

Abstract
Cyclo-oxygenase (COX) and lipoxygenase (LO) share a common substrate, arachidonic acid. Aspirin and related drugs inhibit COX activity. In a subset of patients with asthma aspirin induces clinical symptoms associated with increased levels of certain LO products, a phenomenon known as aspirin-sensitive asthma. The pharmacological pathways regulating such responses are not known.
Here COX-1 and LO activity were measured respectively by the formation of thromboxane B2 (TXB2) or leukotrienes (LT) C4, D4 and E4 in whole blood stimulated with A23187. COX-2 activity was measured by the formation of prostaglandin E2 (PGE2) in blood stimulated with lipopolysaccharide (LPS) for 18 h.
No differences in the levels of COX-1, COX-2 or LO products or the potency of drugs were found in blood from aspirin sensitive vs aspirin tolerant patients. Aspirin, indomethacin and nimesulide inhibited COX-1 activity, without altering LO activity. Indomethacin, nimesulide and the COX-2 selective inhibitor DFP [5,5-dimethyl-3-(2-isopropoxy)-4-(4-methanesulfonylphenyl)-2(5H)-furanone] inhibited COX-2 activity. NO-aspirin, like aspirin inhibited COX-1 activity in blood from both groups. However, NO-aspirin also reduced LO activity in the blood from both patient groups. Sodium salicylate was an ineffective inhibitor of COX-1, COX-2 or LO activity in blood from both aspirin-sensitive and tolerant patients.
Thus, when COX activity in the blood of aspirin-sensitive asthmatics is blocked there is no associated increase in LO products. Moreover, NO-aspirin, unlike other NSAIDs tested, inhibited LO activity in the blood from both aspirin sensitive and aspirin tolerant individuals. This suggests that NO-aspirin may be better tolerated than aspirin by aspirin-sensitive asthmatics.
Keywords: Aspirin, asthma, cyclo-oxygenase, COX-1, COX-2, prostaglandins, leukotrienes, lipoxygenase, whole blood, NSAID
Introduction
Cyclo-oxygenase (COX) and lipoxygenase (LO) are two main pathways involved in the metabolism of arachidonic acid (AA). COX catalyses the synthesis of prostaglandins (PG) and thromboxane (Smith et al., 1991). COX exists in two isoforms, COX-1 is constitutively expressed whilst COX-2 is induced by inflammatory stimuli (Mitchell & Warner, 1999). Non-steroidal anti-inflammatory drugs (NSAIDs), which include aspirin are a group of chemically distinct compounds which inhibit both COX-1 and COX-2 activity. COX-2 is the predominate isoform present in a range of human inflammatory diseases and most recently selective inhibitors of COX-2 (e.g. celecoxib and rofecoxib) have been developed and introduced for the treatment of arthritic disease. By contrast, inhibitors of COX-1 (or COX-2) are not recommended for the treatment of inflammatory airway diseases such as asthma. This is despite the identification of active enzyme in human airway cells (Belvisi et al., 1997; 1998; Mitchell et al., 1994). In fact NSAIDs are counter-indicated for asthmatics and in a sub-population induces symptoms such as rhinorrhea, nasal congestion and severe nasal polyps. Asthmatic patients who are sensitive to aspirin, and possibly other NSAIDs, are commonly referred to as aspirin-sensitive asthmatics.
The biochemical pathways involved in aspirin-sensitive asthma are not fully established. However, there is strong evidence that leukotrienes (LT) C4, D4 and E4 derived from the 5-lipoxygenase (5-LO) pathway are involved. These LTs are pro-inflammatory in nature, causing bronchoconstriction and migration of inflammatory cells into the airway. The levels of LTC4, D4 and E4 are increased after ingestion of drugs such as aspirin in sensitive patients and symptoms are relieved by 5-LO inhibitors as well as by LT antagonists (Christie et al., 1991; Nasser et al., 1994). Furthermore recent studies suggest that asthma could be associated with expression of 5-LO in blood leukocytes (Koshino et al., 1998) and/or mutations in the gene promoter for the 5-LO activating protein (Koshino et al., 1999). There are three ways in which inhibition of COX activity are thought to regulate 5-LO products. Firstly, inhibition of COX may increase the level of free arachidonic acid available in cells after stimulation. Secondly, the blockade of PG (e.g. PGE2) formed by COX is thought to remove an endogenous ‘brake' on the formation of LTs, which has been demonstrated in human eosinophils and neutrophils (Docherty & Wilson, 1987; Tenor et al., 1996). Finally, where COX-2 predominates, aspirin may induce the production of 5-LO products via the modification of metabolites formed (Mitchell & Belvisi, 1997). Thus, aspirin blocks the enzymatic activities of COX-1 irreversibly. By contrast aspirin merely diverts the metabolite formed by COX-2 away from PGH2 and towards 15-hydroxyeicosatetreanoic acid (15-HETE) (Holtzman et al., 1992). This 15-HETE can then be further metabolized by 5-LO to the novel 15-epilipoxins (Claria & Serhan, 1995).
However, none of the above hypotheses generated to explain aspirin-sensitive asthma have been fully tested and thereby proven or disproved. Here we have used a modified whole blood assay to compare the relative sensitivity of COX-1 vs COX-2 in tissue from aspirin-sensitive and aspirin tolerant individuals. In parallel we have studied how inhibiting either COX-1 or COX-2 influences the production of 5-LO products in blood from aspirin sensitive patients and in blood from healthy control donors. Some of these results have been presented in abstract form (Gray et al., 2000).
Methods
Characterization of aspirin sensitive group
All subjects in the ‘aspirin sensitive asthma' group had a history of aspirin-induced reaction. They developed a respiratory type reaction (asthma±rhinoconjunctivitis) following aspirin or NSAID ingestion. These patients had their aspirin-sensitivity confirmed by an intranasal challenge with lysine-aspirin. These were carried out in controlled circumstances, with full resuscitative facilities. Briefly, each patient was challenged with placebo, followed by two incremental doses of lysine-aspirin (4 and 8 mg), in a single-blind fashion. Parameters measured before and after the challenge included nasal symptom score, nasal inspiratory peak flow, peak expiratory flow rate, and acoustic rhinometry (measuring change in nasal minimum cross-sectional area, and volume). A change of 25% (reduction) in nasal cross-sectional area, and volume (between 0–7 cm) was taken as confirmation of a positive challenge. All patients were off their intranasal steroids for 1 week prior to the challenge. All patients had characteristic nasal polyps.
Characterization of control group
Blood donors from the control (aspirin tolerant and non-asthmatic) group had no history of aspirin sensitivity and confirmed that they took aspirin and related drugs routinely.
COX-1 and COX-2 activity in human blood
Venous blood was collected into tubes containing heparin saline (19 U ml−1) and pipetted (100 μl) into individual wells of 96-well plates. For COX-1 assays, blood was treated with NCX4016 (NO-aspirin; 10−11–10−3 M), sodium salicylate (10−11–10−3 M), indomethacin (10−12–10−4 M), nimesulide (10−12–10−4 M), DFP, (5,5-dimethyl-3-(2-isopropoxy)-4-(4-methanesulfonylphenyl)-2(5H)-furanone; 10−12–10−4 M) a selective inhibitor of COX-2 (Warner et al., 1999) or vehicle (dimethyl sulphoxide; DMSO). All drugs were dissolved in DMSO to make stock solutions of 10−1 and 10−2 M and then further diluted in Dulbecco's Modified Eagle Medium for the respective concentration ranges. The final percentage of DMSO in blood in each well was 0.1 or 1%. The appropriate vehicle controls were also performed in each experiment. Plates were placed in an incubator at 37°C for 1 h, and then the blood was stimulated with calcium ionophore A23187 (5×10−5 M). After 30 min, plates were centrifuged (1500×g, 4°C, 5 min), plasma removed and immediately frozen at −20°C until COX and LO products were assayed.
For COX-2 assays, blood was treated with aspirin (12 μg ml−1) to inactivate COX-1 activity. Six hours later, lipopolysaccharide (LPS; 10 μg ml−1) and drugs were added (18 h incubation time) as in the COX-1 protocol. Plates were then spun and plasma removed and frozen. Using radioimmunoassay, concentrations of TXB2 (as a measure of TXA2 formation and COX-1 activity) or PGE2 (as a measure of COX-2 activity) were determined in plasma samples from COX-1 or COX-2 assays, respectively (Warner et al., 1999).
5-LO activity in whole blood
Levels of total LTC4, D4, and E4 were measured by a common enzyme immunoassay, according to the manufacturer's instructions, as an indicator of 5-LO products. The formation of LTs by cells requires both the release of substrate arachidonic acid and activation of 5-LO activating protein (FLAP). Stimulation of whole blood with A23187 provides both of these necessary events. Thus, as expected, samples obtained under identical conditions as for the COX-1 contained clear and consistent elevations in the level of LTs. However, samples taken under conditions identical to those for the COX-2 assay, where A23187 is not added, LTs levels were undetectable. Thus, all drug incubation studies were performed on blood treated under identical conditions to those described for the COX-1 assay.
Materials
Aspirin, sodium salicylate, indomethacin, nimesulide, DMSO and Ca ionophore A23187 were obtained from Sigma Chemical Co (Poole, Dorset, U.K.). NicOx (Sophia Antipolis, France) supplied NO-aspirin (NCX 4016). DFP was a gift from Merck-Frosst Labs (Pointe Claire, PQ, Canada). Heparin was provided by National Veterinary supplies (Stoke on Trent, U.K.). For the radioimmunoassay, PGE2 and TXB2 antisera were obtained from Sigma Chemical Co, U.K. [3H]-PGE2 and [3H]-TXB2 were purchased from Amersham Life Sciences (Bucks, U.K.). LTC, D, E4 enzyme immunoassay was purchased from Biotrak, (Amersham Life Sciences, U.K.).
Statistical analysis
All values in the text, figures and table are expressed as the mean±s.e.mean of n observations. Concentration response curves were fitted using a sigmoidal regression with variable slope and statistical analysis was performed (raw values expressed as a percentage of control) using a two-way analysis of variance (ANOVA) and a Bonferroni post test. Students t-test was used to analyse the IC50s for the concentration curves. A P value of <0.05 was considered statistically significant.
Results
Effects of NSAIDs on COX-1 activity
Unstimulated blood from aspirin-sensitive or healthy donors contained low levels of TXB2 (4±2 vs 3±1 ng ml−1 respectively). However, when stimulated with Ca ionophore A23187, large amounts of TXB2 (control: 56±10 vs aspirin-sensitive: 75±8 ng ml−1; P<0.05 compared to unstimulated blood) were produced. No statistical differences were found between the levels of TXB2 (i.e. COX-1 activity) under unstimulated or A23187-stimulated conditions in blood from aspirin sensitive vs control donors (P>0.05; unpaired Students t-test). Pre-treatment of blood with the following NSAIDs; NO-aspirin (10−11–10−3 M), aspirin (10−11–10−3 M), indomethacin (10−12–10−4 M) or nimesulide (10−12–10−4 M) caused a concentration-dependent inhibition of TXB2 formation by blood donated by both patient groups (Figure 1). No significant differences (P>0.05) were found in the potencies of NO-aspirin, aspirin or indomethacin in blood from either patient group (Figure 1). By contrast nimesulide was slightly more potent (P<0.05 for IC50 values by t-test; Table 1) as an inhibitor of COX-1 activity in blood from control donors vs aspirin sensitive individuals. Neither DFP nor sodium salicylate had any effect on COX-1 activity in blood from either aspirin-sensitive or control donors (Figure 2).
Figure 1.
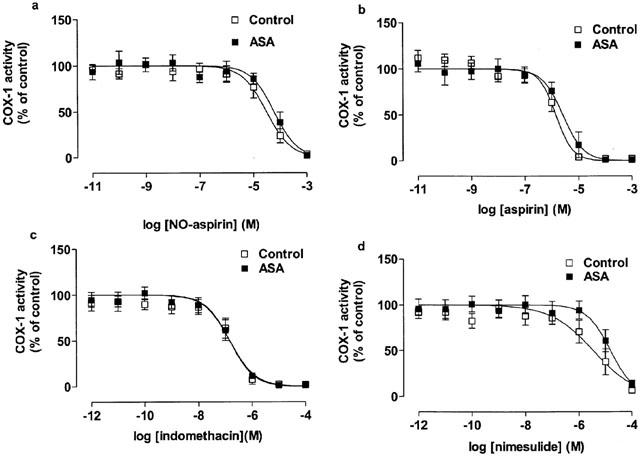
The inhibitory effect of (a) NO-aspirin, (b) aspirin, (c) indomethacin and (d) nimesulide on COX-1 activity in blood from healthy donors and aspirin-sensitive asthmatics (ASA). Data represent enzyme activity calculated as percentage of control response from each group (n=5–6).
Table 1.
Comparison of the IC50s for COX-1 and 2 activities in whole blood from aspirin-sensitive vs control donors

Figure 2.
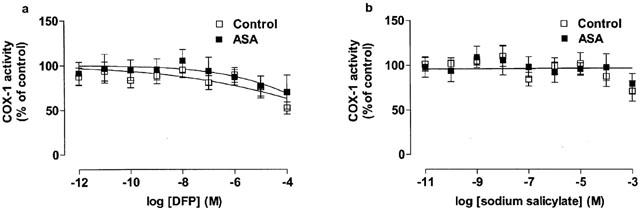
The inhibitory effect of (a) DFP and (b) sodium salicylate on COX-1 activity in blood from healthy donors and aspirin-sensitive asthmatics (ASA). Data represent enzyme activity calculated as percentage of control response from each group (n=5–6).
Effects of NSAIDs on COX-2 activity
In unstimulated blood incubated under sterile conditions for 18 h PGE2 levels were undetectable (detection limit: 0.7 ng ml−1). When challenged with LPS for 18 h, blood from control as well as aspirin-sensitive patients released large amounts of PGE2 (control: 68±26 vs aspirin sensitive donors: 29±8 ng ml−1). The levels of PGE2 (index of COX-2 activity) were lower in LPS stimulated blood from aspirin-sensitive than tolerant donors, however this did not reach statistical significance (P>0.05; unpaired Student t-test). Indomethacin, nimesulide or DFP caused concentration-dependent inhibitions of COX-2 activity in blood from both groups (Figure 3). As was seen for its activity on COX-1, nimesulide was a more potent inhibitor of COX-2 activity in blood from control compared to aspirin-sensitive donors (P<0.05 for IC50 values tested by Students unpaired two-tailed t-test; P<0.05 for whole curve analysis using two-way ANOVA; Table 1, Figure 3). NO-aspirin, aspirin or sodium salicylate induced only weak inhibitions of COX-2 activity in whole blood and no differences were apparent in the inhibition of COX-2 activity in blood from control vs aspirin-sensitive patients (Figure 4).
Figure 3.
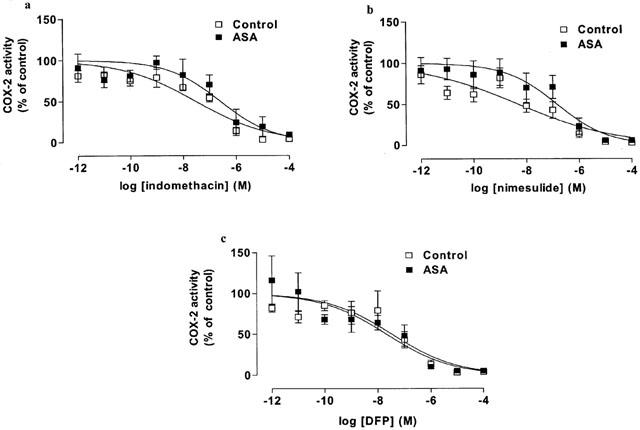
The inhibitory effect of (a) indomethacin, (b) nimesulide and (c) DFP on COX-2 activity in blood from healthy donors and aspirin-sensitive asthmatics (ASA). Data represent enzyme activity calculated as percentage of control response from each group (n=5–6).
Figure 4.
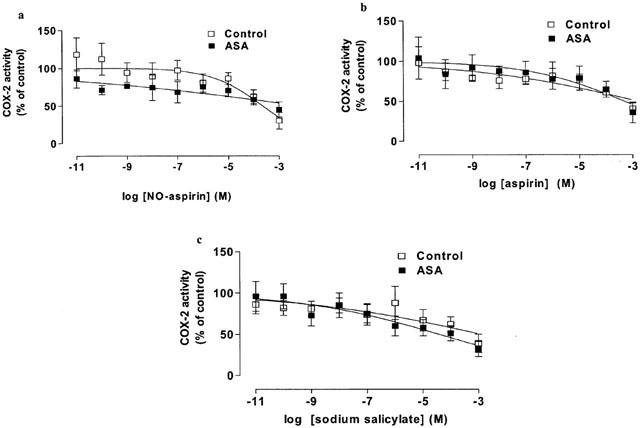
The inhibitory effect of (a) NO-aspirin, (b) aspirin and (c) salicylate on COX-2 activity in blood from healthy donors and aspirin-sensitive asthmatics (ASA). Data represent enzyme activity calculated as percentage response from each group (n=5–6).
LO activity
Basal release of total 5-LO products (LTC, D and E4) in blood from control or aspirin-sensitive donors was undetectable. However, when stimulated with A23187, large amounts of LTs were released by blood from both patient groups (control: 39±3 vs aspirin sensitive asthmatics: 44±4 ng ml−1). In each case, no effect on LT production was seen in paired samples of blood where COX-1 activity had been inhibited by indomethacin, nimesulide or aspirin (n=4; data not shown). The lack of effect of COX inhibition on corresponding LO activity was apparent at all concentrations of the NSAIDs used. By contrast, albeit at high concentrations, when blood was treated with NO-aspirin, 5-LO activity was inhibited. The inhibitory effect of NO-aspirin on LT production was significant (P<0.05 for effect of concentration on whole curve) and of a similar magnitude in blood from both patient groups (Figure 5).
Figure 5.
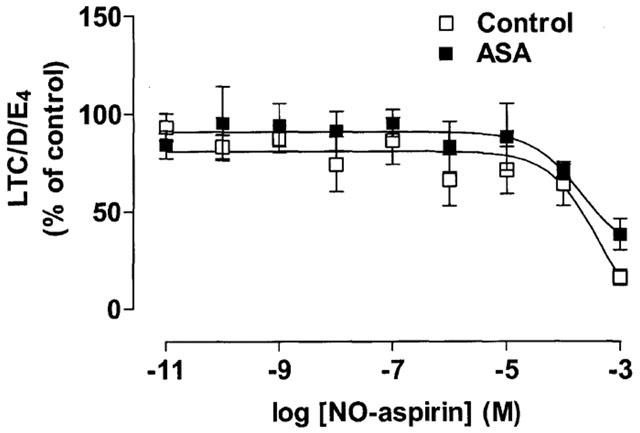
The effect of NO-aspirin on LT production by Ca ionophore A23187 stimulated blood from healthy donors and aspirin-sensitive asthmatics (ASA). Data represent enzyme activity calculated as percentage response from each group (n=4).
When blood was stimulated with LPS to induce COX-2, LT levels did not increase and remained undetectable (n=3; data not shown).
Discussion
Aspirin-sensitive asthma is a significant clinical problem affecting between 5 and 20% of the asthmatic population (Babu & Salvi, 2000). The common consensus of opinion suggests that aspirin-sensitive asthma has a biochemical origin and is mediated by blocking the COX pathway. However, there are still a number of important questions about this condition that remain unanswered or controversial. Thus, the relative relationship between COX-1, COX-2, 5-LO and LTC4 synthase activity in aspirin sensitive asthma is unclear. In the current study we have used the whole blood assay for COx-1 and COX-2 (Warner et al., 1999) and demonstrated no difference in potency of standard NSAIDs (except for nimesulide) or the COX-2 selective inhibitor DFP of activity in blood from aspirin sensitive asthmatics compared to tolerant control subjects. Furthermore, we have shown that when COX activity is blocked with NSAIDs, including aspirin, 5-LO products are not released in excess. Finally, we found that NO-aspirin, unlike authentic aspirin, reduced LT production in blood from aspirin-sensitive and aspirin tolerant individuals.
Aspirin-sensitivity is a complex clinical cohort. The organ systems influenced by aspirin ingestion inclue the lungs, skin, eyes and gastro-intestinal tract. Furthermore, it is not fully established that all forms of aspirin-sensitivity are a true NSAID-class effect or if, in some cases, the response is due to allergy based mechanisms to the drug itself and independent of effects on PG formation. Indeed, the vast majority of studies investigating the mechanisms of aspirin-sensitive asthma use a single NSAID, namely aspirin as the chosen COX inhibitor. Nevertheless, numerous studies have established that, in sensitive individuals, increased levels of the 5-LO product, LTC4 and its derivatives are present in the urine and correlate to the severity of symptoms (Knapp et al., 1992). This observation together with others showing that (i) aspirin-sensitivity is associated with the increased expression of LTC4 synthase and (ii) inhibitors of the LTC4 production and activity limit aspirin-induced responses has firmly established a role for LTs in this condition.
We found that LTC4, D4 and E4 were released in similar levels by A23187-stimulated blood from the two patient groups. Thus we did not observe a predicted elevation in LTC4 synthase products in blood from aspirin-sensitive patients. Similarly no differences in the levels of either TxB2 (for the COX-1 assay) or PGE2 (for the COX-2 assay) were seen in stimulated blood from the two patient groups. Furthermore, we found that when COX-1 activity in whole blood stimulated with A23187 was blocked with aspirin, indomethacin or nimesulide there was no associated increase in LTC4 or its derivative in samples from either aspirin-tolerant control donors or from aspirin-sensitive asthmatic patients. Others have shown using leukocytes isolated from aspirin-sensitive patients that aspirin alone, or in combination with N-formyl-methionyl-leucyl-phenylalanine, does not increase LT release (Pierzchalska et al., 2000). By contrast, May et al. (1999), describing results for a cellular antigen stimulation test, found that aspirin in combination with C5a in vitro caused leukocytes from aspirin intolerant patients to release higher levels of LTs than control subjects. Interestingly, May and co-workers showed that indomethacin but not diclofenac or ibuprofen displayed cross-reactivity with aspirin in their model. Thus, the data from our current study and others (Pierzchalska et al., 2000) suggest that, in peripheral cells at least, aspirin-sensitivity is not associated with a biochemical disruption in the balance between COX and LO products. Whether or not the same is true for cells of the airway remains the subject of investigation.
As mentioned above, the potency of aspirin or indomethacin as inhibitors of COX-1 activity in whole blood was the same in samples from aspirin-sensitive and aspirin tolerant patients. In addition, when COX-2 was induced in blood from aspirin-sensitive asthmatics, the potency of the COX-2 selective inhibitor, DFP was equivalent to that seen in blood from tolerant donors. This observation suggests that there is no anomaly in the biochemical properties of the COX-1 or COX-2 in aspirin-sensitive asthma. By contrast to aspirin, indomethacin or DFP, we found that the potency of nimesulide was slightly, but significantly, reduced in both the COX-1 and COX-2 assays using blood from aspirin-sensitive donors. The reason why the potency of nimesulide, but not other NSAIDs, may be altered in aspirin-sensitive asthma is not clear. Moreover, the relevance of such small differences may not be manifest in an in vivo setting. Nevertheless, the reasons why this observation has been made should be discussed. Nimesulide is not a pro-drug and as such does not require metabolism for activity. However, it may be that nimesulide itself is metabolized or bound in some way that renders it less active in aspirin-sensitive asthma. Interestingly, in a limited number of studies, nimesulide has been given to aspirin-sensitive asthmatics without inducing symptoms. Nimesulide displays a moderate selectivity as an inhibitor of COX-2 over COX-1 (Warner et al., 1999). Recently another COX-2 selective NSAID, rofecoxib, has been given to aspirin-sensitive asthmatics without inducing symptoms (Szczeklik et al., 2001). The tolerability of COX-2 selective (or COX-1 sparing; Vane & Warner, 2000) drugs has led to the suggestion that there is a specific involvement of COX-1 in aspirin-sensitivity (Szczeklik et al., 2001). This concept may appear, at first, counterintuitive since asthma is a chronic inflammatory disease with the certain presence of COX-2 at the site of inflammation (Sousa et al., 1997). However, we have recently shown that, despite the presence of active COX-2 in cytokine-stimulated human airway smooth muscle, the ability of NSAIDs to stimulate the release of granulocyte-macrophage colony stimulating factor (the presence of which is associated with allergic airway disease) correlated with their selectivity as inhibitors of COX-1 (Lazzeri et al., 2001).
In our assay, aspirin appears to produce only a weak inhibition of COX-2; this is because the incubation time required to observe COX-2 activity allows for almost total breakdown of the drug to salicylate (Higgs et al., 1987). Salicylate itself is very easily displaced by endogenous or exogenous substrate (Mitchell et al., 1997). Thus, under these conditions, aspirin (i.e. in salicylate form) as well as authentic salicylate are weak inhibitors of COX-2 activity in the blood of either patient group.
NO-aspirin is a relatively novel pharmaceutical preparation that has been shown to have fewer gastrointestinal side effects in the rat than authentic aspirin (Takeuchi et al., 1998). We found that, like aspirin, NO-aspirin inhibited COX-1 activity in the blood of aspirin-sensitive asthmatics and of aspirin tolerant controls. However, we also found that, unlike any of the other NSAIDs tested, NO-aspirin inhibited the release of LTs by blood from both patient groups. Our understanding of the pharmacology of NO-aspirin is incomplete. However, it has been suggested that the release of NO or a related NO-moiety, is responsible for the gastroprotective effects of this compound. Specifically, NO-NSAIDs are examples of bifunctional donors, where the pharmacological actions of a drug class like NSAIDs are retained and conjugated to a NO-moiety, which conceivably releases NO. There are two classes of NO-NSAIDs depending on whether they contain the nitrate ester functional group, as is the case for NO-aspirin or a S-nitrosothiol. It is thought that cleavage at the ester bond, by esterases releases a NO species. The structure of NO-aspirin consists of several ester groups, acetate, benzoate and nitrate esters. One study has investigated the metabolism of NO-aspirin in vitro, by incubating it with sub-cellular fractions of rat liver (Carini et al., 2001). Salicylate and benzenemethanol-3-hydroxy-α-nitrate (HBN) are the main metabolites. The latter is then rapidly metabolized to an unknown compound and the remainder converted to benzenemethanol-3-hydroxy (HBA). Carini et al. (2001) postulate that NO-aspirin undergoes first pass metabolism in the liver and the biotransformation of HBN, which may lead to the production of excretory nitrates or the conversion of bioactivate NO. This research group has also provided evidence using nitrosylhemoglobin complex (marker of NO formation) that NO-aspirin slowly releases NO, which is detectable in the blood of a rat given orally.
Thus, it is possible that NO-aspirin inhibits LT release by stimulated whole blood via an NO-like pathway. This notion is supported by some reports in the literature. For example, endogenously released NO inhibits the production of 5-LO metabolites in macrophages (Brunn et al., 1997), an effect that may occur at the level of 5-LO activating protein (Coffey et al., 2000). NO has also been shown to interfere with the iron binding site of 5-LO (Kanner et al., 1992). However, the precise mechanisms of action of NO-aspirin on LTs synthesis remains the subject of investigation.
In conclusion, we found no differences in the pharmacology of standard and novel NSAIDs on COX-1 or COX-2 activity in blood from aspirin-tolerant vs aspirin-sensitive individuals. Furthermore, when COX activity was blocked we did not see an associated increase in the level of LTs released. Finally, unlike authentic aspirin, NO-aspirin inhibited the release of LTs by stimulated blood from either patient group. These observations suggest that, in blood borne cells there is (i) no defect in the biochemical function of COX-1 or COX-2, that (ii) 5-LO and/or LTC4 synthase is not rate limiting and that (iii), there is no COX-dependent brake on LT production. NO-aspirin reduced the production of LT in vitro suggesting that this form of aspirin may have a therapeutic relevance for the treatment of inflammatory conditions where LTs are elevated, e.g. asthma. However, this hypothesis would require further and more direct experimentation before firm conclusions could be drawn.
Abbreviations
- AA
arachidonic acid
- Ca
calcium
- COX
cyclo-oxygenase
- DFP
[5,5-dimethyl-3-(2-isopropoxy)-4-(4-methanesulfonylphenyl)-2(5H)-furanone]; 15-HETE, 15-hydroxyeicosatetraenioc acid
- LPS
lipolysaccharide
- LT
leukotrienes
- NO-aspirin
(nitric oxide releasing aspirin NCX 4016)
- NO
nitric oxide, NSAIDs, non-steroidal anti-inflammatory drugs
- PG
prostaglandin
- TxB2
thromboxane B2
References
- BABU K.S., SALVI S.S. Aspirin and asthma. Chest. 2000;118:1470–1476. doi: 10.1378/chest.118.5.1470. [DOI] [PubMed] [Google Scholar]
- BELVISI M.G., SAUNDERS M., YACOUB M., MITCHELL J.A. Expression of cyclo-oxygenase-2 in human airway smooth muscle is associated with profound reductions in cell growth. Br. J. Pharmacol. 1998;125:1102–1108. doi: 10.1038/sj.bjp.0702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELVISI M.G., SAUNDERS M.A., HADDAD EL B., HIRST S.J., YACOUB M.H., BARNES P.J., MITCHELL J.A. Induction of cyclo-oxygenase-2 by cytokines in human cultured airway smooth muscle cells: novel inflammatory role of this cell type. Br. J. Pharmacol. 1997;120:910–916. doi: 10.1038/sj.bjp.0700963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUNN G., HEY C., WESSLER I., RACKE K. Endogenous nitric oxide inhibits leukotriene B4 release from rat alveolar macrophages. Eur. J. Pharmacol. 1997;326:53–60. doi: 10.1016/s0014-2999(97)00136-2. [DOI] [PubMed] [Google Scholar]
- CARINI M., ALDINI G., STEFANI R., ORIOLI M., FACINO R.M. Nitrosylhemoglobin, an unequivocal index of nitric oxide release from nitroaspirin: in vitro and in vivo studies in the rat by ESR spectroscopy. J. Pharm. Biomed. Anal. 2001;26:509–518. doi: 10.1016/s0731-7085(01)00478-2. [DOI] [PubMed] [Google Scholar]
- CHRISTIE P.E., SPUR B.W., LEE T.H. The effect of inhalation of the leukotriene receptor antagonist, SK&F 104353, on leukotriene C4- and leukotriene E4-induced bronchoconstriction in subjects with asthma. J. Allergy Clin. Immunol. 1991;88:193–198. doi: 10.1016/0091-6749(91)90328-l. [DOI] [PubMed] [Google Scholar]
- CLARIA J., SERHAN C.N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. U.S.A. 1995;92:9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COFFEY M.J., PHARE S.M., PETERS-GOLDEN M. Prolonged exposure to lipopolysaccharide inhibits macrophage 5-lipoxygenase metabolism via induction of nitric oxide synthesis. J. Immunol. 2000;165:3592–3598. doi: 10.4049/jimmunol.165.7.3592. [DOI] [PubMed] [Google Scholar]
- DOCHERTY J.C., WILSON T.W. Indomethacin increases the formation of lipoxygenase products in calcium ionophore stimulated human neutrophils. Biochem. Biophys. Res. Commun. 1987;148:534–538. doi: 10.1016/0006-291x(87)90909-0. [DOI] [PubMed] [Google Scholar]
- GRAY P.A., WARNER T.D., VOJNOVIC I., PARIKH A., SCADDING G., DEL SODALTO P., MITCHELL J.A. Leukotriene production in human blood is reduced by NO-aspirin but not aspirin: comparisons between responses in healthy volunteers and aspirin-sensitive asthma. Br. J. Pharmacol. 2000;129 Proceedings Suppl.:P1–P296. [Google Scholar]
- HIGGS G.A., SALMON J.A., HENDERSON B., VANE J.R. Pharmacokinetics of aspirin and salicylate in relation to inhibition of arachidonate cyclooxygenase and antiinflammatory activity. Proc. Natl. Acad. Sci. U.S.A. 1987;84:1417–1420. doi: 10.1073/pnas.84.5.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLTZMAN M.J., TURK J., SHORNICK L.P. Identification of a pharmacologically distinct prostaglandin H synthase in cultured epithelial cells. J. Biol. Chem. 1992;267:21438–21445. [PubMed] [Google Scholar]
- KANNER J., HAREL S., GRANIT R. Nitric oxide, an inhibitor of lipid oxidation by lipoxygenase, cyclooxygenase and hemoglobin. Lipids. 1992;27:46–49. doi: 10.1007/BF02537058. [DOI] [PubMed] [Google Scholar]
- KNAPP H.R., SLADEK K., FITZGERALD G.A. Increased excretion of leukotriene E4 during aspirin-induced asthma. J. Lab. Clin. Med. 1992;119:48–51. [PubMed] [Google Scholar]
- KOSHINO T., TAKANO S., HOUJO T., SANO Y., KUDO K., KIHARA H., KITANI S., TAKAISHI T., HIRAI K., ITO K., MORITA Y. Expression of 5-lipoxygenase and 5-lipoxygenase-activating protein mRNAs in the peripheral blood leukocytes of asthmatics. Biochem. Biophys. Res. Commun. 1998;247:510–513. doi: 10.1006/bbrc.1998.8789. [DOI] [PubMed] [Google Scholar]
- KOSHINO T., TAKANO S., KITANI S., OHSHIMA N., SANO Y., TAKAISHI T., HIRAI K., YAMAMOTO K., MORITA Y. Novel polymorphism of the 5-lipoxygenase activating protein (FLAP) promoter gene associated with asthma. Mol. Cell Biol. Res. Commun. 1999;2:32–35. doi: 10.1006/mcbr.1999.0147. [DOI] [PubMed] [Google Scholar]
- LAZZERI N., BELVISI M.G., PATEL H.J., YACOUB M.H., FAN CHUNG K., MITCHELL J.A. Effects of prostaglandin E2 and cAMP elevating drugs on GM-CSF release by cultured human airway smooth muscle cells. Relevance to asthma therapy. Am. J. Respir. Cell. Mol. Biol. 2001;24:44–48. doi: 10.1165/ajrcmb.24.1.4027. [DOI] [PubMed] [Google Scholar]
- MAY A., WEBER A., GALL H., KAUFMANN R., ZOLLNER T.M. Means of increasing sensitivity of an in vitro diagnostic test for aspirin intolerance. Clin. Exp. Allergy. 1999;29:1402–1411. doi: 10.1046/j.1365-2222.1999.00655.x. [DOI] [PubMed] [Google Scholar]
- MITCHELL J.A., BELVISI M.G. Too many COX (cyclo-oxygenase) spoil the broth: aspirin-sensitive asthma and 5-lypoxygenase. Thorax. 1997;52:933–935. doi: 10.1136/thx.52.11.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITCHELL J.A., BELVISI M.G., AKARASEREENONT P., ROBBINS R.A., KWON O.J., CROXTALL J., BARNES P.J., VANE J.R. Induction of cyclo-oxygenase-2 by cytokines in human pulmonary epithelial cells: regulation by dexamethasone. Br. J. Pharmacol. 1994;113:1008–1014. doi: 10.1111/j.1476-5381.1994.tb17093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITCHELL J.A., SAUNDERS M., BARNES P.J., NEWTON R., BELVISI M.G. Sodium salicylate inhibits cyclo-oxygenase-2 activity independently of transcription factor (nuclear factor kappaB) activation: role of arachidonic acid. Mol. Pharmacol. 1997;51:907–912. doi: 10.1124/mol.51.6.907. [DOI] [PubMed] [Google Scholar]
- MITCHELL J.A., WARNER T.D. Cyclo-oxygenase-2: pharmacology, physiology, biochemistry and relevance to NSAID therapy. Br. J. Pharmacol. 1999;128:1121–1132. doi: 10.1038/sj.bjp.0702897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NASSER S.M., BELL G.S., FOSTER S., SPRUCE K.E., MACMILLAN R., WILLIAMS A.J., LEE T.H., ARM J.P. Effect of the 5-lipoxygenase inhibitor ZD2138 on aspirin-induced asthma. Thorax. 1994;49:749–756. doi: 10.1136/thx.49.8.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIERZCHALSKA M., MASTALERZ L., SANAK M., ZAZULA M., SZCZEKLIK A. A moderate and unspecific release of cysteinyl leukotrienes by aspirin from peripheral blood leukocytes precludes its value for aspirin sensitivity testing in asthma. Clin. Exp. Allergy. 2000;30:1785–1791. doi: 10.1046/j.1365-2222.2000.00953.x. [DOI] [PubMed] [Google Scholar]
- SMITH W.L., MARNETT L.J., DEWITT D.L. Prostaglandin and thromboxane biosynthesis. Pharmacol. Ther. 1991;49:153–179. doi: 10.1016/0163-7258(91)90054-p. [DOI] [PubMed] [Google Scholar]
- SOUSA A., PFISTER R., CHRISTIE P.E., LANE S.J., NASSER S.M., SCHMITZ-SCHUMANN M., LEE T.H. Enhanced expression of cyclo-oxygenase isoenzyme 2 (COX-2) in asthmatic airways and its cellular distribution in aspirin-sensitive asthma. Thorax. 1997;52:940–945. doi: 10.1136/thx.52.11.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZCZEKLIK A., NIZANKOWSKA E., BOCHENEK G., NAGRABA K., MEJZA F., SWIERCZYNSKA M. Safety of a specific COX-2 inhibitor in aspirin-induced asthma. Clin. Exp. Allergy. 2001;31:219–225. doi: 10.1046/j.1365-2222.2001.01075.x. [DOI] [PubMed] [Google Scholar]
- TAKEUCHI K., UKAWA H., KONAKA A., KITAMURA M., SUGAWA Y. Effect of nitric oxide-releasing aspirin derivative on gastric functional and ulcerogenic responses in rats: comparison with plain aspirin. J. Pharmacol. Exp. Ther. 1998;286:115–121. [PubMed] [Google Scholar]
- TENOR H., HATZELMANN A., CHURCH M.K., SCHUDT C., SHUTE J.K. Effects of theophylline and rolipram on leukotriene C4 (LTC4) synthesis and chemotaxis of human eosinophils from normal and atopic subjects. Br. J. Pharmacol. 1996;118:1727–1735. doi: 10.1111/j.1476-5381.1996.tb15598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANE J.R., WARNER T.D. Nomenclature for COX-2 inhibitors. Lancet. 2000;356:1373–1374. doi: 10.1016/s0140-6736(00)02837-3. [DOI] [PubMed] [Google Scholar]
- WARNER T.D., GIULIANO F., VOJNOVIC I., BUKASA A., MITCHELL J.A., VANE J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7563–7568. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]