

Abstract
It is well-established that inhibitors of cyclo-oxygenase (COX) and hence of prostaglandin (PG) biosynthesis reverse inflammatory hyperalgesia and oedema in both human and animal models of inflammatory pain.
Paw oedema and hyperalgesia in rats were induced by injecting carrageenan (250 μg paw−1) into a hindpaw. Both inflammatory responses were followed for 24 h after the injection, measuring hyperalgesia by decreased pain threshold in the paws and oedema by plethysmography.
Three selective inhibitors of cyclo-oxygenase-2 (COX-2), celecoxib, rofecoxib and SC 236, given systemically in a range of doses, before the inflammatory stimulus, abolished carrageenan-induced hyperalgesia with little reduction of oedema. These inhibitors also induced hypoalgesia, increasing nociceptive thresholds in the inflamed paw above normal, non-inflamed levels. This hypoalgesia was lost at the higher doses of the selective inhibitors, although hyperalgesia was still prevented.
In paws injected with saline only, celecoxib, given at the dose inducing the maximum hypoalgesia after carrageenan, did not alter the nociceptive thresholds.
Two non-selective inhibitors of COX-2, indomethacin and piroxicam, abolished hyperalgesia and reduced oedema but did not induce hypoalgesia.
Celecoxib given locally into the paw also abolished inflammatory hyperalgesia and induced hypoalgesia without reducing oedema.
We conclude that hypoalgesia is expressed only over a critical range of COX-2 inhibition and that concomitant inhibition of COX-1 prevents expression of hypoalgesia, although hyperalgesia is still prevented.
Our results suggest a novel anti-nociceptive pathway mediating hypoalgesia, involving COX-2 selectively and having a clear peripheral component. This peripheral component can be further explored for therapeutic purposes.
Keywords: Hypoalgesia, cyclo-oxygenases, COX-1 and COX-2 inhibitors, hyperalgesia, paw oedema
Introduction
Prostaglandins (PGs) are well established as mediators of several components of the inflammatory response. Particularly, the oedema resulting from increased microvascular permeability is a consequence of the vasodilator effect of PGs potentiating the microvascular effects of other mediators such as bradykinin, substance P and histamine (Williams & Morley, 1973; Williams & Peck, 1977). These mediators also induce pain in inflammatory sites and this component is also potentiated by PGs (Ferreira, 1972). Although PGs are not direct algesic agents as are bradykinin, substance P and histamine, they nevertheless induce a state of hyperalgesia in which previously non-painful stimuli are now perceived as painful in both animal models and in human subjects (Ferreira, 1972; 1990). Logically, therefore, inhibitors of PG biosynthesis should reduce PG-induced hyperalgesia to basal levels of pain perception, reflecting the algesic action of the directly acting agents, but not reducing pain perception beyond the normal threshold.
The observed anti-oedema and analgesic effects of the non-steroidal anti-inflammatory drugs (NSAIDs) were attributed over 30 years ago to inhibition of the biosynthesis of PGs catalysed by cyclo-oxygenase (COX) (Vane, 1971). There are now known to be two isoforms of COX, COX-1 and COX-2, with the latter being strongly induced in inflammatory sites (Bakhle & Botting, 1996; Vane et al., 1998). Most of the NSAIDs already used clinically (aspirin, indomethacin, ibuprofen, diclofenac, etc.) are non-selective inhibitors of COX, affecting both isoforms to a variable extent (Vane et al., 1998; Warner et al., 1999). The major side effect of the NSAIDs, gastric or intestinal ulceration, has been attributed to inhibition of COX-1 and this attribution has been the major impetus for the introduction into clinical practice of two selective inhibitors of COX-2, celecoxib and rofecoxib, as ‘NSAIDs without ulcers' (Bombardier, 2002; Hawkey, 1999). These selective COX-2 inhibitors exhibit NSAID-like effects in human disease and, in animal models of inflammation, they decrease oedema and hyperalgesia (Chan et al., 1999; Smith et al., 1998).
We were therefore surprised to observe, in our model of inflammation induced by carrageenan in rat paws, not only the expected loss of hyperalgesia but additionally, a sub-normal pain perception after treatment with celecoxib (Francischi et al., 2002). This was expressed as a raised threshold for nociception and this state is referred to as ‘hypoalgesia'. We have extended these initial observations using both selective COX-2 inhibitors and non-selective NSAIDs, seeking to define and analyse the hypoalgesic effect. We have also measured oedema over the same time course to explore the relation between vascular events and nociception, in terms of COX-inhibition. From our results we would conclude that the hypoalgesic effects of COX inhibitors reflected COX-2 inhibition selectively and that hypoalgesia requires a lesser degree of inhibition of COX-2 than does the reduction in oedema.
Methods
Animals
Male Holtzman rats from the Bioresources Centre of Federal University of Minas Gerais (body weight, 150–200 g) were used throughout this study. The animals were left to adapt for 24 h under controlled experimental conditions (23–26°C, light/dark cycles of 12/12 h with lights on at 0700 h, food and water ad libitum). Ethical guidelines of the International Association for the Study of Pain in conscious animals were followed (Zimmerman, 1983).
Inflammatory reaction to carrageenan
Lambda–carrageenan was injected (100–500 μg paw−1 in 0.1 ml of physiological saline) in the foot pad of hind paws, to induce hyperalgesia and oedema. An intermediate dose of 250 μg paw−1 was chosen as the standard inflammatory stimulus in most of our studies. Contralateral paws received the same volume (0.1 ml) of saline, the vehicle for carrageenan.
Measurement of the state of algesia
Assessment of algesia consisted of measurement of the threshold stimulus for reaction (escape or paw withdrawal) using a weight (maximum limit of 500 g) applied to the pads of hind paws by an experimenter using an Ugo Basile apparatus; this is essentially the method of Randall & Selitto (1957). The threshold for pain sensation was measured before (time zero) and 1, 2, 3, 4 and 24 h after the intraplantar injection of carrageenan. Results are presented as the difference in threshold between the test (carrageenan-injected) paw and the contralateral, control, (saline-injected) paw. These measurements were made without knowledge of any pre-treatments.
Oedema measurements
The volume (in ml) of the hind paws from control and treated animals were measured with a hydroplethysmometer (Ugo Basile 1750) at the same time-points used for hyperalgesia measurements, i.e., time zero and 1, 2, 3, 4 and 24 h after stimulus injection. Results are presented as the difference in volume between the test paw and the control paw for each animal at the time shown.
Pre-treatment with COX inhibitors
Cyclo-oxygenase inhibitors were administered either subcutaneously (s.c.), or intraplantarly (i.pl.). The capsule content or compressed tablet from commercial preparations of celecoxib, rofecoxib, and piroxicam was weighed and crushed into a fine suspension with physiological saline (NaCl, 0.9% w v−1) based on the weight of active substance quoted per tablet or capsule. These suspensions were then diluted further with saline to give appropriate amounts of the active substance. All suspensions were made immediately before use and were not stored. Indomethacin was initially dissolved in Tris buffer (0.1 M, pH=8.0); SC 236 was dissolved in ethanol and then diluted in 5% (v v−1) Tween 80 in water, as described in Guo et al. (2001). Further dilutions were made with saline. All pre-treatments were given 30 min before the injection of carrageenan. Control animals were treated with their respective vehicles at the same times. The standard volume used for s.c. injection was 1 ml kg−1 with the exception of piroxicam, which was given at 2 ml kg−1. For i.pl. injections, 0.1 ml was given to each paw.
Materials
The following commercial preparations of COX inhibitors were used: celecoxib (capsules, 100 or 200 mg, Celebra, Searle & Co, Cáguas, Porto Rico), rofecoxib (compressed tablets, 25 mg, Vioxx, MSD, Campinas, Brazil), and piroxicam (capsules, 20 mg, Feldene, Pfizer, Guarulhos, Brazil). We thank Searle (U.S.A.) for a generous gift of SC236. Indomethacin, lambda-carrageenan and Trizma base were purchased from Sigma (St Louis, MO, U.S.A.). Sodium chloride (‘pro-analysis') was from Reagen (Rio de Janeiro Quimibras Indústrias Químicas S.A., RJ, Brazil).
Statistics
Results are presented either as mean (±s.e.mean) values from n animals (n⩾4) in each treatment group, at each time point (Figures 1, 2 and 6) or as area under the curve (AUC) values. The latter was calculated as the sum, over 4 h, of the individual values for each time point for each animal, using the trapezoidal rule (GWBASIC software). The mean (±s.e.mean) AUC was derived from the AUCs for n animals in each treatment group. Mean values from the treated groups were compared with the mean values from the group receiving the vehicle only as treatment, using Students t-test or Anova t-test (for multiple comparison, when necessary) to determine significant differences, accepting a difference between means when P<0.05. For the assays of nociceptive threshold, negative AUC values (less than zero) represented hyperalgesia and positive values represented hypoalgesia. Thus abolition of hyperalgesia would be shown by a mean AUC value not significantly different from zero and hypoalgesia by a mean value significantly greater than zero.
Figure 1.

Hyperalgesia and oedema induced by carrageenan in rat paws. The effects of three doses of carrageenan measured hourly for 4 h and then again at 24 h, following intraplantar injection are shown separately for hyperalgesia (a) and oedema (b), although both variables were measured in each animal. Hyperalgesia is represented by a decrease in the response threshold below the control value and is expressed as the weight (in g) at which the response was elicited. Oedema is shown as an increase in paw volume above control value. The means (±s.e.mean) values from groups of four rats for each condition are shown. For all doses of carrageenan, the nociceptive threshold was significantly less than that for control (saline injection) for 1–3 h and, for the two higher doses also at 4 h. Paw volume was significantly increased for all doses of carrageenan for all the times shown. *Significantly different from control; P<0.05.
Figure 2.
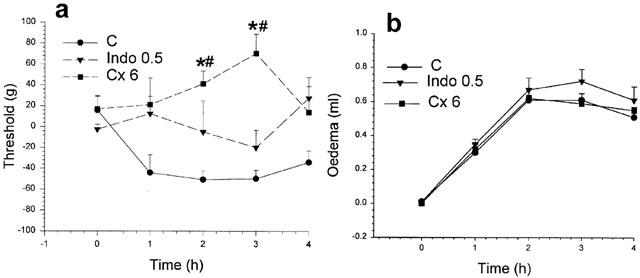
Effects of indomethacin and celecoxib on rat paw hyperalgesia and oedema. In (a) the hyperalgesia induced by carrageenan was less at each time after pre-treatment with indomethacin (0.5 mg kg−1) but these changes were not significant. Celecoxib (6 mg kg−1) pre-treatment prevented the drop in threshold (hyperalgesia) induced by carrageenan and increased the nociceptive threshold above normal, particularly at 2 and 3 h after carrageenan. In (b) the oedema induced by carrageenan was not reduced by either pre-treatment. *Significantly different from corresponding values for carrageenan; #significantly greater than zero time value; P<0.05.
Figure 6.
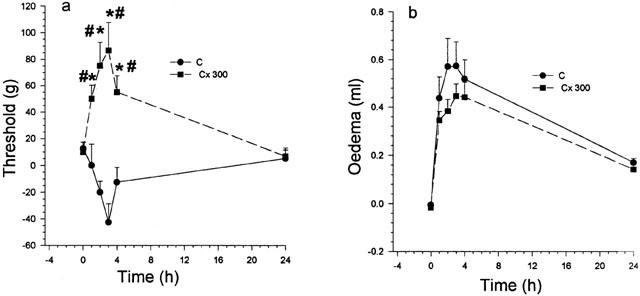
Intraplantar injection of celecoxib. In these experiments, celecoxib (300 μg) was given locally, i.e., into the footpad, 30 min before the carrageenan. Hyperalgesia was completely prevented and the nociceptive threshold raised above normal for at least 4 h, returning to baseline by 24 h. In (b) no moderation of the oedema was observed throughout the 24 h. *Significantly different from corresponding value for carrageenan; #significantly greater than zero time value; P<0.05.
Results
Hyperalgesia and oedema development induced by carrageenan in rat paws
Intraplantar administration of carrageenan (100–500 μg per paw) dose-dependently reduced the weight threshold required for the animals' response over 4 h, clearly characterising the development and resolution of hyperalgesia (Figure 1a). This hyperalgesia, expressed as a decrease in the threshold weight, reached a maximum value between 2–3 h, was clearly fading by 4 h (Figure 1a) and had returned to basal levels at 24 h following carrageenan injection. After carrageenan injection, paw volume also increased over about the same time as hyperalgesia (Figure 1b). This oedema reached a maximum value at 3 h, persisted beyond 4 h but resolved by 24 h. The hyperalgesia and oedema induced by the intermediate dose of 250 μg of carrageenan were adequate to show either increase or decrease after treatment and this level was therefore chosen as the standard inflammatory stimulus in our subsequent experiments.
Modification of these standard inflammatory responses by inhibition of PG biosynthesis by pre-treatment with a single dose of a non-selective (indomethacin; 0.5 mg kg−1) or of a selective COX-2 inhibitor (celecoxib; 6 mg kg−1; Penning et al., 1997) is shown in Figure 2. In Figure 2a, the effect of indomethacin was to reduce the mean threshold values at each time to between those induced by carrageenan alone and those induced by saline alone, ie, no hyperalgesia. By contrast, treatment with celecoxib not only prevented the hyperalgesia but also allowed the animal to withstand a greater than normal weight before withdrawing the paw under test. This increase of the weight threshold above normal levels, very clear at 2 h and at 3 h after carrageenan injection, we have called a state of ‘hypoalgesia'. The anti-hyperalgesic effect of celecoxib was, like that of a low indomethacin dose, not accompanied by reductions in oedema (Figure 2b). These two types of anti-hyperalgesic responses were examined further as described below, for both selective and non-selective inhibitors of COX-2.
Effect of selective COX-2 inhibitors on hyperalgesia and oedema development induced by carrageenan
The effects of pre-treatment with two selective, clinically used, COX-2 inhibitors celecoxib and rofecoxib and one experimental selective inhibitor, SC 236 (Penning et al., 1997) on hyperalgesia and oedema induced by carrageenan were studied over a range of concentrations. The results from these experiments are presented, for brevity, as the mean ‘area under the curve' (AUC) for each treatment group. The AUC was calculated for each animal by summing the values measured at each time point and the mean calculated from these individual AUC values, as described in the methods.
In Figure 3a, celecoxib (3–12 mg kg−1) dose-dependently prevented hyperalgesia and then displayed a hypoalgesic effect, very marked at 12 mg kg−1, which was lost at the highest dose, 30 mg kg−1. These large changes in the hyperalgesic response to carrageenan were not accompanied by any reduction in oedema (Figure 3b), until the highest dose level, when a modest reduction in oedema was recorded over the 4 h period. In the absence of the inflammatory stimulus, i.e., in paws injected with saline only, there was no hyperalgesia and no oedema. Pre-treatment with celecoxib at the dose giving the most marked hypoalgesia (12 mg kg−1) did not induce any changes in either nociceptive threshold or paw volume, under these conditions (Table 1).
Figure 3.
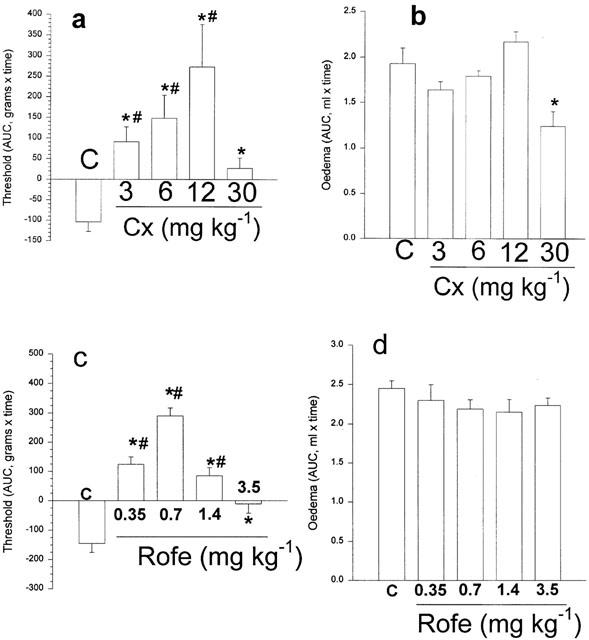
Effects of the selective COX-2 inhibitors, celecoxib or rofecoxib on rat paw hyperalgesia and oedema. Here the results are shown as the mean area under the curve for the time course over 4 h for each group of animals (see methods). In (a) the hyperalgesia induced by carrageenan (bar marked C) was totally prevented by all levels of celecoxib. Hypoalgesia, measured as an increase of nociceptive threshold above the normal was shown for all except the highest dose of celecoxib. In (b), oedema was unchanged, except after the highest dose of celecoxib when it was reduced by about 30%. In (c) pre-treatment with rofecoxib reversed the hyperalgesia and induced hypoalgesia. The loss of hypoalgesia with increasing doses of rofecoxib is clearly shown here with the highest dose preventing hyperalgesia but showing no hypoalgesia. Note that none of the doses of this selective COX-2 inhibitor reduced paw oedema (d). *Significantly different from carrageenan only; #significantly different from zero; P<0.05.
Table 1.
Lack of effect of celecoxib (12 mg kg−1; s.c.) on nociceptive threshold and paw oedema responses in rat paws injected with saline

Rofecoxib, over a 10-fold range of doses, showed very similar effects on the responses to carrageenan (Figure 3c,d), with abolition of hyperalgesia throughout the dose range, marked hypoalgesia at an intermediate dose (1.4 mg kg−1) and loss of hypoalgesia at the highest dose tested. One clear difference between the two selective inhibitors was that oedema was not affected by any of the doses of rofecoxib used here. Because these two clinically used selective COX-2 inhibitors were used in our experiments as suspensions of the tablet or capsule preparations, it was possible that these effects might be related to the excipients involved. We therefore extended our analysis to the experimental selective inhibitor SC236 (Penning et al., 1997) which was obtained as the pure substance. With this inhibitor, given s.c., over a 2 fold dose range, essentially the same results were obtained. Thus SC236 showed decreased hyperalgesia at the lower dose, and then at the higher dose, hypoalgesia (Figure 4a). There were no reductions in oedema at either of the doses of SC236 used (Figure 4b).
Figure 4.

Effects of the selective COX-2 inhibitor, SC-236, on rat paw hyperalgesia and oedema. In (a) the hyperalgesia was totally prevented by the lower dose (1 mg kg−1) and a marked hypoalgesia induced by the higher dose of SC-236 (12 mg kg−1). In (b) neither dose reduced oedema. *Significantly different from carrageenan only; #significantly different from zero; P<0.05.
Effects of non-selective inhibitors of COX-2 on carrageenan hyperalgesia and oedema
The non-selective inhibitors of COX-2 also inhibit the constitutive isoform COX-1, usually more potently than they inhibit COX-2 (Warner et al., 1999; Vane et al., 1998). Indomethacin, as already shown (Figure 2), decreased hyperalgesia without reduction of oedema. Piroxicam, at two doses (3 and 20 mg kg−1), abolished hyperalgesia and markedly reduced oedema, but did not show any signs of hypoalgesia (Figure 5a,b).
Figure 5.
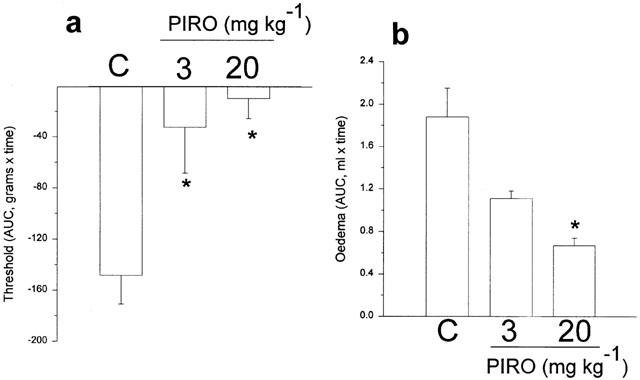
Effects of the non-selective COX inhibitor, piroxicam, on rat paw hyperalgesia and oedema. Both doses of piroxicam (3 or 20 mg kg−1) prevented the hyperalgesia but did not cause hypoalgesia and here oedema was also reduced. *Significantly different from carrageenan; P<0.05.
Effects of celecoxib given locally (intraplantar, i.pl.) on carrageenan-induced hyperalgesia and oedema
Pre-treatment with the selective COX-2 inhibitor, celecoxib (300 μg), given into the paw 30 min before the carrageenan, abolished the subsequent hyperalgesia and induced a clear hypoalgesia, as shown in Figure 6a. However, this local treatment with celecoxib did not change the level of oedema (Figure 6b).
Discussion
Our experiments reported here have confirmed and extended our earlier observations, in inflamed raw paws, of hypoalgesia induced by celecoxib, a selective inhibitor of COX-2 (Francischi et al., 2002). We have used different selective COX-2 inhibitors over a range of doses and showed that this hypoalgesia was induced at doses that did not reduce oedema, another inflammatory response. Two non-selective inhibitors of COX (indomethacin and piroxicam) were able to reduce paw oedema but were only able to prevent hyperalgesia without inducing hypoalgesia.
Some analysis and definitions of the terms, hypo- and hyperalgesia, is needed as a preliminary to discussing our results. For many years, the analgesic effects of COX inhibitors (NSAIDs) have been explained by postulating PGs as algesic potentiators, whose action was to increase the algesic effects of the directly acting algesic agonists, such as bradykinin (Ferreira, 1990). In this paradigm, PGs have no overt or direct, algesic action. The sensitization of peripheral sensory nerve receptors by PGs to direct chemical and mechanical stimulation was demonstrable in experimental conditions and in human studies (Collier & Schneider, 1972; Ferreira, 1972). This potentiation of nociception was termed hyperalgesia. A defining feature of the analgesic action of the NSAIDs is that they are effective only in nociception associated with PG production, i.e., only in inflammatory pain. The response to painful stimuli in non-inflamed sites is not altered by NSAIDs and this contrasts crucially with other types of analgesics such as the local anaesthetics or opioids with very different modes of action. An essential corollary of the status of PGs as nociceptive sensitisers is that it is not logically possible for inhibition of PG production to alter nociceptive thresholds to levels above the normal state, i.e., to act as local anaesthetics. In the present context, we have referred to a state of higher than normal nociceptive thresholds as hypoalgesia. Accordingly, inhibitors of PG biosynthesis (COX inhibitors) should only abolish hyperalgesia but not cause hypoalgesia.
From our results, the magnitude of the hypoalgesia, its demonstration with three selective COX-2 inhibitors and with two modes of administration, systemic or local, all establish hypoalgesia in this model as a real phenomenon. We sought in the subsequent experiments to define the hypoalgesia further and to explore mechanisms underlying this phenomenon. Some possible mechanisms can be readily eliminated.
Stress-induced analgesia is not likely to have contributed to our results as other COX inhibitors (indomethacin, piroxicam) used under identical conditions did not exhibit hypoalgesia. The possibility that the selective inhibitors were acting like local anaesthetics can be eliminated as the normal (non-inflamed) pathways of nociception were not altered by celecoxib since it did not induce hypoalgesia in paws injected with saline, following either local or systemic administration. In most of our experiments, suspensions of the commercial preparations of the selective COX-2 inhibitors were used so that the active substance was accompanied by a variety of excipients and these excipients may have altered the response of the model. We feel this is unlikely as we also used piroxicam from the commercial preparation without observing hypoalgesia. The different excipients used in the tablets, which are designed to be without pharmacological effect, would have to exert effects that separated the selective (celecoxib, rofecoxib) from the non-selective inhibitor (piroxicam). More convincingly, our experiments with the selective inhibitor SC236 and the non-selective inhibitor, indomethacin, used the pure substance and gave results essentially identical to those with the suspensions of the commercial preparations. Furthermore the dose-dependency of hypoalgesia strongly suggests that this effect was mediated by some biological activity shared by the three compounds, celecoxib, rofecoxib and SC 236.
The simplest explanation would be to attribute the hypoalgesia to selective inhibition of COX-2 in the inflamed paw. Support for this would be provided by the doses of the three selective inhibitors causing hypoalgesia. In our experiments there was about a 10 fold difference in dose between the two clinically used inhibitors, with 0.7 mg kg−1 of rofecoxib producing an increase of about 100 g above the normal nociceptive threshold, an effect reproduced by between 6 and 12 mg kg−1 of celecoxib. The clinically effective doses of these two COX-2 inhibitors and their potency in vitro against COX-2 are of this order (Chan et al., 1999). The non-selective COX inhibitors also inhibit the other isoform, COX-1, but usually with greater potency than COX-2. Piroxicam is a more potent inhibitor of COX-1, with a potency ratio of about 600 and indomethacin is less so with a potency ratio of about 60 (Warner et al., 1999; Chan et al., 1999; Vane et al., 1998). However, neither was able to induce hypoalgesia over a range of doses although, as expected, both decreased hyperalgesia markedly. It must also be remembered that although non-selective COX inhibitors such as piroxicam are potent inhibitors of COX-1, their anti-inflammatory effects (analgesia and oedema reduction) are nevertheless attributed to inhibition of COX-2. The crucial difference between selective COX-2 inhibitors and non-selective inhibitors, at anti-inflammatory doses, is that with the latter, COX-1 is also inhibited. This interpretation would suggest that, with the non-selective inhibitors, inhibition of COX-1, in some way, prevented the expression of the hypoalgesic effects of concomitant COX-2 inhibition. This apparent interaction between the isoforms contrasts with the results of Smith et al. (1998) who, in a similar model of inflammation, found that inhibition of COX-1 was not relevant to either loss of hyperalgesia or to reduction of oedema. However, Ballou et al. (2000) concluded that both isoforms were involved in PG mediated hyperalgesia in their models. These comparisons with previous work have to be moderated by important differences in the details such as animal lineage, nociceptive stimulus, time of assay, etc, of the experimental procedures. Nonetheless, our results have clearly shown an anti-hyperalgesic effect for both selective and non-selective inhibitors, but with the particular effect of hypoalgesia restricted to the selective inhibitors of COX-2.
A distinctive feature of the hypoalgesia was that it was exhibited at doses that did not bring about the other classical anti-inflammatory effect, decreased oedema. All three selective inhibitors caused both decreased hyperalgesia and then induced hypoalgesia, without a significant reduction in oedema. This lack of effect on oedema is probably related to the degree of inhibition of COX-2 as the highest dose of celecoxib did reduce oedema and all three selective inhibitors are well known to reduce oedema in this model (Penning et al., 1997; Chan et al., 1999). Further, the non-selective inhibitor, indomethacin, was also able to reduce hyperalgesia without reducing oedema, although it, too, is known to reduce oedema in this model. It may be that marked inhibition of COX-2 is needed to prevent the potentiation of oedema by PGs (Williams & Peck, 1977) but that a lesser inhibition is enough to prevent the sensitization of nociceptors. However this explanation would not account entirely for the hypoalgesia, which, as mentioned earlier, is not compatible with the present concepts of PG action, only as a sensitizer of sensory neurons.
Another characteristic of the hypoalgesic effect was its bell-shaped, dose-effect relationship, shown by all three selective inhibitors. With celecoxib, the lowest dose decreased hyperalgesia, intermediate doses produced hypoalgesia and at the highest dose there was loss of the hypoalgesia but still abolition of hyperalgesia. Similarly, three doses of rofecoxib exhibited hypoalgesia and the highest dose (3.5 mg kg−1) returning to normal nociceptive thresholds. From this observation it would appear that hypoalgesia is expressed only at a particular level of COX-2 inhibition and that level did not reduce oedema. Such a window of COX-2 inhibition could also explain why hypoalgesia has not been previously reported in the extensive studies on these three compounds. Because there would be no reduction of oedema, this degree of COX-2 inhibition, i.e. this dose of inhibitor, would be classified as ineffective, in anti-inflammatory terms. Therefore, higher doses would be used to modify oedema and, as we have shown, at these higher doses, hyperalgesia was still abolished (the expected anti-inflammatory effect) buthypoalgesia disappeared.
Although COX-2 is expressed constitutively in brain (Yamagata et al., 1993; Vane et al., 1998), it is the COX-2 in the spinal cord that is important for nociception and the analgesic actions of selective COX-2 inhibitors (Svensson & Yaksh, 2002; Samad et al., 2001; Vanegas & Schaible, 2001). There is evidence for the involvement of COX-1, also in the spinal cord, which would be particularly relevant to analgesia induced by non-selective inhibitors (Mazario et al., 2001; Ballou et al., 2000). Our results with local intraplantar injection of the selective COX-2 inhibitor, celecoxib, would nevertheless suggest a major peripheral site of action of COX-2 inhibitors in mediating the hypoalgesia we observed.
In summary, our results in a model of acute inflammation have disclosed a hypoalgesic effect of COX inhibition which appears to be independent of oedema reduction and correlated with selective inhibition of COX-2. We believe the hypoalgesic mechanism is mediated by COX-2 as it was not observed in the absence of inflammation and was invoked by three different, selective, inhibitors of COX-2. Its novelty lies in the hypoalgesia observed, the lack of a concomitant effect on oedema and the bell-shaped dose-response relation. These features were not shown by the non-selective inhibitors or in earlier work with the selective COX-2 inhibitors and appear to be incompatible with the present concepts of PGs acting merely as potentiators of normal nociceptive pathways and of the reversal of such potentiation by inhibition of COX. It must be emphasized that models of inflammatory hyperalgesia are many and various; our report seeks to establish hypoalgesia with selective inhibitors of COX-2 in one model as an incentive to others to look for the same phenomenon in their own models. Nevertheless, although our findings are still far from being fully elucidated, they raise important questions about the pathophysiological role of PGs and the COX isoforms in the mediation and perception of inflammatory pain and may necessitate new paradigms of PG action in nociception.
Acknowledgments
We thank Webster G.P. Reis, for his excellent technical assistance and Fapemig, CNPq and CAPES for their financial support.
Abbreviations
- AUC
area under the curve
- COX
cyclo-oxygenase
- i.pl.
intraplantar
- NSAID
non-steroid anti-inflammatory drug
- PG
prostaglandin
References
- BAKHLE Y.S., BOTTING R.M. Cyclooxygenase-2 and its regulation in inflammation. Mediat. Inflamm. 1996;5:305–323. doi: 10.1155/S0962935196000452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALLOU L.R., BOTTING R.M., GOORHA S., ZHANG J., VANE J.R. Nociception in cyclooxygenase isozyme-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10272–10276. doi: 10.1073/pnas.180319297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOMBARDIER C. An evidence-based evaluation of the gastrointestinal safety of coxibs. Am. J. Cardiol. 2002;89:3D–9D. doi: 10.1016/s0002-9149(02)02231-2. [DOI] [PubMed] [Google Scholar]
- CHAN C.C., BOYCE S., BRIDEAU C. Rofecoxib (Vioxx, MK-0966; 4-(4′-methylsulfonylphenyl)-3-phenyl-2(5H)-furanone: a potent and orally active cyclooxygenase inhibitor. Pharmacological and biochemical profiles. J. Pharm. Exp. Ther. 1999;290:551–560. [PubMed] [Google Scholar]
- COLLIER H.O., SCHNEIDER C. Nociceptive response to prostaglandins and analgesic actions of aspirin and morphine. Nature New Biology. 1972;236:141–143. doi: 10.1038/newbio236141a0. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H. Prostaglandins, Aspirin-like drugs and Analgesia. Nature New Biology. 1972;240:200–203. doi: 10.1038/newbio240200a0. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H.A classification of peripheral analgesics based upon their mode of action Migraine: a spectrum of ideas 1990New York: Oxford University Press; 59–72.In: Sander, M., Collins, G.M. (eds) [Google Scholar]
- FRANCISCHI J.N., CHAVES C.T., LIMA A.S., BAKHLE Y.S. A new anti-nociceptive pathway mediated by cyclooxygenase-2 in rat paw inflammation induced by carrageenan. Br. J. Pharmacol. 2002;135:159P. [Google Scholar]
- GUO X., LIU E.S., KO J.K., WONG B.C., YE T., LAM S., CHO C. Protective role of cyclooxygenase inhibitors in the adverse action of passive smoking on the initiation of experimental colitis in rats. Eur. J. Pharmacol. 2001;411:193–203. doi: 10.1016/s0014-2999(00)00914-6. [DOI] [PubMed] [Google Scholar]
- HAWKEY C.J. COX-2 Inhibitors. Lancet. 1999;353:307–314. doi: 10.1016/s0140-6736(98)12154-2. [DOI] [PubMed] [Google Scholar]
- MAZARIO J., GAITAN G., HERRERO J.F. Cyclooxygenase-1 vs. cyclooxygenase-2 inhibitors in the induction of antinociception in rodent withdrawal reflexes. Neuropharmacology. 2001;40:937–946. doi: 10.1016/s0028-3908(01)00020-x. [DOI] [PubMed] [Google Scholar]
- PENNING T.D., TALLEY J.J., BERTENSHAW S.R., CARTER J.S., COLLINS P.W., DOCTER S., GRANETO M.J., LEE L.F., MALECHA J.W., MIYASHIRO J.M., ROGERS R.S., ROGIER D.J., YU S.S., ANDERSON G.D., BURTON E.G., COGBURN J.N., GREGORY S.A., KOBOLDT C.M., PERKINS W.E., SEIBERT K., VEENHUISEN A.W., ZHANG Y.Y., ISAKSON P. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-Methylphenyl-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib) J. Med. Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- RANDALL L.D., SELITTO J.J. A method for measurement of analgesic activity on inflamed tissues. Arch. Int. Pharmacodyn. 1957;113:233–249. [PubMed] [Google Scholar]
- SAMAD T.A., MOORE K.A., SAPIRSTEIN A., BILLET S., ALLCHORNE A., POOLE S., BONVENTRE J.V., WOOLF C.J. Interleukin-1beta mediated induction of COX-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- SMITH J.M., ZHANG Y., KOBOLDT C.M., MUHAMMAD J., ZWEIFEL B.S., SHAFFER A.F., TALLEY J.J., MASFERRER J., SEIBERT K., ISAKSON P. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SVENSSON C.I., YAKSH T.L. The spinal phospholipase-cyclooxygenase-prostanoid cascade in nociceptive processing. Annu. Rev. Pharmacol. Toxicol. 2002;42:553–583. doi: 10.1146/annurev.pharmtox.42.092401.143905. [DOI] [PubMed] [Google Scholar]
- VANE J.R. Inhibition of prostaglandin synthesis as a mechanism of action for the aspirin-like drugs. Nature. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- VANE J.R., BAKHLE Y.S., BOTTING R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- VANEGAS H., SCHAIBLE H.G. Prostaglandins and cyclooxygenases in the spinal cord. Prog. Neurobiol. 2001;64:327–363. doi: 10.1016/s0301-0082(00)00063-0. [DOI] [PubMed] [Google Scholar]
- WARNER T.D., GIULIANO F., VOJNOVIC I., BUKASA A., MITCHELL J.A., VANE J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7563–7568. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLIAMS T.J., MORLEY J. Prostaglandins as potentiators of increased vascular permeability in inflammation. Nature. 1973;246:215–217. doi: 10.1038/246215a0. [DOI] [PubMed] [Google Scholar]
- WILLIAMS T.J., PECK M.J. Role of prostaglandin-mediated vasodilatation in inflammation. Nature. 1977;270:530–532. doi: 10.1038/270530a0. [DOI] [PubMed] [Google Scholar]
- YAMAGATA K., ANDREASSON K.I., KAUFMAN W.E., BARNES C.A., WORLEY P.F. Expression of a mitogen-inducible cyclooxygenase in brain neurons; regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- ZIMMERMAN M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]