

Abstract
The effects of pharmacological preconditioning with U50488H (U50), a selective κ-opioid receptor agonist, on Ca2+ homeostasis in rat ventricular myocytes subjected for 9 min to metabolic inhibition (MI) and anoxia (A), consequences of ischaemia, were studied and compared with those of preconditioning with brief periods of MI/A.
Precondition with 30 μM of U50 for three cycles of 1 min each cycle separated by 3 min of recovery (UP) significantly increased the percentage of non-blue cells following MI/A. The effect of UP is the same as that of preconditioning with an inhibitor of glycolysis and an oxygen scavenger for three 1-min cycles separated by three-minute recovery (MI/AP). The results indicate that like MI/AP, UP also confers cardioprotection.
MI/A increased intracellular Ca2+ ([Ca2+]i) and reduced the amplitude of caffeine-induced [Ca2+]i transients, an indication of Ca2+ content in the sarcoplasmic reticulum (SR). MI/A also reduced the electrically-induced [Ca2+]i transient, that indicates Ca2+-release during excitation-contraction coupling, and Ca2+ sparks in unstimulated myocytes, that indicates spontaneous Ca2+-release from SR. It also prolonged the decline of the electrically-induced [Ca2+]i transient and slowed down the recovery of the electrically-induced [Ca2+]i transient after administration of caffeine. In addition, MI/A prolonged the decline of caffeine induced [Ca2+]i transient, an indication of Na+–Ca2+ exchange activity, and UP prevented it. So UP, that confers cardioprotection, prevented the changes induced by MI/A. With the exception of Ca2+-spark, which was not studied, the effects of MI/AP are the same as those of UP.
It is concluded that pharmacological preconditioning with U50, that confers immediate cardioprotection, prevents changes of Ca2+ homeostasis altered by MI/A in the rat heart. This may be responsible, at least partly, for the cardioprotective action.
The study also provided evidence that MI/A causes mobilization of Ca2+ from SR to cytoplasm causing Ca2+-overload which may be due to reduced Ca2+-uptake by SR. MI/A also reduces spontaneous and electrically induced Ca2+ release from SR.
Keywords: Anoxia, metabolic inhibition, Ca2+ homeostasis, ventricular myocyte, Ca2+ spark, preconditioning, κ-opioid receptor, U50488H, sarcoplasmic reticulum
Introduction
Since the demonstration of cardioprotection of ischaemic preconditioning in 1986 (Murry et al., 1986), subsequent studies have shown that membrane receptors to humoral substances such as adenosine (Liu et al., 1991), acetylcholine (Richard et al., 1994), δ-opioid (Schulz et al., 1998) and noradrenaline (Wilson et al., 1996) mediate the cardioprotection of preconditioning. It has also been shown that one of the consequencies of ischaemia such as metabolic inhibition (MI) (Wu et al., 1999) or hypoxia (Gray et al., 1997) also confers similar cardioprotection. Recently we have found that pretreatment with a κ-opioid receptor agonist, U50488H (U50), a selective κ-opioid receptor agonist, confers similar cardioprotection as, while blockade of the κ-opioid receptor with a selective κ-opioid receptor antagonist attenuates significantly the cardioprotection of, ischaemic preconditioning (IP) (Wang et al., 2001) or metabolic inhibition preconditioning (MIP) (Wu et al., 1999), indicating that κ-opioid receptor mediates cardioprotection of IP and MIP. In addition to causing myocardial injury, ischaemia increases intracellular Ca2+ ([Ca2+]i) (Steenbergen et al., 1990, 1993a, 1993b; Bersohn et al., 1997; Ylitalo et al., 2000) while IP, that confers cardioprotection, attenuates the elevation in [Ca2+]i (Steenbergen et al., 1993b; Ylitalo et al., 2000; Smith et al., 1996), suggesting that ischaemia causes Ca2+ overload, while IP prevents the elevation of [Ca2+]i. The restoration of [Ca2+]i may be responsible at least partly for cardioprotection of preconditioning. We hypothesized that pretreatment or pharmacological preconditioning with a κ-opioid receptor agonist, that confers cardioprotection, prevents changes of Ca2+ homeostasis altered by ischaemic or anoxic or MI stress the same way as physiological preconditioning with ischaemia or anoxia or MI.
It has been shown that following myocardial ischaemia, Ca2+-uptake by the sarcoplasmic reticulum (SR), and the activity and phosphorylation of the SR Ca2+ ATPase are significantly reduced (Osada et al., 2000), indicating a reduction in Ca2+-uptake by its intracellular Ca2+ store. Since Ca2+ uptake by SR accounts for over 80% of the increased Ca2+ after excitation-contraction (E–C) coupling in the heart (Janezewski & Lakatta, 1993), the reduction in Ca2+-uptake by SR may be mainly responsible for the increased [Ca2+]i. Prior exposure to a brief period or periods of ischaemia, that confers cardioprotection against subsequent and more severe ischaemic insults, reverses the inhibitory effects of ischaemia on SR Ca2+-uptake, and the activity and phosphorylation of the SR Ca2+ ATPase (Osada et al., 2000). The effects of ischaemia with and without preconditioning on Ca2+ content in SR and Ca2+ release from SR in unstimulated state and during E–C coupling are, however, not known. Although there is evidence that the extrusion of Ca2+ by Na+–Ca2+ exchange activity is reduced upon ischaemia (Steenbergen et al., 1993a, 1993b; Bersohn et al., 1997), the effect of preconditioning on Na+–Ca2+ exchange activity is not known either.
The purpose of the present study was 3 fold. First we determined the immediate cardioprotective action of pharmacological preconditioning with U50 (UP) and compare with the effect of physiological preconditioning with MI and anoxia (MI/AP). Secondly, we tested the hypothesis that prior activation of κ-opioid receptor would prevent changes of Ca2+ homeostasis altered by a MI and anoxia (MI/A) stress in a similar way as MI/AP. Thirdly we determined the effects of UP or MI/AP on Ca2+ homeostasis with particular emphasis on Ca2+ content in SR and release of Ca2+ from SR in unstimulated state and during E–C coupling, and Na+–Ca2+ exchange activities in myocytes subjected to MI/A. We employed an isolated ventricular myocytes preparation subjected to a glucose-free Krebs–Ringer solution containing an inhibitor of glycolysis (deoxyglucose) and an oxygen scavenger (sodium dithionite), which causes MI (Macianskiene et al., 2001; Overend et al., 2001; Sugiyama et al., 2001) and anoxia (McKenna et al., 1991; Otter & Austin, 2000; Tateishi et al., 2001). We preconditioned the myocytes with U50 or with the glycolysis inhibitor and the oxygen scavenger according to Seki & Macleod (1995) and Wu et al. (1999). We measured the [Ca2+]i and its transients induced by caffeine or electrical stimulation with spectrofluorometry. The caffeine induced [Ca2+]i transient is an indication of Ca2+ content in SR as caffeine depletes Ca2+ of SR (Janezewski & Lakatta, 1993), while the electrically-induced [Ca2+]i transient represents the influx of Ca2+ across the sarcolemma and release of Ca2+ from SR following electrical stimulation (Janezewski & Lakatta, 1993). We also measured the decay of the electrically-induced [Ca2+]i transients and the recovery of the electrically induced [Ca2+]i transients after the washout of caffeine that indicate the uptake of Ca2+ back to SR from cytoplasm (Chossat et al., 2001; Trafford et al., 2001). In addition we measured the Ca2+ spark with laser scanning confocal microscopy in unstimulated ventricular myocytes, which indicates spontaneous Ca2+ release from SR. To gain an insight into the Na+–Ca2+ exchange activity, we analysed the decline of the caffeine-induced [Ca2+]i transient as an indicator of the activity (Terracciano & Macleod, 1994; Seki & Macleod, 1995; Sham et al., 1995). Results from the study showed that pharmacological preconditioning with U50 confers immediate cardioprotection and prevents changes in Ca2+ homeostasis altered by MI/A in a similar way as MI/AP does. Mobilization of Ca2+ from SR to cytoplasm, which causes Ca2+ overload following MI/A, is due to reductions in Ca2+-uptake by SR and Ca2+ extrusion via the Na+–Ca2+ exchange. Both the spontaneous and electrically stimulated Ca2+ release is also reduced following MI/A due to reduced Ca2+ content in SR.
Methods
Isolation of ventricular myocytes
The study was approved by the Committee on the Use of Live Animals in Teaching and Research of the University of Hong Kong. Cardiac ventricular myocytes were isolated from the hearts of adult male Sprague-Dawley rats (190–230 g), using a collagenase perfusion method described previously (Dong et al., 1993). After isolation, they were allowed to stabilize for at least 30 min before experiments.
Experimental protocols
Ventricular myocytes were divided into five groups as shown in Figure 1. In the vehicle-pretreatment control group (VP), ventricular myocytes were superfused with Krebs solution for 13 min and then with glucose-free Krebs solution for 9 min containing 10 mM 2-deoxy-D-glucose (2-DOG), an inhibitor of glycolysis (Macianskiene et al., 2001; Overend et al., 2001; Sugiyama et al., 2001), and 10 mM sodium dithionite (Na2S2O4), an oxygen scavenger (McKenna et al., 1991; Otter & Austin, 2000; Takeishi et al., 2001), which causes MI and anoxia, two consequences of ischaemia. We adopted the method from previous work (Do et al., 1995; Seki & Macleod, 1995; Cumming et al., 1996; Wu et al., 1999) with modification. For the study on Ca2+ spark, 1 mM Na2S2O4 was used as 10 mM Na2S2O4 completely suppressed spontaneous release of Ca2+. This was followed by superfusion with normal Krebs solution only for 10 min –reperfusion. There were two groups of ventricular myocytes subjected to preconditioning, one group with U50, and the other with 2-DOG+Na2S2O4. For both groups, cells were subjected to three cycles of 1 min, each cycle of superfusion either with U50 at 30 μM or with 2-DOG+Na2S2O4 both at 10 mM each in glucose-free Krebs solution, separated by 3 min of superfusion with normal Krebs solution. Then the ventricular myocytes were subjected to 10 mM 2-DOG and 10 mM Na2S2O4 in glucose-free Krebs solution for 9 min followed by reperfusion of 10 min as in the VP group. These two groups were named UP and MI/AP, respectively. In addition, there were two groups of cells subjected to UP or MI/AP in the presence of 5 μM of nor-binaltorphimine (nor-BNI), a selective antagonist of κ-opioid receptor (Portoghese et al., 1994; Blake et al., 1997). Nor-BNI was added 5 min before and during preconditioning. These two groups were NBI-UP and NBI-MI/AP, respectively.
Figure 1.
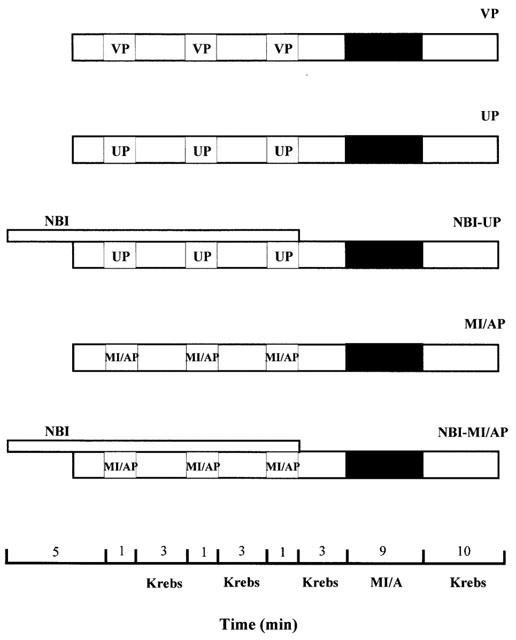
Experimental design. After isolation, ventricular myocytes were allowed to stabilize for 30 min before treatment. Myocytes were then subjected to either pharmacological preconditioning with 30 μM U50 (UP) or metabolic inhibition/anoxia preconditioning (MI/AP) with 10 mM Na2S2O4+10 mM 2-DOG in glucose-free Krebs for three 1-min cycles separated by three min superfusion with normal Krebs solution. A group of cells were incubated with saline in Krebs as vehicle-preconditioning control (VP). Another two groups of myocytes were subjected to UP or MI/AP in the presence of 5 μM nor-BNI, which was administered 5 min before and throughout the preconditioning period. These two groups were NBI-UP and NBI-MI/AP, respectively. Then the ventricular myocytes in all groups were subjected to MI/A with 10 mM Na2S2O4+10 mM 2-DOG in glucose-free Krebs solution for 9 min followed by superfusion with normal Krebs solution for 10 min–reperfusion.
Trypan blue exclusion for cell viability study
Trypan blue exclusion was used as an index of cell viability (Zhou et al., 1996; Hiebert & Ping, 1997). Ventricular myocytes were incubated with 0.4% trypan blue dye for 3 min. The unstained myocytes, called non-blue cells, are viable myocytes as they are able to exclude the dye. Cells were counted in a haemocytometer chamber during reperfusion after MI/A under a light microscope. The percentage ratio of non-blue cells in total cells was used as indication of cell viability.
Measurement of [Ca2+]i
A spectrofluorometric method with Fura-2/AM as the Ca2+ indicator was used for measurement of [Ca2+]i (Dong et al., 1993). Loading of cells with Fura-2/AM was performed as described previously (Dong et al., 1993). Ventricular myocytes were incubated with 4 μM Fura-2/AM for 30 min in a MEM solution containing 1.25 mM CaCl2. Fluorescent signals obtained at 340 nm (F340) and 380 nm (F380) excitation wavelengths were recorded and stored in a computer for data processing and analysis. The F340/F380 ratio was used to represent cytosolic [Ca2+]i in the ventricular myocyte.
Measurement of [Ca2+]i transients induced by caffeine and electrical stimulation
10 mM caffeine dissolved in normal Krebs solution containing 1.25 mM Ca2+ was applied directly to the ventricular myocyte as described previously (Sheng & Wong, 1996). To measure electrically induced [Ca2+]i transient, myocytes were subjected to 0.2 Hz electrical field stimulation with 15 ms pulses at 60 V as described previously (Sheng & Wong, 1996).
Since previous studies demonstrated that the slope in the relaxation phase of the electrically induced [Ca2+]i transients is one of the most direct physiological effect of Ca2+-uptake from cytoplasm to SR by SR Ca2+-ATPase (SERCA) (Giordano et al., 1997; He et al., 1997; Baker et al., 1998; Loukianov et al., 1998; Hajjar et al., 1999), we measured the decay of the electrically induced [Ca2+]i transient. The time constant of the monoexponential decline of the electrically induced [Ca2+]i transient, tau (τ), was determined according to the equation: [Ca](t)=[Ca] peak·e−t/τ+[Ca] baseline, where t represents the continuous variable of time. The fits were analysed using GraphPad software. Only fits with a γ2>0.95 were used.
To further determine the uptake of Ca2+ in SR, the electrically-induced [Ca2+]i transient was determined in the ventricular myocyte for at least 50 s during caffeine washout and the time constant of recovery of the amplitude of the electrically-induced [Ca2+]i transient was used as indication of uptake/relocation of Ca2+ by SR (Chossat et al., 2001; Trafford et al., 2001).
To determine the Na+–Ca2+ exchange activity, the decay of the caffeine induced [Ca2+]i transient was analysed according to Terracciano & Macleod (1994) and Sham et al. (1995). Since caffeine keeps the Ryanodine receptor open, the decay of its transient represents only the Ca2+ extrusion via the sarcolamma, about 80% of which take place via the Na+–Ca2+ exchange.
Measurement of Ca2+ spark
Ca2+ sparks were visualized using the membrane permeable Ca2+-sensitive fluorescent dye, Fluo-3 acetoxymethyl ester (Fluo-3/AM). Ventricular myocytes were loaded with 5–10 μM Fluo-3/AM (dissolved in DMSO with 20% pluronic acid) in normal Tyrode solution for 15 min at room temperature (22°C). Cells were then washed thoroughly with 2 μM Ca2+ Tyrode solution to remove extracellular Fluo-3/AM, and rested for 15–30 min in a cell chamber to allow for complete de-esterification of cytosolic dye. Confocal images were acquired using a Zeiss LSM-510 inverted confocal microscope (Carl Zeiss Inc., Germany) with a Zeiss Plan-Neofluor 40× oil immersion objective (NA=1.3). The confocal pinhole was set to render a spatial resolution of 0.4 μm in the X–Y axis, and 0.9 μm in the Z-axis. Fluo-3/AM was excited by the 488 nm light of an argon laser, and fluorescence was measured at >505 nm. Images were acquired in the linescan mode (digital zoom rendering a 38 μm scan line), scanning at 0.075 μm pixel−1, 512 pixel line−1 at 2 ms intervals for 512 lines image−1 from the same cells once every minute before and during metabolic inhibition. Photobleaching and laser damage to the cells were minimized by attenuating the laser to ∼1% of its maximum power (25 mW) with an acousto-optical tunable filter. All experiments were performed at room temperature.
Spontaneous Ca2+ sparks were detected by an automated detection algorithm custom-written using the Interactive Data Language (IDL; Research Systems Inc., Boulder, CO, U.S.A.) to minimize subjective selection bias by identifying Ca2+ sparks on the basis of their statistical deviation from background noise, similar to that described previously by Cheng et al. (1993) and Remillard et al. (2002).
In brief, the linescan images were first taken in the presence of a cell. Then the background fluorescence was estimated by taking a linescan image in the absence of a cell. The background fluorescence was then subtracted from the fluorescence images. The images of Ca2+ sparks were further quantified as F/Fo, where F was the fluorescence intensity of a spark, and Fo was the resting fluorescence intensity when no spark was detected. The mean (m) and variance (σ2) of the confocal image were estimated. Ca2+ sparks were then identified based on local fluorescence intensity greater than m+3✓σ2. The amplitude of sparks being selected was expressed as ÄF/F0; in some cases, F/F0 was calibrated to absolute [Ca2+] by a pseudo-ratio method, using the following equation: [Ca2+]i=(K·R)/[((K/[Ca2+]rest)+1)−R], where R is F/F0, KD of Fluo-3 is 400 nM, and resting Ca2+ ([Ca2+]rest) is assumed to be 100 nM. The temporal and spatial dimensions of Ca2+ sparks were quantified as the full-duration-half-maximum (FDHM) and full-width-half-maximum (FWHM), respectively. The spark frequency of each cell was defined as number of sparks detected per second in the scan-line.
Drugs and chemicals
U50488H (trans–(±)-3, 4-dichloro-N-methyl-N-(2-[1-pyrroli-dinyl] cyclohexyl)-benzeneacetamide methanesulphonate salt), 2-deoxy-D-glucose, fura-2/AM, fluo-3/AM, type 1 collagenase, caffeine and trypan blue dye were purchased from Sigma, sodium dithionite and nor-BNI were from Merck and Tocris Cookson Ltd, respectively. All chemicals were dissolved in distilled water or Krebs solution, except fura-2/AM and fluo-3/AM, which were dissolved in dimethyl sulphoxide (DMSO), and DMSO with 20% pluronic acid, respectively, at a final DMSO concentration <0.1%, at which no effect can be observed (Wu et al., 1999). The concentrations of 2-DOG (Do et al., 1995; Cumming et al., 1996), U50 (Wu et al., 1999), nor-BNI (Wu et al., 1999), caffeine (Sheng & Wong, 1996; Chossat et al., 2001), trypan blue (Zhou et al., 1996; Hiebert & Ping, 1997) used in the present study was according to the previous studies. The concentration of sodium dithionite for [Ca2+]i transient study was according to Tateishi et al. (2001), Otter & Austin (2000) and McKenna et al. (1991); while that for Ca2+ spark was according to Cumming et al. (1996) and Do et al. (1995). The concentration for [Ca2+]i transient study was 10 times that of Ca2+ spark study simply because at only the higher concentration, the inhibition of the [Ca2+]i transients by the inhibitor were great enough for comparison with preconditioning.
Statistical analysis
Data were expressed as mean±s.e.mean. One-way ANOVA was used to determine the differences among the multiple groups. For analysis of drug effects, the non-parametric Kruskal–Wallis test was used. P<0.05 was considered statistically significant.
Results
Effects of UP or MI/AP on viability of ventricular myocytes subjected to MI/A
As shown in Figure 2, 69.4% of the total cells were non-blue in the Krebs control group, i.e. myocytes in this group were not subjected to any treatment. After MI/A for 9 min with 10 mM 2-DOG, an inhibitor of glycolysis and 10 mM Na2S2O4, an oxygen scavenger, only 28.0% of the cells were non-blue (Figure 2), which is consistent with our previous observation that MI with cyanide and 2-DOG causes cell death (Wu et al., 1999). In the myocytes preconditioned with a protocol of three one-minute exposures to 30 μM U50 (UP) or with 10 mM 2-DOG and 10 mM Na2S2O4 alternating with 3-min of recovery period (MI/AP), the percentages of non-blue cells following MI/A were 58 and 53%, respectively, which were significantly (P<0.001) greater than that of the VP group (Figure 2). The result indicates immediate cardioprotection of UP or MI/AP, which is consistent with the previous observation of cardioprotection of UP or IP in the isolated perfused rat heart (Wang et al., 2001).
Figure 2.
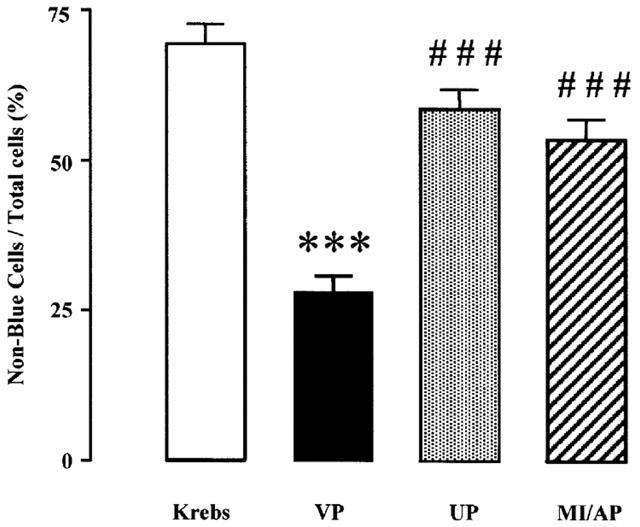
Effects of UP or MI/AP on viability in rat ventricular myocytes subjected to prolonged MI/A. The viability of the cells was determined with trypan blue exclusion method following prolonged MI/A. Myocytes were put in the glucose-free Krebs solution containing 10 mM Na2S2O4+10 mM 2-DOG for 9 min (prolonged MI/A), then washed by and kept in normal Krebs solution for determination of viability. Non-blue cells are live cells. Values, presented as non-blue cells/total cells, are mean±s.e.mean. n=7 experiments. In each experiment, about 200 ventricular myocytes from three rats were used. ***P<0.001 vs Krebs control; ###P<0.001 vs VP group.
Effects of UP or MI/AP on [Ca2+]i and its transients of ventricular myocytes subjected to MI/A
The resting [Ca2+]i concentration in rat ventricular cells superfused with a normal Krebs solution with 1.25 mM Ca2+ at room temperature was 179±32 nM. MI/A for 9 min caused a significant increase in [Ca2+]i by 22.5% (Figure 3A,B). UP or MI/AP significantly attenuated the elevation of [Ca2+]i by MI/A (Figure 3B). The observation indicates that preconditioning prevents at least partially the elevation in [Ca2+]i by MI/A.
Figure 3.
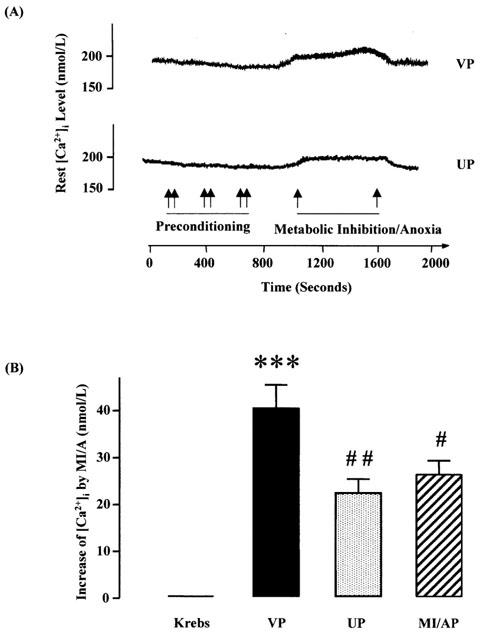
Effects of UP or MI/AP on [Ca2+]i in single ventricular myocytes subjected to prolonged MI/A. (A) Representative tracings showing the effects of MI/A and UP (B) Group results showing increases values in [Ca2+]i during prolonged MI/A using 10 mM Na2S2O4+10 mM 2-DOG in glucose-free Krebs solution for 9 min. Values are mean±s.e.mean. n=16 in VP and MI/AP groups, n=15 in UP group; and the value of Krebs control as zero increase, is an average level during the stable period of all cells. The myocytes were obtained from 7–9 rats in each group. ***P<0.001 vs Krebs control; #, ##P<0.05, 0.01 vs VP group, respectively.
The amplitude of the [Ca2+]i transient induced by electrical stimulation, an indication of the release of Ca2+ during E–C coupling (Janezewski & Lakkata, 1993), was significantly decreased during MI/A (Figure 4). Similarly the amplitude of the [Ca2+]i transient induced by caffeine, an indication of Ca2+ content in SR (Janezewski & Lakkata, 1993), was also significantly decreased following MI/A (Figure 5). These results are in agreement with the effects of MI or hypoxia as reported in the previous study (Seki & Macleod, 1995). Like the change in [Ca2+]i, the decreases in the [Ca2+]i transients were significantly attenuated by UP or MI/AP. The attenuating effects of UP and MI/AP were blocked by nor-BNI (Figures 4 and 5B), a selective κ-opioid receptor antagonist (Portoghese et al., 1994; Blake et al., 1997), which itself had no effect on the [Ca2+]i transients. The observations indicate that MI/A caused a reduction in Ca2+ content in SR, thus reducing the Ca2+ released during E–C coupling and that preconditioning restored the Ca2+ content in SR, thus restoring the electrically induced release of Ca2+.
Figure 4.
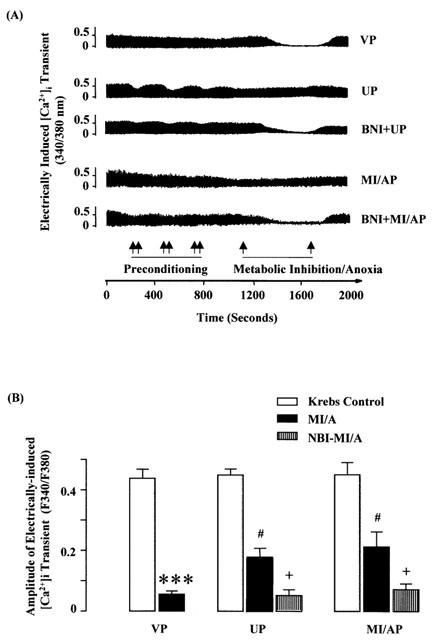
Effects of UP or MI/AP on the amplitude of electrically-induced [Ca2+]i transient in single ventricular myocytes subjected to severe MI/A with or without nor-BNI added in pretreatment. Ventricular myocytes were perfused with glucose-free Krebs solution containing 10 mM Na2S2O4+10 mM 2-DOG for 9 min (prolonged MI/A), and then reperfused with normal Krebs solution for 10 min. (A) Representative tracings: double arrows indicate periods of preconditioning. (B) Group results of the fluorescence signal values. Values are mean±s.e.mean. n=12 cells in VP group, 8 for MI/AP, 9 in NBI-MI/AP, 11 in UP and 9 in NBI-UP group. The myocytes were obtained from 6–8 rats for each group. ***P<0.001 vs Krebs control value in the same group; #P<0.05 vs corresponding value in VP group, +P<0.05 vs the corresponding value without nor-BNI within the group.
Figure 5.
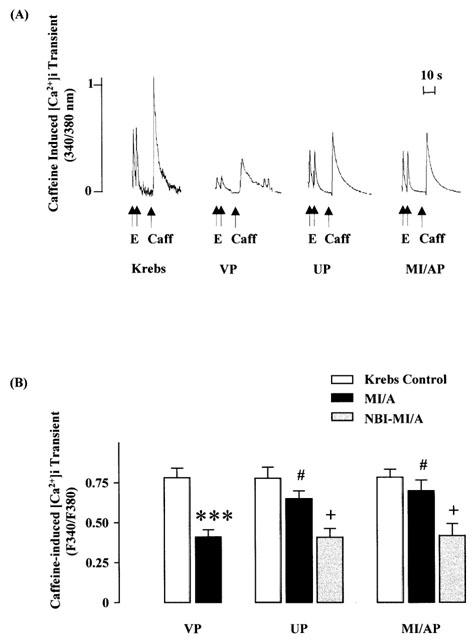
Effects of UP or MI/AP on the amplitude of caffeine-induced [Ca2+]i transient in single ventricular myocytes subjected to prolonged MI/A with or without the presence of nor-BNI. Severe MI/A was induced by 10 mM Na2S2O4+10 mM 2-DOG in glucose-free Krebs solution for 9 min. (A) Representative tracings. (B) Group results of the fluorescence signal values. Values are mean±s.e.mean. n=10 myocytes in VP group, and n=9 in MI/AP, n=11 in UP, n=6 cells in NBI-MI/AP and n=7 in NBI-UP groups. The cells were obtained from 6–7 rats for each group. E=electrical stimulation; Caff=caffeine. ***P<0.001 vs Krebs control value in the same group; #P<0.05 vs corresponding value in VP group, +P<0.05 vs the corresponding value without nor-BNI within the group.
Effects of UP or MI/AP on decay of the electrically induced [Ca2+]i transient and recovery of the electrically induced [Ca2+]i transient following caffeine treatment in ventricular myocytes subjected to MI/A
In order to determine the uptake of Ca2+ by SR, we first measured the decay of the electrically-induced [Ca2+]i transients. The rate of the decay in the electrically-induced [Ca2+]i transient of myocytes as indicated by the time constant of transient decay, tau (τ), according to Chossat et al. (2001) and Trafford et al. (2001), was 725±45 ms in the untreated control (Table 1), while the corresponding value after MI/A in the VP group was significantly prolonged by 22.4% (Table 1). In UP and MI/AP groups the τ values were restored back to the level of the untreated control, and were significantly lower than that of the VP group (Table 1).
Table 1.
Decay of electrically-induced [Ca2+]i transient

To further determine the uptake of Ca2+ by SR, the recovery of the electrically induced [Ca2+]i transient following caffeine was measured according to the previous workers (Chossat et al., 2001; Trafford et al., 2001). Immediately after administration of 10 mM of caffeine, the amplitude of the electrically induced [Ca2+]i transient was decreased markedly followed by gradual recovery (Figure 6A). The time constant of the recovery curve (Figure 6B), which indicates the rate of recovery, was determined as an indicator of SR Ca2+ recovery (Ullich et al., 2000; Hung et al., 2002). The time constant of the untreated control was 2.252±0.59 (n=8). MI/A caused a significant prolongation of the time constant to 3.934+0.28 (n=8).
Figure 6.
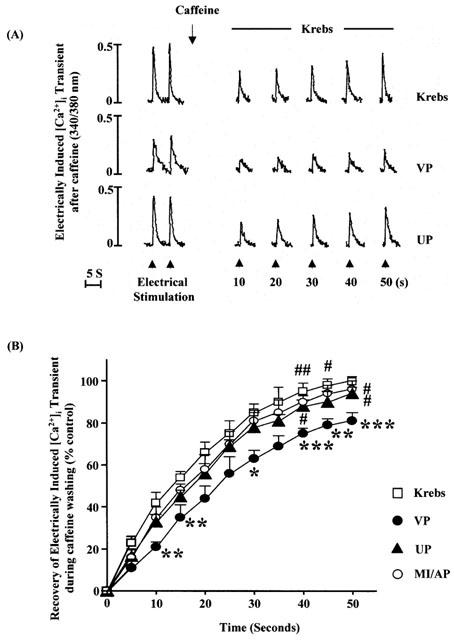
Effects of UP or MI/AP on recovery of electrically-induced [Ca2+]i transient during washout of caffeine. 10 mM caffeine was administered first followed 1 min later by electrical stimulation for 50 s. (A) Representative tracings. (B) Group results. The values, which were normalized by dividing the values by its value in corresponding original control period, are mean±s.e.mean. n=8 cells for Krebs control, VP and UP groups each, and n=9 in MI/AP group. These myocytes were from five rats for each group. *,**,***P<0.05, 0.01, 0.001 vs corresponding value in Krebs control at the same moment; #,##P<0.05, 0.01 vs corresponding value in VP group at the same time point.
Following UP or MI/AP the time constants were 2.814±0.35 (n=8) and 2.712±0.34 (n=9), respectively, which were significantly smaller (P<0.05) than that of the VP group. At 50th second, the amplitude of the electrically induced [Ca2+]i transient of the VP group was 81% of that of the original control while the corresponding values in the UP and AP groups were 96 and 94%, respectively (Figure 6B), which were significantly different (P<0.05) from that of VP group.
The results indicate that the Ca2+-uptake by SR was reduced by MI/A, and that preconditioning with either U50 or MI/A prevented at least partly the reduced Ca2+ uptake by SR.
Effects of UP on Ca2+ spark in unstimulated ventricular myocytes subjected to MI/A
To determine the spontaneous release of Ca2+ from SR, spontaneous Ca2+ sparks were recorded from unstimulated ventricular myocytes before, during and after exposure to MI/A induced by 10 mM 2-DOG and 1 mM Na2S2O4 (Figure 7A). As shown in Figure 7B, in VP group, under control conditions the spark frequency was 5.7±0.9 s−1, and a significant, time-dependent decrease in spark frequency was apparent starting after 4–6 min of MI/A and complete obliteration of Ca2+ spark was observed in two-thirds of the myocytes at the end of MI/A and during reperfusion. In addition to the spark frequency, we also compared the changes in amplitude, and spatial and temporal parameters of the Ca2+ spark during MI/A. Since there were too few Ca2+ sparks for analysis during 7–9 min in the un-preconditioned myocytes, only data obtained from 4–6 min of MI/A were presented and compared. MI/A also caused a significant reduction in spark amplitude (Figure 8), but had no effect on the spatial and temporal parameters of Ca2+ sparks at the same period (Figure 8). The reduction of spontaneous Ca2+ sparks during MI/A indicates that the spontaneous leak of Ca2+ from SR via Ryanodine receptors was reduced during MI/A.
Figure 7.
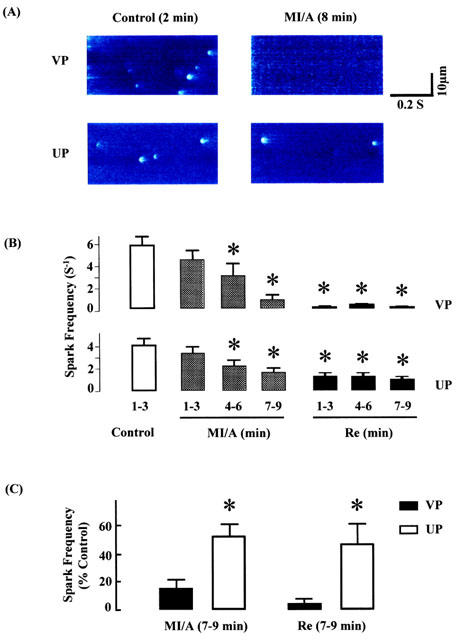
Effects of UP on spontaneous Ca2+ sparks from unstimulated ventricular myocytes subjected to MI/A and reperfusion (Re). A glucose-free Krebs solution containing 1 mM Na2S2O4+10 mM of 2-DOG was used as MI/A treatment, and this was followed by superfusion with normal Krebs solution for 10 min as reperfusion. (A) Representative line-scan images generated from isolated ventricular cells in control (left) or MI/A (right) conditions. (B) Results of time course in Ca2+ sparks frequency obtained from the myocytes during control, MI/A and Re periods, respectively. *P<0.05 vs control period (empty bar). (C) Comparison in Ca2+ sparks frequency between VP and UP during MI/A and Re periods. Data obtained at 7–9 min in MI/A or reperfusion periods, were normalized by dividing the values by its value in control period. All of the data were obtained from 369 sparks in 21 cells for VP group, and 311 sparks in 26 cells in UP groups, respectively. *P<0.05 vs corresponding value in VP group.
Figure 8.
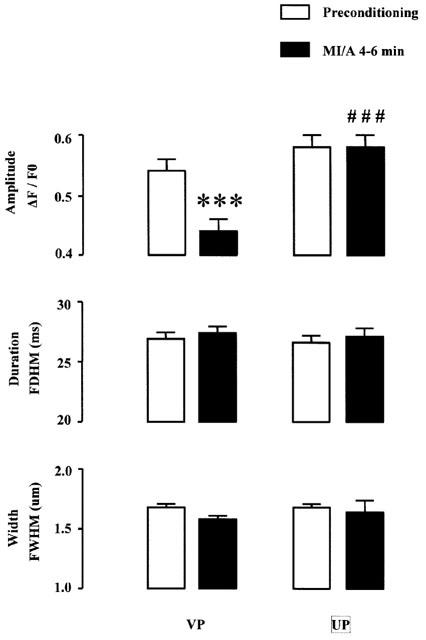
Effects of UP on spark amplitude (ΔF/F0), duration (FDHM, ms) and width (FWHM, μm) of spontaneous Ca2+ sparks from unstimulated ventricular myocytes subjected to MI/A. Only results before MI/A (1 mM Na2S2O4+10 mM 2-DOG in glocose-free Krebs solution) and at 4–6 min of MI/A were presented. n=21 cells for total 369 sparks in VP group, and n=26 cells for total 311 sparks in UP groups, respectively. ***P<0.001 vs the corresponding preconditioning value within the group, and ###P<0.001 vs the corresponding value in VP group.
UP itself did not cause significant changes in spark frequency, amplitude, duration and width (data not shown). Exposure of these preconditioned myocytes to MI/A also caused a time-dependent decrease in spark frequency (Figure 7B). However, the attenuation in spark frequency at the end of MI/A and during reperfusion was both significantly less after UP than in the VP group (Figure 7C). The amplitude of Ca2+ spark was restored to normal in preconditioned myocytes at 4–6 min of MI/A (Figure 8).
These results indicate that UP partially alleviated the attenuating effects of MI/A on spontaneous Ca2+ sparks in ventricular myocytes.
Effects of UP or MI/AP on decay of the caffeine induced [Ca2+]i transient in ventricular myocytes subjected to MI/A
Since caffeine keeps the Ryanodine receptors open during the period of its application, the decline of the caffeine induced [Ca2+]i transient depends only on Ca2+ extrusion out of the myocyte, and this happens mainly through the Na+–Ca2+ exchange (Sham et al., 1995). So we determined the value of t-50, the time of 50% for decay, in the caffeine induced [Ca2+]i transient as an indicator of Na+–Ca2+ exchange activity as described previously (Terracciano & Macleod, 1994; Sham et al., 1995). As shown in Table 2, MI/A significantly increased the t-50 value in agreement with the previous finding (Seki & Macleod, 1995). Both UP and MI/AP prevented the change (Table 2). The observation indicates that MI/A inhibits the Ca2+ extrusion via Na+–Ca2+ exchange while preconditioning with either U50 or MI/A reverses the inhibitory effect of MI/A.
Table 2.
Decay of caffeine-induced [Ca2+]i transient

Discussion
Previous studies in our laboratory have shown that UP confers immediate cardioprotection against ischaemic insult (Wang et al., 2001) and delayed cardioprotection against MI (Wu et al., 1999). In agreement with the previous findings, the present study has also shown that UP confers cardioprotection against MI/A. More importantly the present study has provided first evidence that UP also prevents changes of Ca2+ homeostasis altered by MI/A in rat ventricular myocytes. The alterations induced by MI/A included [Ca2+]i, SR Ca2+ content, release and uptake of Ca2+ by SR and Na+–Ca2+ exchange activity. The prevention of changes of Ca2+ homeostasis may be responsible, at least partly, for the cardioprotection of preconditioning. With the exception of Ca2+ spark, which was not determined in ventricular myocytes preconditioned with MI/A in the present study, the effects of pharmacological preconditioning with U50 were the same as those of MI/AP.
Previous studies have shown that ischaemia causes cell Ca2+ overload, which is partly prevented by IP (Steenbergen et al., 1993b; Bersohn et al., 1997; Ylitalo et al., 2000). In agreement with the observation we also observed that the Ca2+ overload, caused by MI/A was attenuated by preconditioning with either U50 or MI/A. We also found that the decay of the [Ca2+]i transients induced by electrical stimulation was significantly delayed and the recovery of the electrically induced [Ca2+]i transient following caffeine reduced after MI/A, suggesting a reduction in Ca2+-uptake by SR. Both were reversed towards normal by UP or MI/AP. The observations are in agreement with the previous finding that IP restores the activity and phosphorylation of SERCA, the enzyme that pumps Ca2+ back to SR (Osada et al., 2000). Both studies indicated that the Ca2+-uptake by SR was reduced by ischaemic insults, which were partly prevented by preconditioning. In addition there are two new findings regarding the Ca2+ handling by the SR. The first is that the caffeine induced [Ca2+]i transient, an indication of the content of Ca2+ in SR, which was significantly reduced by MI/A, was restored towards normal by preconditioning. The second is that both electrically induced [Ca2+]i transient in the electrically stimulated ventricular myocyte and the Ca2+ spark in the unstimulated cells were reduced following MI/A and reversed close to normal by preconditioning, indicating that spontaneous and electrically-stimulated release of Ca2+ were restored towards normal by preconditioning. With the new findings, the Ca2+ handling by SR in ventricular myocytes subjected to ischaemic insults with or without preconditioning is now better understood. When the heart is subjected to ischaemia or MI/A, the Ca2+-uptake by SR is reduced, thus reducing the Ca2+ content in SR. Together with the reduced Na+–Ca2+ exchange activity (Steenbergen et al., 1993a, 1993b; Bersohn et al., 1997), the cytoplasmic [Ca2+]i is increased, causing Ca2+ overload, a precipitating cause of tissue injury. The reduced Ca2+ in SR also reduces the availability of Ca2+ for spontaneous release and release during E–C coupling. In short previous and present studies it was showed that preconditioning prevented the changes of Ca2+ handling tasking place in SR in myocytes subjected to MI/A.
In the present study we also found that the decay of the caffeine induced [Ca2+]i transient, which is an indication of Na+–Ca2+ exchange activity (Terracciano & Macleod, 1994; Sham et al., 1995), was significantly prolonged following MI/A, indicating a reduction in Na+–Ca2+ exchange activity, which is in agreement with previous observations that ischaemia reduces the Na+–Ca2+ exchange activity (Steenbergen et al., 1993a, 1993b; Seki & MacLeon, 1995; Bersohn et al., 1997). More importantly, UP and MI/AP prevented the recovery of the caffeine induced [Ca2+]i transient caused by MI/A. So UP and MI/AP restored the Na+–Ca2+ exchange activity as they did on SR functions.
It is well established that during myocardial ischaemia (Schumacher et al., 1998) and MI (Ferrari, 1998), the cardiac contractility is reduced and preconditioning partially prevents it. As reported in previous studies (Yu et al., 1998; Hohl & Li, 1991), the alterations in cardiac contractility are directly related to the electrically induced [Ca2+]i transient, an indication of the release of Ca2+ during E–C coupling. In the present study we have also shown that the caffeine induced [Ca2+]i transient, an indication of Ca2+ content in SR, is reduced by MI/A and the reduction is prevented by preconditioning. Since the content of Ca2+ in SR for release during MI/A is reduced, the availability of Ca2+ for release is reduced as a result of reduced SR Ca2+ content, which leads to reduced electrically induced [Ca2+]i transient and cardiac contractility. Preconditioning restores the SR Ca2+ content and availability of Ca2+ for release during E–C coupling, thus restoring both the electrically induced [Ca2+]i transient and cardiac contractility.
In our previous studies (Wu et al., 1999; Wang et al., 2001), we have shown that pharmacological preconditioning with U50 confers the same cardioprotection and shares the same secondary messengers, namely PKC and KATP, as physiological preconditioning with ischaemic insults. In the present study we have shown that pharmacological preconditioning also shares the same mechanisms in regulation of intracellular Ca2+. The observations indicate that pharmacological preconditioning, which may be more convenient than preconditioning with ischaemia or MI or anoxia in certain situations, may be used for cardioprotection in clinical practice.
In conclusion, the present study has provided novel evidence that pharmacological preconditioning with U50, that confers cardioprotection, prevents changes of Ca2+ homeostasis in the heart subjected to MI/A. The study has also provided evidence for the first time that in addition to [Ca2+]i and Ca2+ uptake by SR, preconditioning helps to maintain the Ca2+ content in SR and spontaneous and electrically induced release of Ca2+ from SR in ventricular myocytes. It also prevents the inhibition of the Na+–Ca2+ exchange activity upon ischaemic insult.
Acknowledgments
The study was supported by The Research Grants Council, Hong Kong and a Cardiovascular Physiology Research Fund donated by L.C.S.T. (Holding) Ltd. We thank Dr E.G. Lakatta for advice and Mr C.P. Mok for technical assistance. J.C.S. Ho and K.W.L. Kam were supported by Postgraduate Studentships of the University of Hong Kong.
Abbreviations
- [Ca2+]i
intracellular Ca2+; 2-DOG, 2-deoxy-D-glucose
- E–C coupling
excitation-contraction coupling
- IP
ischaemic preconditioning
- κ-opioid receptor
kappa-opioid receptor
- MI/A
metabolic inhibition/anoxia
- MI/AP
metabolic inhibition/anoxia preconditioning
- nor-BNI
nor-binaltorphimine
- SERCA
SR Ca2+-ATPase
- SR
sarcoplasmic reticulum
- U50
U50488H
- UP
U50-preconditioning
References
- BAKER D.L., HASHMOTO K., GRUPP I.L., JI Y., REED T., LOUKIANOV E., GRUPP G., BHAGWHAT A., HOIT B., WALSH R., MARBAN E., PERIASAMY M. Targeted overexpression of the sarcoplasmic reticulum Ca2+-ATPase increases cardiac contractility in transgenic mouse hearts. Circ. Res. 1998;83:1205–1214. doi: 10.1161/01.res.83.12.1205. [DOI] [PubMed] [Google Scholar]
- BERSOHN M.M., MOREY A.K., WEISS R.S. Sarcoplasmic calcium transporters in myocardial ischemia. J. Mol. Cell. Cardiol. 1997;29:2525–2532. doi: 10.1006/jmcc.1997.0487. [DOI] [PubMed] [Google Scholar]
- BLAKE A.D., BOT G., LI S., FREEMAN J.C., REISINE T. Differential agonists regulation of the human κ-opioid receptor. J. Neurochem. 1997;68:1846–1852. doi: 10.1046/j.1471-4159.1997.68051846.x. [DOI] [PubMed] [Google Scholar]
- BONNET S., BELUS A., HYVELIN J.M., ROUX E., MARTHAN R., SAVINEAU J.P. Effect of chronic hypoxia on agonist-induced tone and calcium signaling in rat pulmonary artery. Am. J. Physiol. 2001;281:L193–L201. doi: 10.1152/ajplung.2001.281.1.L193. [DOI] [PubMed] [Google Scholar]
- CHENG H., LEDERER W.J., CANNELL M.B. Calcium spark: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- CHOSSAT N., GRISCELLI F., JOURDON P., LOGEART D., RAGOT T., HEIMBURGER M., PERRICAUDET M., LOMPRE A.M., HATEM S., MERCADIER J.J. Adenoviral SERCA1a gene transfer to adult rat ventricular myocytes induces physiological changes in calcium handling. Cardiovasc. Res. 2001;49:288–297. doi: 10.1016/s0008-6363(00)00234-0. [DOI] [PubMed] [Google Scholar]
- CUMMING D.V., HEADS R.J., BRAND N.J., YELLON D.M., LATCHMAN D.S. The ability of heat stress and metabolic precdonditioning to protect primary rat cardiac myocytes. Basic. Res. Cardiol. 1996;91:79–85. doi: 10.1007/BF00788868. [DOI] [PubMed] [Google Scholar]
- DO E., BAUDET S., GOW I.F., ELLIS D., NOIREAUD J. Intracellular pH during hypoxia in normal and hypertrophied right ventricle of ferret heart. J. Mol. Cell. Cardiol. 1995;27:927–939. doi: 10.1016/0022-2828(95)90043-8. [DOI] [PubMed] [Google Scholar]
- DONG H., SHENG J.Z., LEE C.M., WONG T.M. Calcium antagonistic and antiarrhythymic action of CPU23, a substituted tetrahydroisoquinoline. Br. J. Pharmacol. 1993;109:113–119. doi: 10.1111/j.1476-5381.1993.tb13539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERRARI R. Metabolic disarrangement in ischemic heart disease and its therapeutic control. Rev. Port. Cardiol. 1998;17:667–684. [PubMed] [Google Scholar]
- GIORDANO F.J., HE H., MCDONOUGH P., MEYER M., SAYEN M.R., DILLMANN W.H. Adenovirus-mediated gene transfer reconstitutes depressed sarcoplasmic reticulum Ca2+-ATPase levels and shortens prolonged cardiac myocyte Ca2+ transients. Circulation. 1997;96:400–403. doi: 10.1161/01.cir.96.2.400. [DOI] [PubMed] [Google Scholar]
- GRAY M.O., KARLINER J.S., MOCHLY-ROSEN D. A selective å-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J. Biol. Chem. 1997;272:30945–30951. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- HAJJAR R.J., KANG J.X., GWATHMEY J.K., ROSENZWEIG A. Physiological effects of adenoviral gene transfer of sarcoplasmic reticulum calcium ATPase in isolated rat myocytes. Circulation. 1999;95:423–429. doi: 10.1161/01.cir.95.2.423. [DOI] [PubMed] [Google Scholar]
- HE H., GIORDANO F.J., DANDAN R.H., CHOI D.J., ROCKMAN H.A., MCDONOUGH P.M., BLUHM W.F., MEYER M., SAYEN M.R., SWANSON E., DILLMANN W.H. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and caediac relaxation. J. Clin. Invest. 1997;100:380–389. doi: 10.1172/JCI119544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIEBERT L., PING T. Protective effect of dextrant sulfate and heparin on adult rat cardiomyocytes damaged by free radicals. J. Mol. Cell. Cardiol. 1997;29:229–235. doi: 10.1006/jmcc.1996.0267. [DOI] [PubMed] [Google Scholar]
- HOHL C.M., LI Q. Compartmentation of cAMP in adult canine ventricular myocytes: relation to single free Ca2+ transient. Circ. Res. 1991;69:1369–1379. doi: 10.1161/01.res.69.5.1369. [DOI] [PubMed] [Google Scholar]
- HUNG M.J., CHERNG W.J., KUO L.T., WANG C.H., CHERN M.S. Analysis of left atrial volume change rate during left ventricular diastolic phase with M-mode echocardiography for differentiation between normal and pseudonormal mitral inflow. Am. J. Cardiol. 2002;89:552–556. doi: 10.1016/s0002-9149(01)02295-0. [DOI] [PubMed] [Google Scholar]
- JANEZEWSKI A.M., LAKATTA E.G. Buffering of calcium influx by sarcoplasmic reticulum during the action potential in guinea pig ventricular myocytes. J. Physiol. 1993;471:343–363. doi: 10.1113/jphysiol.1993.sp019904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU G.S., THORNTON J., VAN WINKIE D.M., STANLEY A.W.H., OLSSON R.A., DOWNEY J.M. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–356. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- LOUKIANOV E., JI Y., GRUPP I.L., KIRKPATRICK D.L., BAKER D.L., LOKIANOV T., GRUPP G., LYTTON J., WALSH R.A., PERIASAMY M. Enhanced myocardial contractility and increased Ca2+ transport function in transgenic hearts expressing the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase. Circ. Res. 1998;83:889–897. doi: 10.1161/01.res.83.9.889. [DOI] [PubMed] [Google Scholar]
- MACIANSKIENE R., MATEJOVIC P., SIPIDO K., FLAMENG W., MUBAGWA K. Modulation of the extracellular divalent cation-inhibited non-selective conductance in cardiac cells by metabolic inhibition and by oxidants. J. Mol. Cell. Cardiol. 2001;33:1371–1385. doi: 10.1006/jmcc.2001.1401. [DOI] [PubMed] [Google Scholar]
- MCKENNA C.E., GUTHEIL W.G., SONG W. A method for preparing analytically pure sodium dithionite- Dithionite quality and observed nitrogenase-specific activities. Biochim. Biophys. Acta. 1991;1075:109–117. doi: 10.1016/0304-4165(91)90082-r. [DOI] [PubMed] [Google Scholar]
- MURRY C.E., JENNINGS R.B., REIMER K.A. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- OSADA M., NETTICADAN T., KAWABATA K., TAMURA K., DHALLA N.S. Ischemic preconditioning prevents I/R-induced alterations in SR calcium-calmodulin protein kinase II. Am. J. Physiol. 2000;278:H1791–H1798. doi: 10.1152/ajpheart.2000.278.6.H1791. [DOI] [PubMed] [Google Scholar]
- OTTER D., AUSTIN C. Hypoxia, metabolic inhibition, and isolated rat mesenteric tone: influence of arterial diameter. Microvasc. Res. 2000;59:107–114. doi: 10.1006/mvre.1999.2212. [DOI] [PubMed] [Google Scholar]
- OVEREND C.L., EISNER D.A., O'NEILL S.C. Altered cardiac sarcoplasmic reticulum function of intact myocytes of rat ventricle during metabolic inhibition. Circ. Res. 2001;88:181–187. doi: 10.1161/01.res.88.2.181. [DOI] [PubMed] [Google Scholar]
- PORTOGHESE P.S., LIN C.E., FAROUZ-GRANT G.F., TAKEMORI A.E. Structure-activity relationship of N17′-substituted norbinaltorphimine congeners: role of the N17 basic group in the interaction with a putative address subsite on the κ-opioid receptor. J. Med. Chem. 1994;37:1495–1500. doi: 10.1021/jm00036a015. [DOI] [PubMed] [Google Scholar]
- REMILLARD C.V., ZHANG W.M., SHIMODA L.A., SHAM J.S.K. Physiological properties and functionas of Ca2+ sparks in rat intra-pulmonary arterial smooth muscle cells. Am. J. Physiol., Lung Cell Mol. Physiol. 2002;283:L433–L444. doi: 10.1152/ajplung.00468.2001. [DOI] [PubMed] [Google Scholar]
- RICHARD V., KAEFFER N., TRON C., THUILLEZ C. Ischemic preconditioning protects against coronary endothelial dysfunction induced by ischemia and reperfusion. Circulation. 1994;89:1254–1261. doi: 10.1161/01.cir.89.3.1254. [DOI] [PubMed] [Google Scholar]
- SCHULZ J.J., HSU A.K., GROSS G.J. Ischemic preconditioning in the intact rat heart is mediated by κ- but not δ- or μ-opioid receptors. Circulation. 1998;97:1282–1289. doi: 10.1161/01.cir.97.13.1282. [DOI] [PubMed] [Google Scholar]
- SCHUMACHER C.A., BAARTSCHEER A., CORONEL R., FIOLET J.W. Energy dependent transport of calcium to the extracellular space during acute ischemia of the rat heart. J. Mol. Cell. Cardiol. 1998;30:1631–1642. doi: 10.1006/jmcc.1998.0728. [DOI] [PubMed] [Google Scholar]
- SEKI S., MACLEOD K.T. Effects of anoxia on intracellular Ca2+ and contraction in isolated guinea pig cardiac myocytes. Am. J. Physiol. 1995;268:H1045–H1052. doi: 10.1152/ajpheart.1995.268.3.H1045. [DOI] [PubMed] [Google Scholar]
- SHAM J.S.K., HATEM S.N., MORAD M. Species differences in the Na+–Ca2+ exchange in mammalian cardiac myocytes. J. Physiol. 1995;488:623–631. doi: 10.1113/jphysiol.1995.sp020995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHENG J.Z., WONG T.M. Chronic U50488H abolishes inositol 1,4,5-trisphosphate and intracellular Ca2+ elevation evoked by ê-opioid in rat myocytes. Eur. J. Pharmacol. 1996;307:323–329. doi: 10.1016/0014-2999(96)00280-4. [DOI] [PubMed] [Google Scholar]
- SMITH G.B., STEFENELLI T., WU S.T., WIKMAN C.J., PARMLEY W.W., ZAUGG C.E. Rapid adaptation of myocardial calcium homeostasis to short episodes of ischemia in isolated rat hearts. Am. Heart. J. 1996;131:1106–1112. doi: 10.1016/s0002-8703(96)90084-8. [DOI] [PubMed] [Google Scholar]
- STEENBERGEN C., FRALIX T.A., MURPHY E. Role of increased cytosolic free calcium concentration in myocardial ischemic injury. Basic Res. Cardiol. 1993a;88:456–470. doi: 10.1007/BF00795412. [DOI] [PubMed] [Google Scholar]
- STEENBERGEN C., MURPHY E., WATTS J.A., LONDON R.E. Correlation between cytosolic free calcium, contraction, ATP, and irreversible ischemic injury in perfused rat heart. Circ. Res. 1990;66:135–146. doi: 10.1161/01.res.66.1.135. [DOI] [PubMed] [Google Scholar]
- STEENBERGEN C., PERLMAN M.E., LONDON R.E., MURPHY E. Mechanism of preconditioning-ionic alterations. Circ. Res. 1993b;72:112–125. doi: 10.1161/01.res.72.1.112. [DOI] [PubMed] [Google Scholar]
- SUGIYAMA S., SATOH H., NOMURA N., TERADA H., WATANABE H., HAYASHI H. The importance of glycolytically-derived ATP for the Na+/H+exchange activity in guinea pig ventricular myocytes. Mol. Cell. Biochem. 2001;217:153–161. doi: 10.1023/a:1007261322878. [DOI] [PubMed] [Google Scholar]
- TATEISHI N., SUZUKI Y., CICHA I., MAEDA N. O(2) release from erythrocytes flowing in a narrow O(2)-permeable tube: effects of erythrocyte aggregation. Am. J. Physiol. 2001;281:H448–H456. doi: 10.1152/ajpheart.2001.281.1.H448. [DOI] [PubMed] [Google Scholar]
- TERRACCIANO C.M.N., MACLEOD K.T. Effects of acidosis on Na+–Ca2+ exchange and consequences for relaxation in guinea pig cardiac myocytes. Am. J. Physiol. 1994;267:H477–H487. doi: 10.1152/ajpheart.1994.267.2.H477. [DOI] [PubMed] [Google Scholar]
- TRAFFORD A.W., DIAZ M.E., EISNER D.A. Coordinated control of cell Ca2+ loading and triggered release from the sarcoplasmic reticulum underlies the rapid inotropic response to increased L-type Ca2+ current. Circ. Res. 2001;88:195–201. doi: 10.1161/01.res.88.2.195. [DOI] [PubMed] [Google Scholar]
- ULLRICH R., SCHERRER-CROSBIE M., BLOCH K.D., ICHINOSE F., NAKAJIMA H., PICARD M.H., ZAPOL W.M., QUEZADO Z.M. Congenital deficiency of nitric oxide synthase-2 protects against endotoxin-induced myocardial dysfunction in mice. Circulation. 2000;102:1440–1446. doi: 10.1161/01.cir.102.12.1440. [DOI] [PubMed] [Google Scholar]
- WANG G.Y., WU S., PEI J.M., YU X.C., WONG T.M. κ- but not δ-opioid receptors mediate effects of ischemic preconditioning on both infarct and arrhythmia in rats. Am. J. Physiol. 2001;280:H384–H391. doi: 10.1152/ajpheart.2001.280.1.H384. [DOI] [PubMed] [Google Scholar]
- WILSON S., WU S., KAROLY K., RAVINGEROVA T., VEGH A., PAPP J., TOMISAWA S., PARRATT J.R., PYNE N.J. Delayed cardioprotection is associated with the sub-cellular relocalisation of ventricular protein kinase C epsilon, but not p42/44MAPK. Mol. Cell. Biochem. 1996;160–161:225–230. doi: 10.1007/BF00240053. [DOI] [PubMed] [Google Scholar]
- WU S., LI H.Y., WONG T.M. Cardioprotection of preconditioning by metabolic inhibition in the rat ventricular myocyte, involvement of κ-opioid receptor. Circ. Res. 1999;84:1388–1395. doi: 10.1161/01.res.84.12.1388. [DOI] [PubMed] [Google Scholar]
- YLITALO K.V., ALA-RAMI A., LIIMATTA E.V., PEUKURINEN K.J., HASSINEN E. Intracellular free calcium and mitochondrial menbrane potential in ischemia/reperfusion and preconditioning. J. Mol. Cell. Cardiol. 2000;32:1223–1238. doi: 10.1006/jmcc.2000.1157. [DOI] [PubMed] [Google Scholar]
- YU X.C., LI H.Y., WANG H.X., WONG T.M. U50488H inhibits effects of norepinephrine in rat cardiomyocytes, cross-talk between kappa opioid and beta adrenergic receptors. J. Mol. Cell. Cardiol. 1998;30:405–413. doi: 10.1006/jmcc.1997.0604. [DOI] [PubMed] [Google Scholar]
- ZHOU X., ZHAI X., ASHRAF M. Direct evidence that initial oxidative stress triggered by preconditioning contributes to second window of protection by endogenous antioxidant enzyme in myocytes. Circulation. 1996;93:1177–1184. doi: 10.1161/01.cir.93.6.1177. [DOI] [PubMed] [Google Scholar]