-2-methylpropionyl)-L-cystein), an anti-rheumatoid drug, has been used clinically primarily for patients with rheumatoid arthritis (RA). Tsuji et al. (1999) reported that bucillamine inhibited NF-κB activation and tumour necrosis factor-alpha (TNF-α) mRNA and protein expression in the human monocytic leukaemia cell line THP-1. Furthermore, bucillamine had significant suppressive effects on interleukin (IL-6) and IL-8 production by synovial cells in vitro (Matsuno et al., 1998). In synovial cells, bucillamine inhibited IL-1-induced vascular endothelial growth factor (VEGF) production (Tsuji et al., 1999). Despite these reports of the drug's effect on the induction of various cytokines, its effect on hypoxic-induction of VEGF has not been investigated to date.
VEGF is a highly endothelial cell-specific mitogen and vasopermeability factor (Berse et al., 1992; Leung et al., 1989) whose expression is dramatically increased by hypoxia (Shweiki et al., 1992), which is a primary stimulus of ocular neovascularization. VEGF is also a key regulator of pathologic angiogenesis in other ischaemic and inflammatory diseases including cancer, rheumatoid arthritis, and skin pyrogenic granulomas and is a target for treating such diseases. Complications from pathologic angiogenesis underlie most eye diseases that cause severe and irreversible loss of vision. These diseases include diabetic retinopathy, age-related macular degeneration, retinopathy of prematurity, and retinal vein occlusion, of which recent studies have implicated VEGF as a possible mediator (Adamis et al., 1996; Aiello et al., 1994; 1995; Amin et al., 1997; Kliffen et al., 1997; Lopez et al., 1996; Lutty et al., 1996; Robinson et al., 1996). Accordingly, VEGF might be a viable target for pharmacological intervention in ischemic retinal conditions and choroidal neovascular diseases. In the study described herein, we investigated the inhibitory mechanism of bucillamine on hypoxia-induced VEGF expression in cultured bovine retinal microcapillary endothelial cells and bovine aortic endothelial cells.
Methods
Cell cultures
Bovine retinal endothelial cells (BREC) were isolated by homogenization of retinas and a series of filtration steps, as previously described (King et al., 1996). Briefly, under sterile conditions, the retinas were isolated and extensively rinsed in Dulbecco's modified Eagle's medium (DMEM, Gibco, Grand Island, NY, U.S.A.) and suspended in an enzyme solution containing collagenase (500 μg ml−1; Nitta Gelatin, Osaka, Japan) in 10 mM phosphate-buffered saline (PBS; Sigma, St. Louis, MO, U.S.A.). Pieces of adherent retinal pigment epithelium were dissected, and the retinas were minced and incubated with shaking at 37°C. Sequential sieving was performed over 210- and 52-μm nylon mesh, and the remaining retinal capillary fragments were plated on fibronectin (Sigma, St. Louis, MO, U.S.A.) coated 35 mm-dishes (Iwaki Glas Inc., Tokyo, Japan) containing DMEM with 5.5 mM glucose, 10% plasma-derived horse serum (PDHS, Wheaton, Pipersville, PA, U.S.A.), 50 mg l−1 heparin and 50 u l−1 endothelial cell growth factor (Boehringer Mannheim, Indianapolis, IN, U.S.A.). Bovine aortic endothelial cells (BAEC) were isolated by peeling of bovine aorta, as previously described (King et al., 1985). Primary BAEC were grown on Type I collagen-coated 35 mm-dishes (Iwaki Glas Inc., Tokyo, Japan) containing DMEM with 5.5 mM glucose, 10% PDHS. Cells were cultured in 5% CO2 at 37°C, and media were changed every 3 days. After the cells reached confluence, cells from passages 6–9 were used for these experiments. Endothelial cell homogeneity was confirmed by immunoreactivity with anti-factor VIII antibodies analysed by confocal microscopy, and cell characteristics such as shape and growth rate were carefully observed until passage 15, and remained unchanged throughout the observation period.
Drugs
Bucillamine was purchased from Santen Pharmaceutical (Osaka, Japan). N-acetylcysteine and D-penicillamine were purchased from Wako Pure Chemical (Osaka, Japan). These compounds were dissolved in 25% ethanol/PBS solution and regulated to final ethanol concentration of less than 0.25%.
Analysis of cell viability
BREC viability was determined by measuring DNA content, as previously described (Takagi et al., 1996). The cells were plated sparsely (∼10,000 cells per well) in 24-well culture dishes (Iwaki Glass, Tokyo, Japan) overnight in DMEM containing 10% PDHS to maintain the cell viability. The medium was then changed to DMEM with 10% calf serum containing various concentrations of bucillamine. After incubation at 37°C for 24 h, the cells were lysed in 0.1% sodium dodecylsulphate and DNA content was measured using Hochst-33258 dye and a fluorometer (model TKO-100, Hoefer Scientific Instruments, San Francisco, CA, U.S.A.).
Hypoxia studies
BREC were cultured under the hypoxic conditions, as previously described (Koyama et al., 1999). Confluent cell monolayers on 100-mm tissue culture plates were exposed to 1±0.5% oxygen using a water-jacketed mini-CO2/multi-gas incubator with reduced oxygen control (model BL-40M, Jujikagaku, Tokyo, Japan). All cells were maintained at 37°C in a constant 5% carbon dioxide atmosphere with oxygen deficit induced by nitrogen replacement. Bucillamine, N-acetylcysteine and D-penicillamine were added to the culture media just at the beginning of the induction of hypoxia. Four-ml of hypoxia-conditioned medium was collected from confluent cultures on 100-mm tissue culture plates after 24 or 48 h, and filtered before use to remove cellular components. Medium from dishes cultured under normoxic conditions (95% air, 5% CO2) was used as a control. Furthermore, the conditioned media were concentrated by 20 fold concentration using Centricon YM-30 (Millipore, Bedford, MA, U.S.A.).
Northern blot analysis
Total RNA was isolated from individual tissue culture plates using guanidium thiocyanate (Chomczynski & Sacchi, 1987). Northern blot analysis was performed on 20 μg total RNA after 1% agarose-2 M formaldehyde gel electrophoresis and subsequent capillary transfer to Biodyne nylon membranes (Pall Bio Support, East Hills, NY, U.S.A.) and ultraviolet crosslinking using a FUNA-UV-LINKER (FS-1500, Funakoshi Inc., Tokyo, Japan). Radioactive probes were generated using Amersham Megaprime labelling kits and [α32P]-dCTP (Amersham, Arlington Heights, IL, U.S.A.). Blots were prehybridized, hybridized and washed in 0.5×SSC (75 mM sodium chloride, 7.5 mM sodium citrate, pH 7.0), 5% SDS at 65°C with four changes over 1 h in a rotating hybridization oven (TAITEC, Koshigaya, Japan). All signals were analysed using a densitometer (BAS-2000II, Fuji Photo Film, Tokyo, Japan) and lane-loading differences were normalized using a 36B4 cDNA probe, which hybridizes to acidic ribosomal phosphoprotein PO (Liang & Pardee, 1992). Bovine HIF1A cDNA was made by a polymerase chain reaction (PCR) (Hara et al., 1999).
mRNA stability assay
To determine whether the increase in VEGF mRNA was caused by an increase in transcription, BREC were stimulated by normoxic or hypoxic conditions for 24 h. Then de novo mRNA transcription was inhibited by the addition of 4 μM actinomycin D. BREC were exposed to actinomycin D after 2 and 4 h of incubation under normoxic or hypoxic conditions. Total RNA was then extracted, and Northern blot analysis was performed.
Enzyme-linked immunosorbent assay (ELISA)
The concentration of VEGF was determined using an ELISA assay kit (R&D Systems, Minneapolis, MN, U.S.A.); the assay was performed as specified by the supplier. Samples of conditioned medium (200 μl) were added to the antibody-coated plate. Plates were incubated for 2 h at room temperature. After thorough washing, polyclonal anti-human VEGF antibody was dispensed into each well and incubated for another 2 h. Plates were rinsed again and substrate and amplifier solutions were added to each well at 20 min intervals. The reaction product was detected and quantified using a plate reader (450 nm). The amount of VEGF in each sample was calculated using a standard curve that demonstrates a direct relationship between optical density (OD) and VEGF concentration. After removal of conditioned media, cellular protein of each plate was determined by Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA, U.S.A.). The results were expressed in pg μg−1 protein.
Reporter gene assay
A series of plasmid constructs was made from a genomic DNA clone of the human VEGF gene (Tischer et al., 1991), which contained approximately 2.5 kb of the 5′-flanking region with the putative promoter and 1 kb of 5′-untranslated region (generously provided by Eiji Ikeda et al., Keio University, Tokyo, Japan). These constructs have a series of deletion constructs in the region from 80 bp up to 3.2 kb upstream of the translation start site of the VEGF gene and subcloned upstream of the luciferase gene in the promoterless luciferase reporter vector pGL2-basic vector (Promega, Madison, WI, U.S.A.). As a control plasmid, we used renilla luciferase pRL-SV40 vector (Toyo Inc, Tokyo, Japan). Plasmids were transfected into BREC by LipofectAMINE reagent (Life Technologies, Gaithersburg, MD, U.S.A.). BREC were seeded in 35-mm-diameter culture dishes (Iwaki) and incubated until they became subconfluent. A total of 1.5 μg test plasmid and 0.05 μg control plasmid was mixed with LipofectAMINE and added to the cells. After 5 h incubation, the mixture was replaced by normal growth medium and incubated for an additional 20 h. The cells were serum-deprived for 24 h and then stimulated by hypoxia or normoxia for 18 h. Cell extracts were then prepared by Lysis Buffer (Toyo Inc), and luciferase and renilla luciferase activity were measured by Luminoscan (Labsystems, Helsinki, Finland) with a Luciferase Dual Assay System (Toyo Inc). To standardize the transfection efficiency, luciferase activity was divided by renilla luciferase activity, and the degree of induction by hypoxia for each test plasmid was determined as the ratio of standardized luciferase activity in hypoxia-conditioned cells to that in normoxia-treated cells.
Nuclear extract preparation
Cells were washed with ice-cold phosphate-buffered saline, scraped from the plate with a rubber spatula, and suspended in buffer A (in mM: HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid) (pH 7.9) 10, KCl 10, EDTA 0.1, EGTA (ethylene glycol-bis (β-aminoethyl ether)-N, N, N′, N′-tetraacetic acid) 0.1, dithiothreitol 1, and phenylmethylsulfonyl fluoride 0.5). The cells were allowed to swell on ice for 15 min; then Nonidet P-40 was added to a final concentration of 0.5%. After centrifugation, the nuclear pellet was washed once in buffer A, then resuspended in ice-cold buffer C (in mM: HEPES (pH 7.9) 20, EDTA 1, EGTA 1, phenylmethylsulfonyl fluoride 1, dithiothreitol 1, NaCl 0.4 M, 1 μg ml−1 pepstatin, and 1 mg ml−1 leupeptin) and again allowed to swell on ice for 30 min. The nuclear extract was clarified by centrifugation at 12,000×g for 5 min, and the supernatant was frozen at −80°C.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts (4 μg of protein) were incubated in a 20-μl reaction mixture containing in mM: HEPES (pH 7.9) 10, glycerol 4%, dithiothreitol 1, EDTA 1, bovine serum albumin 0.1 μg, 1 μg each of poly (dI-dC) and 5× and 50× cold oligonucleotides as described, with modifications (Forsythe et al., 1996). The consensus oligonucleotides for Sp1 (5′-ATTCGATCGGGGCGGGGCGAGC-3′), AP1 (5′-CGCTTGATGAGTCAGCCGGAA-3′) and AP2 (5′-GATCGAACTGACCGCCCGCGGCCCGT-3′) were purchased from Promega (Madison, WI, U.S.A.). The oligonucleotide probe for the HIF-1 binding element in the VEGF gene (5′-TGCATACGTGGGCTCCAACAG-3′) was from Takara (Tokyo, Japan) (Forsythe et al., 1996). The hypoxia responsive element in the VEGF promoter was previously described by Tischer et al. (1991). The 32P-labelled oligonucleotide (2 μl) was added to the reaction mixture and incubated for 30 min at 25°C. Then 2 μl of 10× agarose loading buffer dye (50% glycerol, 0.25% bromophenol blue, 0.25% xylene cyanol) was added, and the samples were electrophoresed on a 4% polyacrylamide gel (polyacrylamide/bisacrylamide ratio, 80 : 1) at 4°C with TGE in mM: Tris base 25, glycine 190, EDTA (pH 8.5) 1. The gel was subsequently analysed by autoradiography.
Statistical analysis
Determinations were performed in triplicate, and experiments were performed at least three times. Results are expressed as mean±standard error unless otherwise indicated. For multiple treatment groups, a factorial ANOVA followed by Dunnett's least significant difference test was performed. Statistical significance was accepted at P<0.05.
Results
Bucillamine inhibits hypoxia-induced increase of VEGF mRNA and protein in BREC and BAEC
Northern blot analysis demonstrated that bucillamine at 10 and 100 μM inhibited the hypoxia-induced increase of VEGF mRNA levels (2.20±0.04 times compared to normoxia, P<0.0001) by 29.6% (P<0.01) and 54.5% (P<0.001), respectively (Figure 1). Bucillamine also inhibited the basal level of VEGF mRNA by 26.6% at a concentration of 100 μM (P<0.01). Analysis using culture supernatant revealed that bucillamine significantly inhibited the hypoxia-induced increase of VEGF secretion by 29.0% (P<0.01) at 100 μM after 24 h (Figure 2A) and that the compound at 10 and 100 μM inhibited VEGF secretion by 21.5% (P<0.01) or 34.5% (P<0.001), respectively, after 48 h (Figure 2B). Monosulfhydryl compounds, N-acetylcysteine and D-penicillamine, did not affect the hypoxia-induced VEGF mRNA expression and VEGF protein induction at a concentration of 100 and 1000 μM (data not shown). In BAEC, a type of vascular endothelial cells derived from large vessels, bucillamine at 100 μM inhibited the observed hypoxia-induced VEGF induction by 32.3% (P<0.05) (data not shown). No cytotoxic effect of bucillamine was observed at a concentration up to 100 μM on both types of vascular endothelial cells (data not shown). The following experiments were performed only in BREC.
Figure 1.
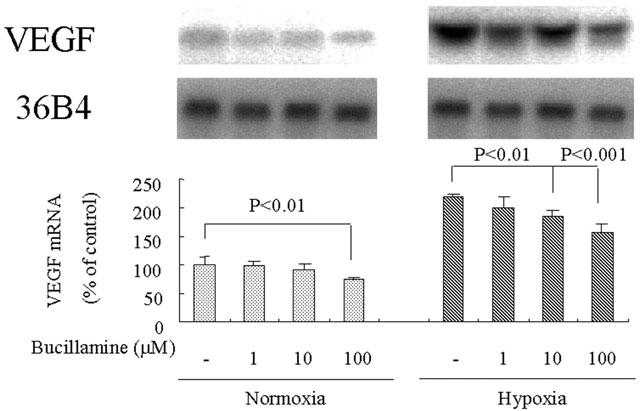
Effect of bucillamine on hypoxia-induced VEGF mRNA expression in bovine retinal endothelial cells. Northern blot analysis of total RNA (20 μg) isolated at 24 h after stimulation by hypoxic or normoxic conditions with or without bucillamine at the indicated concentrations. Representative Northern blots and control 36B4 (top) and quantitation of multiple experiments after normalization to the control signal are shown (bottom).
Figure 2.
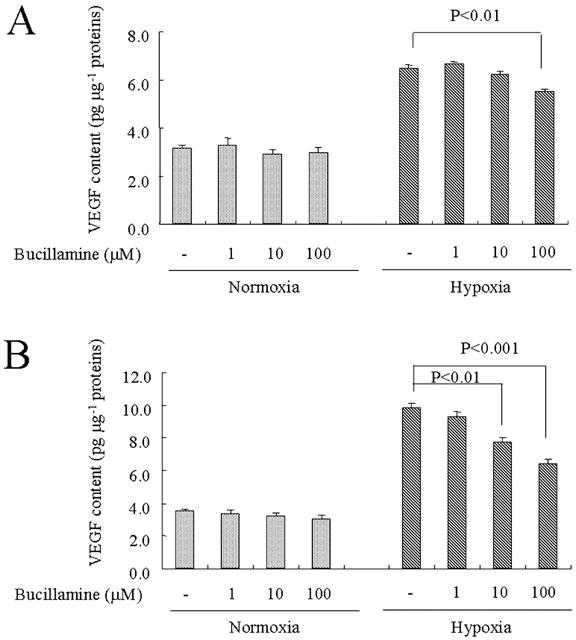
Effect of bucillamine on hypoxia-induced VEGF protein secretion in the culture supernatant of endothelial cells. The culture supernatant was obtained from bovine retinal endothelial cells after treatment of bucillamine. ELISA of media conditioned at 24 (A) or 48 h (B) after stimulation by hypoxic or normoxic conditions with or without bucillamine at the indicated concentrations.
Bucillamine does not affect VEGF mRNA stability in BREC
To evaluate the effects of bucillamine on VEGF mRNA stability, we measured the half-lives of VEGF mRNA in BREC cultured under normoxic and under hypoxic conditions, in both the presence and absence of bucillamine. Half-life values for VEGF mRNA were as follows: under normoxia without bucillamine, 2.71 h; under normoxia with bucillamine, 2.74 h; under hypoxia without bucillamine, 4.28 h; under hypoxia with bucillamine, 3.88 h (Figure 2). We could not detect any significant differences between the half-lives of VEGF mRNA in BREC cultured with or without bucillamine.
Reporter gene analysis
As bucillamine had no effect on VEGF mRNA stability, we determined whether it instead suppresses hypoxia-induced transcription. Therefore, we tested individually six different reporter constructs containing 5′-flanking and 5′-untranslated sequences from the human VEGF gene (Figure 3) for hypoxic responsiveness in a transient transfection assay. They were: 131-bp fragment from ApaI site (−1169 nt; numbered from the transcription start site) downstream to the transcription start site (−1038) that contained the minimal DNA sequence required for the basal transcription of the human VEGF gene (Tischer et al., 1991); 185-bp ApaI–NheI fragment (−1169 nt to −984); a 1.1-kbp ApaI–NarI fragment (−1169 nt to −81); an 844-bp PstI–NheI fragment (−1828 nt to −984); a 1.7-kbp PstI–NarI fragment (−1828 nt to −81); a 941-bp BanI–NheI fragment (−1925 nt to −984). None of them elicited a hypoxic response because they did not contain a 100-bp fragment, hypoxia responsive element (HRE; −1882 to −1782). On the other hand, either a 3.2-kbp KpnI–NarI fragment (−3317 to −81), a 2.8-kbp SpeI–NarI fragment (−2848 to −81) or a 1.2-kbp SacI–NheI fragment (−2218 to −984) contained a HRE element and accordingly elicited a hypoxic response. Bucillamine inhibited the hypoxia-induced reporter activity in these three fragments, suggesting an interaction between HRE and bucillamine in eliciting its inhibitory effect on hypoxic induction of promoter activity. Bucillamine also had an inhibitory effect on promoter activity in the minimal sequence required for basal promoter activity and other all promoter sequences in the normoxic condition. These results suggest that this drug also has an inhibitory effect on basal promoter activity.
Figure 3.
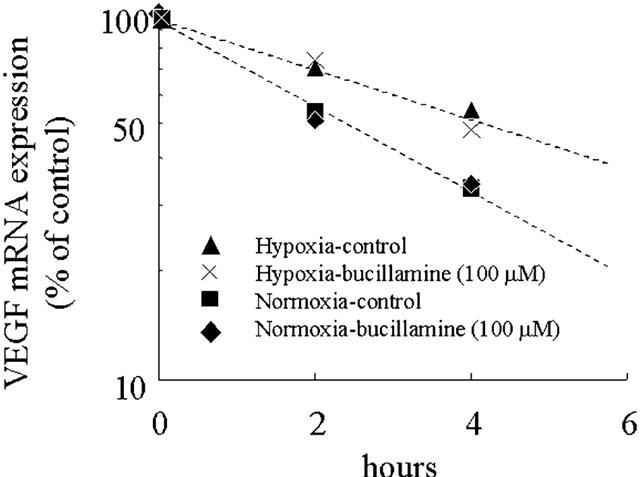
Effect of actinomycin D on VEGF mRNA expression in response to normoxic or hypoxic conditions in bovine retinal endothelial cells (BREC). BREC were stimulated by normoxic or hypoxic conditions for 24 h, and de novo mRNA transcription was inhibited by the addition of 4 μM actinomycin D. Total RNA was extracted at 2 and 4 h after administration of actinomycin D. Northern blot analysis was performed, and normalized to the control signal. Each plot is a percentage of the 0-h value in logarithmic scale.
Effect of bucillamine on hypoxia-induced activation of DNA-binding proteins HIF-1, Sp1, AP-1, and AP-2
Based on promoter analysis, HRE is thought to be involved in the inhibitory effect of bucillamine on hypoxic induction of VEGF and the minimal DNA sequence for the basal VEGF gene transcription, including Sp1. We investigated bucillamine's effect on these transcription factor-binding sites using EMSA (Figure 4). The binding activity of BREC nuclear protein to the HRE element of the VEGF gene increased under hypoxic conditions for 4 h, while binding activity to the Sp1, AP-1 and AP-2 elements was not affected by hypoxia. Bucillamine inhibited the hypoxia-stimulated binding activity to the HRE element in BREC nuclear protein. In normoxia, binding activity to HIF-1 and Sp1 were inhibited by bucillamine, while binding to AP-1 and AP-2 elements was not affected by bucillamine.
Figure 4.
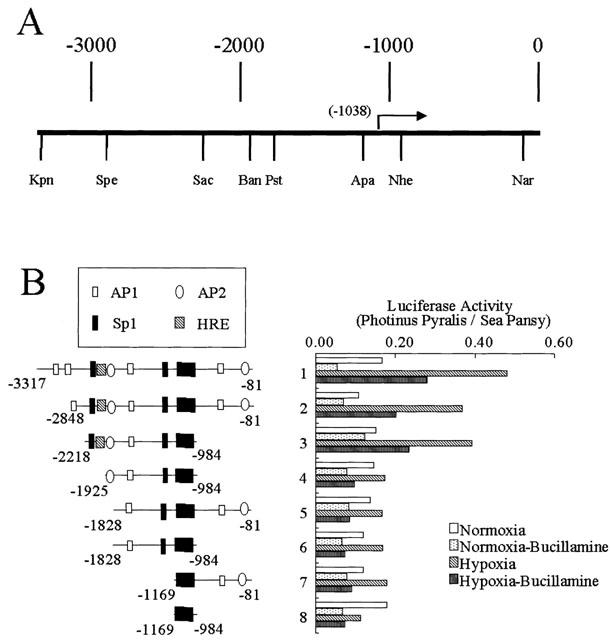
Deletion constructs containing regions from the human VEGF gene tested for responsiveness to hypoxia with and without bucillamine (100 μM). (A) Diagram of the 5′-flanking and 5′-untranslated regions of the human VEGF gene. Nucleotides are numbered from the translation start site, and the transcription start site is indicated with an arrow (−1038). (B) VEGF-luciferase deletion contracts and degrees of induction by hypoxia and suppression by bucillamine (100 μM). These constructs were transfected into BREC, and the degree of induction was determined. Restriction sites are: Kpn, KpnI; Spe, SpeI; Sac, SacI; Ban, BanI; Pst, PstI, Apa, ApaI; Nhe, NheI; Nar, NarI. Transcription factor binding sites are indicated as: hatched square, HRE; filled square, Sp1; open square, AP1; open circle, AP2.
Bucillamine inhibits hypoxia-induced HIF-1α gene expression
VEGF has been shown to be induced through the activation of an ischaemia-induced gene, HIF-1 (Ikeda et al., 1995), so we studied the inhibitory effect of bucillamine on hypoxia-induced gene regulation of HIF-1α. HIF-1α mRNA level of BREC cultured in hypoxia was significantly increased compared to BREC cultured in normoxia for 24 h (2.30±0.21 times, P<0.0001). Bucillamine at 10 and 100 μM inhibited the increased hypoxia-induced mRNA levels by 50.0% (P<0.001) and 73.1% (P<0.001), respectively (Figure 5). Bucillamine at 1 and 10 μM did not affect physiologic expression of HIF-1α mRNA, but did inhibit its expression at 100 μM (P<0.05). Figure 6
Figure 5.
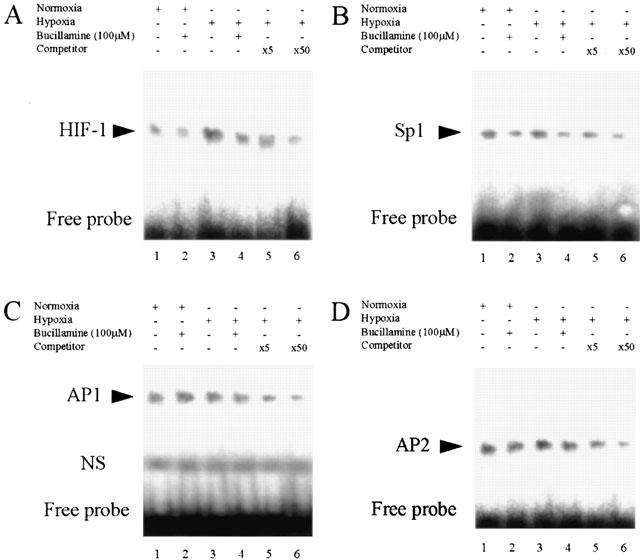
EMSA of BREC nuclear extracts for binding of HIF-1, Sp1, AP1 and AP2 to VEGF sequences. Effect of bucillamine (100 μM) on DNA-binding activity recognized by HIF-1 (A), Sp1 (B), AP1 (C) or AP2 (D) consensus oligonucleotide. Nuclear protein from normoxic conditioned BREC (lanes 1 and 2); nuclear protein from hypoxic conditioned BREC (lanes 3 to 6). Oligonucleotide (1 μg) was 32P-labelled and incubated with 4 μg aliquots of nuclear extract from BREC. BREC were treated by bucillamine (100 μM) for normoxic and hypoxic incubation (lanes 2 and 4). For competition assays (lanes 5 and 6), 5 or 50 ng of unlabelled oligonucleotide was included in the binding reaction mixture as indicated: ×5 or ×50, respectively.
Figure 6.
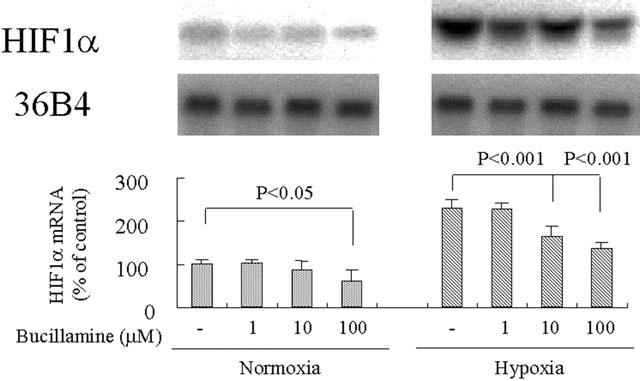
Effect of bucillamine on hypoxia-induced HIF-1α mRNA expression in BREC. Northern blot analysis of total RNA (20 μg) isolated at 24 h after stimulation by hypoxic or normoxic conditions with and without bucillamine at the indicated concentrations. Representative Northern blots and control 36B4 (top) and quantitation of multiple experiments after normalization to the control signal are shown (bottom).
Discussion
In this study, we showed that bucillamine inhibits VEGF mRNA and protein expression in retinal microcapillary endothelial cells. Other sulfhydryl drugs, N-acetylcysteine and D-penicillamine, did not affect hypoxia-induced VEGF production in BREC at concentrations up to 1000 μM whereas bucillamine's inhibitory effect was observed at concentrations from 10 μM. This observation suggests a specificity of the bucillamine's effect on VEGF induction by hypoxia. Tsuji et al. (2000) reported a similar observation that bucillamine inhibited IL-1-induced VEGF production in human synovial stromal cells, while other sulfhydryl compounds, N-acetylcysteine, D-penicillamine, tiopronin and bucillamine-like disulfhydryl compound, scarcely affected VEGF production.
Bucillamine at 10 μM showed significant inhibitory effect both on gene expression and protein secretion of VEGF while 1000 μM is the toxic margin for both types of the vascular endothelial cells. Although there seems to be two logs between the margin of the pharmacological and toxic effects, the margin is not so wide for these cell types and care should be taken for this limit of the compound.
Ikeda et al. (1995), Forsythe et al. (1996), and Goldberg & Schneider (1994) have previously presented evidence that the hypoxia-induced increase of VEGF gene expression is due to both transcriptional activation and increased mRNA stability. We have now demonstrated the effect of bucillamine on these mechanisms in retinal microcapillary endothelial cells. We measured half-lives of VEGF mRNA in BREC under normoxic and hypoxic conditions, and found that the half-life of VEGF mRNA after 24 h of hypoxia treatment was prolonged (about 2.0 fold compared to normoxic conditions). However, bucillamine did not affect VEGF mRNA stability under either normoxia or hypoxia.
Therefore, we investigated the effect of bucillamine on the transcriptional molecular mechanisms in BREC, and tested its effect on the 5′-flanking region of the human VEGF gene by transient transfection with a series of deletion constructs. The full-length gene contains hypoxia responsive elements (−2023 to −1977) in a 293-bp SacI–BanI fragment (−2218 to −1926) in the 5′-flanking region (Ikeda et al., 1995; Tischer et al., 1991), and contains six GC box sequences (−2138 to −2134, −1277 to −1272, −1133 to −1128, −1123 to −1118, −1112 to −1107 and −1096 to −1091), which are potential binding sites for transcription factor Sp1; it also contains four potential AP1 binding sites (−2930 to −2924, −2265 to −2258, −1528 to −1522 and −620 to −614) and two potential AP2 binding sites (−1875 to −1868 and −135 to −128) (Tischer et al., 1991). Since the deletion of HRE affects the promoter activity in response to hypoxia, this element, which is known to bind HIF-1, is responsible for the hypoxia-dependent activation of transcription (Wang et al., 1995).
We demonstrated that bucillamine suppresses the activation of the hypoxia-induced promoter constructs that contain the HRE element. This suggests that bucillamine's inhibitory effect on the hypoxia-dependent transcription is associated with an HRE-mediating mechanism. Furthermore, EMSA analysis revealed that bucillamine suppresses binding to the HRE element in BREC nuclear protein. HIF expression itself has been shown to be upregulated in response to hypoxia (Zhong et al., 1998). Accordingly, we determined the effect of bucillamine on HIF expression. Northern blot analysis revealed that bucillamine not only decreased mRNA levels of HIF under normoxic conditions but also suppressed its induction by hypoxia. These data indicate that bucillamine suppresses hypoxic induction of VEGF by inhibition of HIF induction and binding to the HRE element in BREC.
Regarding spontaneous expression of VEGF, bucillamine at high concentration also decreased the VEGF mRNA level and VEGF protein secretion into culture media. Since mRNA stability was not altered by the drug under normoxic conditions, its effect on promoter activity was tested. A 131-bp fragment from the ApaI site (−1169; numbered from the transcription start site) to the transcription start site (−1038) contained the minimal DNA sequence required for basal transcription of the human VEGF gene, which contains the Sp1 binding site (Tischer et al., 1991; Ikeda et al., 1995; Finkenzeller et al., 1997). Under normoxic conditions, bucillamine suppressed all construct-dependent promoter activity, including the minimal sequence construct for basal transcriptional activity. This suggests that bucillamine has an inhibitory effect on minimal sequence-dependent transcriptional activity. To investigate transcription factors related to the bucillamine's effect, EMSA assay was performed. In normoxia, binding activity to HRE element of VEGF promoter, Sp1 binding site, AP1 and AP2 binding sites was observed. Bucillamine suppressed binding activity to HRE and Sp1 binding site. In contrast, we did not observe any effect on binding of AP1 and AP2. These data suggest that bucillamine suppresses spontaneous VEGF expression by inhibiting the binding activity of HIF and Sp1 under normoxic conditions.
Taken together, these results suggest that bucillamine inhibits hypoxic induction of VEGF not only by inhibition of binding activity of HIF-1, but also by inhibiting expression of the human HIF-1α gene itself. In addition, this study has revealed the effect of bucillamine on physiologic expression of VEGF. It suppressed the binding activity of HIF-1 and Sp1 in normoxia, and thereby suppressed spontaneous transcription and expression of VEGF.
Acknowledgments
The authors thank Dr Eiji Ikeda (Keio University, Tokyo, Japan), Dr Werner Risau and Dr Judith Abraham (Scios Inc., Sunnyvale, CA, U.S.A.) for the series of plasmid constructs, that contained the 5′-flanking region and 5′-untranslated region of the VEGF gene. The authors also thank Dr Peter S. Reinach for helpful discussion.
Abbreviations
- bFGF
basic fibroblast growth factor
- BAEC
bovine aortic endothelial cell
- BREC
bovine retinal microcapillary endothelial cell
- BSA
bovine serum albumin
- DMEM
Dulbecco's modified Eagle's medium
- ELISA
enzyme-linked immunosorbent assay
- EMSA
electrophoretic mobility shift assay
- HIF
hypoxia inducible factor
- HRE
hypoxia responsive element
- IL
interleukin
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PDGF
platelet derived growth factor
- PDHS
plasma derived horse serum
- RA
rheumatoid arthritis
- SDS
sodium dodecylsulphate
- SSC
sodium citrate-sodium chloride
- TGF
transforming growth factor
- TNF-α
tumour necrosis factor-alpha
- VEGF
vascular endothelial growth factor
References
- ADAMIS A.P., SHIMA D.T., TOLENTINO M.J., GRAGOUDAS E.S., FERRARA N., FOLKMAN J., D'AMORE P.A., MILLER J.W. Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization in a nonhuman primate. Arch. Ophthalmol. 1996;114:66–71. doi: 10.1001/archopht.1996.01100130062010. [DOI] [PubMed] [Google Scholar]
- AIELLO L.P., AVERY R.L., ARRIGG P.G., KEYT B.A., JAMPEL H.D., SHAH S.T., PASQUALE L.R., THIEME H., IWAMOTO M.A., PARK J.E., NGUYEN H.V., AIELLO L.M., FERRARA N., KING G.L. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- AIELLO L.P., PIERCE E.A., FOLEY E.D., TAKAGI H., CHEN H., RIDDLE L., FERRARA N., KING G.L., SMITH L.E. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc. Natl. Acad. Sci. U.S.A. 1995;92:10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AMIN R.H., FRANK R.N., KENNEDY A., ELIOTT D., PUKLIN J.E., ABRAMS G.W. Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 1997;38:36–47. [PubMed] [Google Scholar]
- BERSE B., BROWN L.F., VAN DE WATER L., DVORAK H.F., SENGER D.R. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol. Biol. Cell. 1992;3:211–220. doi: 10.1091/mbc.3.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOMCZYNSKI P., SACCHI N. Single-step method of RNA isolation by acid guanidinium thiocyanate-PhOH-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- FINKENZELLER G., SPARACIO A., TECHNAU A., MARME D., SIEMEISTER G. Sp1 recognition sites in the proximal promoter of the human vascular endothelial growth factor gene are essential for platelet-derived growth factor-induced gene expression. Oncogene. 1997;15:669–676. doi: 10.1038/sj.onc.1201219. [DOI] [PubMed] [Google Scholar]
- FORSYTHE J.A., JIANG B., IYER N.V., AGANI F., LEUNG S.W., KOOS R.D., SEMENZA G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLDBERG M.A., SCHNEIDER T.J. Similarities between the oxygen-sensing mechanisms regulating the expression of vascular endothelial growth factor and erythropoietin. J. Biol. Chem. 1994;269:4355–4359. [PubMed] [Google Scholar]
- HARA S., KOBAYASHI C., IMURA N. Molecular cloning of cDNAs encoding hypoxia-inducible factor (HIF)-1α and -2α of bovine arterial endothelial cells. Biochim. Biophys. Acta. 1999;1445:237–243. doi: 10.1016/s0167-4781(99)00048-2. [DOI] [PubMed] [Google Scholar]
- IKEDA E., ACHEN M.G., BREIER G., RISAU W. Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J. Biol. Chem. 1995;270:19761–19766. doi: 10.1074/jbc.270.34.19761. [DOI] [PubMed] [Google Scholar]
- KING G.L., GOODMAN S., BUZNEY S., MOSES A., KAHN R.C. Receptors and growth-promoting effects of insulin and insulin-like growth factors on cells from bovine retinal capillaries and aorta. J. Clin. Invest. 1985;75:1028–1036. doi: 10.1172/JCI111764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KING G.L., KUNISAKI M., NISHIO Y., INOGUCHI T., SHIBA T., XIA P. Biochemical and molecular mechanisms in the development of diabetic vascular complications. Diabetes. 1996;45 Suppl. 3:S105–S108. doi: 10.2337/diab.45.3.s105. [DOI] [PubMed] [Google Scholar]
- KLIFFEN M., SHARMA H.S., MOOY C.M., KERKVLIET S., DE JONG P.T. Increased expression of angiogenic growth factors in age-related maculopathy. Br. J. Ophthalmol. 1997;81:154–162. doi: 10.1136/bjo.81.2.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOYAMA S., TAKAGI H., OTANI A., SUZUMA K., NISHIMURA K., HONDA Y. Tranilast inhibits protein kinase C-dependent signalling pathway linked to angiogenic activities and gene expression of retinal microcapillary endothelial cells. Br. J. Ophthalmol. 1999;127:537–545. doi: 10.1038/sj.bjp.0702564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEUNG D.W., CACHIANES G., KUANG W.J., GOEDDEL D.V., FERRARA N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- LIANG P., PARDEE A.B. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- LOPEZ P.F., SIPPY B.D., LAMBERT H.M., THACH A.B., HINTON D.R. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest. Ophthalmol. Vis. Sci. 1996;37:855–868. [PubMed] [Google Scholar]
- LUTTY G.A., MCLEOD D.S., MERGES C., DIGGS A., PLOUET J. Localization of vascular endothelial growth factor in human retina and choroid. Arch. Ophthalmol. 1996;114:971–977. doi: 10.1001/archopht.1996.01100140179011. [DOI] [PubMed] [Google Scholar]
- MATSUNO H., SUGIYAMA E., MURAGUCHI A., NEZUKA T., KUBO T., MATSUURA K., TSUJI H. Pharmacological effects of SA96 (bucillamine) and its metabolites as immunomodulating drugs – the disulfide structure of SA96 metabolites plays a critical role in the pharmacological action of the drug. Int. J. Immunopharmacol. 1998;20:295–304. doi: 10.1016/s0192-0561(98)00012-5. [DOI] [PubMed] [Google Scholar]
- ROBINSON G.S., PIERCE E.A., ROOK S.L., FOLEY E., WEBB R., SMITH L.E. Oligodeoxynucleotides inhibit retinal neovascularization in a murine model of proliferative retinopathy. Proc. Natl. Acad. Sci. U.S.A. 1996;93:4851–4856. doi: 10.1073/pnas.93.10.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHWEIKI D., ITIN A., SOFFER D., KESHET E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- TAKAGI H., KING G.L., AIELLO L.P. Identification and characterization of vascular endothelial growth factor receptor (Flt) in bovine retinal pericytes. Diabetes. 1996;45:1016–1023. doi: 10.2337/diab.45.8.1016. [DOI] [PubMed] [Google Scholar]
- TISCHER E., MITCHELL R., HARTMAN T., SILVA M., GOSPODAROWICZ D., FIDDES J.C., ABRAHAM J.A. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 1991;266:11947–11954. [PubMed] [Google Scholar]
- TSUJI F., MATSUOKA H., AONO H., TAKAI M., HORIUCHI M., NISHIMURA K., MITA S. Effects of sulfhydryl compounds on interleukin-1-induced vascular endothelial growth factor production in human synovial stromal cells. Biol. Pharm. Bull. 2000;23:663–665. doi: 10.1248/bpb.23.663. [DOI] [PubMed] [Google Scholar]
- TSUJI F., MIYAKE H., AONO Y., KAWASHIMA Y., MITA S. Effects of bucillamine and N-acetyl-L cysteine on cytokine production and collagen-induced arthritis (CIA) Clin. Exp. Immunol. 1999;115:26–31. doi: 10.1046/j.1365-2249.1999.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG G.L., JIANG B.H., SEMENZA G.L. Effect of altered redox states on expression and DNA-binding activity of hypoxia-inducible factor 1. Biochem. Biophys. Res. Commun. 1995;212:550–556. doi: 10.1006/bbrc.1995.2005. [DOI] [PubMed] [Google Scholar]
- ZHONG H., AGANI F., BACCALA A.A., LAUGHNER E., RIOSECO-CAMACHO N., ISAACS W.B., SIMONS J.W., SEMENZA G.L. Increased expression of hypoxia inducible factor-1a in rat and human prostate cancer. Cancer Res. 1998;58:5280–5284. [PubMed] [Google Scholar]