

Abstract
Inducible nitric oxide (iNOS) is thought to involve in host defence and tissue damage in inflammatory loci. In previous study, we have found that the endonuclease inhibitor aurintricarboxylic acid (ATA) can protect macrophages from cell death induced by bacterial lipopolysaccharide. This action is through the interruption with signalling pathways for NF-κB and AP-1 activation, and thus iNOS expression. In this study we have addressed the effects of ATA on JAK-STAT signalling pathways.
In murine RAW 264.7 macrophages, IFN-γ-mediated NO production and iNOS expression were concentration-dependently reduced by the presence of 3–100 μM ATA.
IFN-γ-induced STAT1 activation, as assessed from its tyrosine phosphorylation, nuclear translocation, binding to specific DNA response element and evoked IRF-1 reporter gene assay, were concomitantly inhibited by ATA. However, ATA did not alter IFN-γ binding to RAW 264.7 cells.
The activities of JAK1 and JAK2, the upstream kinases essential for STAT1 signalling in response to IFN-γ, were also reduced by ATA.
Moreover, IL-4, IL-10, GM-CSF and M-CSF elicited tyrosine phosphorylation of STAT3, STAT5 and/or STAT6 in macrophages were diminished by the presence of ATA.
Taken together, we conclude that ATA can interfere JAK-STAT signalling pathways in response to cytokines. This action contributes to the inhibition of IFN-γ-induced iNOS expression.
Keywords: Aurintricarboxylic acid, JAK, STAT, interferon-γ, cytokines, iNOS
Introduction
Aurintricarboxylic acid (ATA), a negatively charged triphenylmethane derivative (473 Da), has been demonstrated to prevent apoptosis in a variety of cell models (Escargueil-Blanc et al., 1997; Rosenbaum et al., 1998; Rui et al., 1998; Andrew et al., 1999; Vollgraf et al., 1999; Heiduschka & Thanos, 2000; Viergutz et al., 2000). The primary anti-apoptotic mechanism of ATA results from its function as an endonuclease inhibitor (Hallick et al., 1977; Batistatou & Greene, 1991). However, recent studies have further shown some endonuclease-independent actions of ATA. These include the inhibition of topoisomerases (Catchpoole & Stewart, 1994; Benchokroun et al., 1995), interferon-α receptors and NMDA receptors (Roberts-Lewis et al., 1993; Zeevald et al., 1993), but activation of mitogen-activated protein kinase (MAPK) (Okada & Koizumi, 1995; Rui et al., 1998), and erbB4 receptor (Okada & Koizumi, 1997). Recently, we also observed a novel action of ATA to inhibit inducible nitric oxide synthase (iNOS) expression in macrophages induced by bacterial lipopolysaccharide (LPS). This iNOS abrogation appears resulting from its non-specific action on the upstream signalling molecules, such as IκB kinase (IKK), extracellular signal-regulated kinase (ERK) and p38 MAPK (Tsi et al., 2002).
Cytokines comprise a large family of secreted glycoproteins that regulate fundamental biological processes including proliferation, differentiation, survival and specialized functions in host resistance to pathogens (Schindler & Darnell, 1995; Bromberg & Darnell, 2000). The functions of cytokines primarily relay on their binding to cell surface receptors and activating intracellular signal transduction cascades, primarily of the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway. The JAK-STAT pathway represents an extremely rapid membrane-to-nucleus signalling system. To date, four mammalian JAKs and seven mammalian STAT family members have been identified, which may be activated individually or in combination (Leonard & O'shea, 1998; Bowman et al., 2000; Seidel et al., 2000). The divergent and combinational coupling between JAKs, and STATs clarify part of the basis for the specificity of signals that are induced by a variety of cytokines.
Interferon-γ (IFN-γ) is well known to play crucial roles in several aspects of the immune responses (Boehm et al., 1997), and one action responsible for its inflammatory function results from activation of iNOS promoter and iNOS gene expression (Lowenstein et al., 1993; Sekine et al., 2000). To achieve pleiotropic biologic activities, including iNOS induction, signalling cascades initiated from receptors associated with JAK-STAT pathway is critical (Shui, 1994; Leonard & O'shea, 1998; Stark et al., 1998; Ramana et al., 2000). This pathway involves the cell membrane-localized JAK1 and JAK2 for tyrosine (Y701) phosphorylation and activation of latent cytoplasmic STAT1. After tyrosine phosphorylation of Y701, STAT1 forms a homodimer, and then translocates to the nucleus and binds to gamma-activated sequence (GAS) elements, which drive the expression of nearby target gene (Ramana et al., 2000). One of STAT1-induced gene products is interferon regulatory factor-1 (IRF-1). IRF-1 is a transcription factor, strongly inducible after IFNγ stimulation and participates in the transcription of many IFN-regulated secondary genes whose promoters contain IFN-stimulated regulatory elements (ISRE) (Sims et al., 1993; Kamijo et al., 1994). The well-characterized 1.7-kb fragment of the 5′-flanking region of the murine iNOS gene contains at least 10 copies of IFN-γ response elements (IRE), three copies of the GAS and two copies of the ISRE (Lowenstein et al., 1993; Xie et al., 1993). It has been found that the transcriptional induction of iNOS by IFN-γ depends on JAK/STAT activation (Lowenstein et al., 1993; Heitmeier et al., 1999), and IRF-1 is essential for iNOS induction by IFN-γ (Kamijo et al., 1994; Martin et al., 1994).
Following our findings showing that ATA can inhibit iNOS induction by LPS in macrophages (Tsi et al., 2002), it is interesting to further understand the effects of ATA on IFN-γ-induced iNOS expression. We, herein, have not only studied the effect of ATA on IFN-γ-stimulated iNOS induction, but also investigated its effects on multiple JAK-STAT signalling pathways elicited by IFN-γ, interleukin-6 (IL-6), interleukin-4 (IL-4), granulocyte-macrophage colony-stimulating factor (GM-CSF), and macrophage colony-stimulating factor (M-CSF). We present evidence showing that ATA can inhibit different JAK-STAT signalling pathways, and one of these actions leads to the inhibition of IFN-γ-mediated iNOS gene expression and NO production from macrophages.
Methods
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin, and streptomycin were obtained from Gibco BRL (Grand Island, NY, U.S.A.). Rabbit polyclonal antibody against iNOS was purchased from Transduction Laboratories (Lexington, KY, U.S.A.). Rabbit polyclonal antibodies against active (phosphorylated) STAT1 (Y701), STAT3 (Y705), STAT5 (Y694) and STAT6 (Y641) were purchased from New England Biolabs (Beverly, MA, U.S.A.). IFN-γ, IL-4, IL-10, M-CSF and GM-CSF were purchased from R&D (Minneapolis, MN, U.S.A.). [α-32P]ATP (3000 Ci/mmol) and [γ-32P]ATP (6000 Ci/mmol), HRP-coupled anti-mouse and anti-rabbit antibodies and the ECL detection agent were purchased from Amersham Pharmacia Biotech (Arlington Heights, IL, U.S.A.). Rabbit polyclonal antibodies specific for STAT1, STAT3, STAT5, STAT6, JAK1, JAK2, β-tubulin, and protein A/G agarose beads were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). The luciferase reporter construct of human IRF-1 gene promoter (−1.3 kilobase pairs) containing one of each GAS and κB element was kindly provided by Dr Yoshihiro Ohmori (Cleveland Clinic Foundation, Cleveland, OH, U.S.A.). Oligonucleotides of SIE (5′-ATCGTTCATTTCCCGTAAATCCCTA-3′) was synthesized by a PS250 CRUACHEM DNA synthesizer. ATA and other chemicals were purchased from Sigma (St. Louis, MO, U.S.A.). All materials for SDS–PAGE were obtained from Bio-Rad Laboratories (Hercules, CA, U.S.A.).
Cell culture
Murine RAW 264.7 macrophages and human colon adenocarcinoma cell line HT-29 obtained from American Type Culture Collection (Manassas, VA, U.S.A.) were grown at 37°C in 5% CO2 using DMEM containing 10% FBS, 100 U/ml penicillin and 100 μg ml−1 streptomycin.
NO assay
Measurement of nitrite production as an assay of NO release was performed. Accumulation of nitrite in the medium was determined by colorimetric assay with Griess reagent. The cells were treated with IFN-γ, ATA and/or the indicated agents for different intervals. Aliquots of conditioned media were mixed with an equal volume of Griess reagent (1% sulfanilamide and 0.1% N-(-naphthyl)-ethylenediamine in 5% phosphoric acid). Nitrite concentrations were determined by comparison with the OD550 using standard solutions of sodium nitrite prepared in cell culture medium. Each experiment was performed in duplicate and repeated for at least three times.
Western blot analysis
After incubation with IFN-γ or ATA, cells were washed twice in ice-cold PBS, and then solubilized in buffer containing 1% Triton X-100, 125 mM NaCl, 1 mM MgCl2, 25 mM β-glycerophosphate, 50 mM NaF, 100 μM sodium orthovanadate, 1 mM PMSF, 10 μg ml−1 aprotinin, 10 μg ml−1 leupeptin and 20 mM Tris-HCl, pH 7.5. Samples of equal amounts of protein (80 μg) were separated through SDS-10% polyacrylamide gels. After electrophoresis, proteins were transferred to nitrocellulose paper by semi-dry electrophoretic transfer. The membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 (TBST), and 5% nonfat milk for 1 h. Proteins were visualized by specific primary antibodies followed by peroxidase-labelled secondary antibodies. Immunoreactivity was detected by ECL reagents following the manufacturer's instructions, and exposure to X-ray film.
JAK assay
JAK1 and JAK2 autophosphorylation was assessed by immunoprecipitation kinase assay as we previously described (Tsi et al., 2002). Briefly, cells after drug treatment were washed twice with PBS (pH 7.4) containing 1 μM sodium orthovanadate and lysed in 1 ml of lysis buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 1% NP-40, 0.1% SDS, 1 mM EDTA (pH 7.4), 1 mM PMSF, 1 μg ml−1 leupeptin, 2 μg ml−1 aprotinin, 20 mM β-glycerophosphate, and 1 mM sodium orthovanadate. Total cell extracts were immunoprecipitated with anti-JAK1 or JAK2 antibody and proteinA/G-agarose beads at 4°C overnight. The precipitates were washed twice with lysis buffer, and then once with kinase buffer (25 mM HEPES, pH 7.5, 20 mM MgCl2, 100 μM sodium orthovanadate and 2 mM DTT). One half of the pellets were resuspended in 20 μl of kinase buffer containing 40 mM HEPES, 10 mM MgCl2, 10 mM MnCl2, 200 μM Na3VO4, 2 mM DTT, 21 μM ATP and 4 μCi [γ-32P]ATP, and then incubated at 30°C for 30 min. Beads were boiled in SDS sample buffer for 5 min. One half of the solubilized proteins were resolved by SDS–PAGE and quantified by phosphorimaging. The other half of the pellets were rendered by immunoblotting and measured with JAK1 or JAK2 antibody.
Reverse transcription-polymerase chain reaction (RT–PCR)
The expression of iNOS mRNA was determined by RT–PCR analysis. Macrophages were homogenized with 1 ml of RNAzol B reagent (Gibco), and total RNA was extracted by acid guanidinium thiocyanate-phenol-chloroform extraction. RT was performed using StrataScript RT–PCR Kit, and 10 μg of total RNA was reverse transcribed to cDNA following the manufacturer's recommended procedures. RT-generated cDNA encoding iNOS and β-actin genes were amplified using PCR. The oligonucleotide primers used correspond to the mouse macrophages iNOS (5′CCC TTC CGA AGT TTC TGG CAG CAG C-3′ and 5′-GGC TGT CAG AGC CTC GTG GCT TTG G-3′) and mouse β-actin (5′GAC TAC CTC ATG AAG ATC CT-3′ and 5′-CCA CAT CTG CTG GAA GGT GG-3′). PCR was performed in a final volume of 50 μl containing: Taq DNA polymerase buffer, all four dNTPs, oligonucleotide primers, Taq DNA polymerase, and RT products. After an initial denature for 2 min at 94°C, 35 cycles of amplification (94°C for 45 s, 65°C for 45 s, and 72°C for 2 min) were performed followed by a 10-min extension at 72°C. PCR products were analysed on 2% agarose gel. The mRNA of β-actin served as an internal control for sample loading and mRNA integrity.
Preparation of nuclear extracts and EMSA
Nuclear extracts were prepared as previously described (Hsu et al., 2001), and incubated in mixtures containing 0.25 μg poly (dI-dC), 20,000 r.p.m. of 32P-labelled STAT probe, 10 mM Tris, pH 7.5, 1 mM EDTA, 4% Ficoll, 1 mM DTT and 75 mM KCl. After 30 min incubation at room temperature, the DNA-protein complex was resolved in 5% polyacrylamide gel. The gels were dried and exposed to X-ray films.
Transfection and reporter gene assay
For transfection assays, 2×105 HT-29 cells were seeded into 12-well plates. Cells were transfected on the following day by lipofectamine 2000 (LF2000, Gibco). Premix DNA with Opti-MEM and LF2000 with Opti-MEM respectively for 5 min. Then the mixture was incubated for 25 min at room temperature, and were added into each well. After 24 h incubation, transfection was complete, and cells were incubated with the indicated concentrations of ATA and/or IFN-γ. After another 8 h incubation, the media were removed, and the cells were washed once with cold PBS. To prepare lysates, 50 μl of reporter lysis buffer (Promega) were added to each well, cells were scraped from dishes. The supernatant was collected after centrifugation at 13,000 r.p.m. for 30 s. Aliquots of cell lysates (5 μl) containing equal amounts of protein (10–20 μg) were placed into wells of an opaque black 96-well microplate. An equal volume of luciferase substrate (Promega) was added to all samples, and the luminescence was measured in a microplate luminometer (Meriden, CT, U.S.A.). Luciferase activity value was normalized to transfection efficiency monitored by the cotransfected β-galactosidase expression vector (pCR3lacZ; Pharmacia, Sweden). The level of induction of luciferase activity was determined as a ratio in comparison to cells with no stimulation.
Flow cytometric analysis of FITC-IFN-γ binding
To prepare FITC-labeled IFN-γ, 20 μg IFN-γ was added to 10 μg FITC (with a molar ratio of fluorescein to protein of 24 : 1) in buffer containing 10 mM NaHCO3 and 20 mM NaCl, pH 9.5. After incubation for 90 min at room temperature in the dark, FITC-IFN-γ conjugates were passed over a sephadex-G50 column to remove free FITC. RAW 264.7 cells were suspended in staining buffer (50 mM Na3PO4, pH 7.5, 100 mM KCl, 150 mM NaCl, 5% glycerol, 0.2% BSA) containing 2 μg FITC-IFN-γ for 60 min at 37°C in the presence of 100 μM ATA or not. After staining, cells were pelleted (400 g, 5 min), washed twice with PBS, and immediately analysed using FACScan and the Cellquest program (Becton Dickinson).
MTT assay
Cell viability was measured by colorimetric MTT assay. Mitochondria of viable cells are able to metabolize 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) to an insoluble formazan precipitate. Cells after drug treatment was incubated with 1 mg ml−1 MTT for 1 h at 37°C, followed by solubilization in DMSO. The coloured formazan product was measured by the absorbance at 550 nm on a microplate reader.
Statistical evaluation
Values were expressed as the mean±s.e.m. of at least three experiments, which were performed in duplicate. Analysis of variance (ANOVA) was used to assess the statistical significance of the differences, and a P value of less than 0.05 is considered statistically significant.
Results
IFN-γ-induced NO formation is attenuated by ATA at transcription level
Upon 1–10 ng ml−1 IFN-γ stimulation for 24 h, RAW 264.7 macrophages released a large amount of NO, as detected from nitrite formation (Figure 1a). In the presence of ATA (30 μM), the concentration-dependent NO increase induced by IFN-γ was diminished. The NO inhibition exhibited a concentration-dependency within 3–100 μM of ATA tested, and was accompanied by the reduced expression of iNOS protein (Figure 1b). To understand whether this action is associated with iNOS gene regulation, we performed RT–PCR analysis to determine iNOS mRNA level. Accordingly, iNOS mRNA induced by IFN-γ (3 ng ml−1) was inhibited by ATA in a concentration-dependent manner (Figure 1c). At these concentrations tested, ATA plus IFN-γ treatment had no significant effect on cell viability as assessed by MTT assay, which is an indicator of mitochondrial function for live cells (data not shown).
Figure 1.
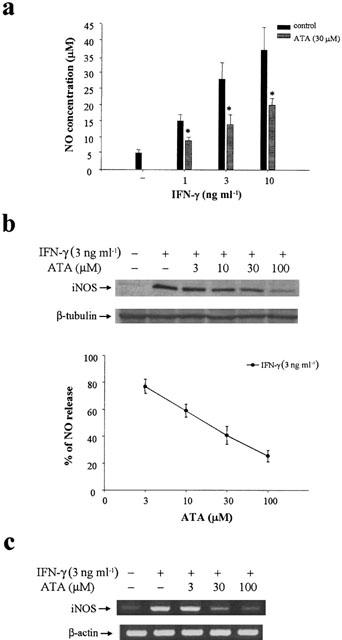
ATA inhibits IFN-γ-induced NO production, iNOS mRNA and protein expression in RAW264.7 macrophages. RAW264.7 cells were pretreated with ATA at the concentrations indicated for 20 min followed by the stimulation with IFN-γ at the concentrations indicated for 24 h (a, b) or 12 h (c). The stable NO metabolite nitrite present in the medium was analysed by the Griess method (a, b). Cell lysates were harvested, and an equal amount of protein was subjected to SDS–PAGE and detected with iNOS antibody (b). In some experiments, total mRNA from cells treated with ATA plus IFN-γ for 12 h was isolated, and RT–PCR for iNOS mRNA was performed (c). The results shown in (a) and (b) are the mean±s.e.m. from at least three independent experiments, each performed in duplicate. *P<0.05, indicates the significant inhibition by ATA treatment. The changes of iNOS protein and mRNA respectively shown in (b) and (c) are representative of three independent experiments with similar results. Internal controls of β-tubulin protein and β-actin mRNA were detected.
To further confirm that ATA inhibition of IFN-γ-induced iNOS induction occurs at the transcriptional level, we determined the time dependency of ATA action. The results indicated that the maximal inhibition of NO production (Figure 2b) and iNOS protein induction (Figure 2a) was obtained upon simultaneous treatment of ATA with IFN-γ. Delayed addition of ATA by 2 or 4 h still resulted in a partial inhibition, while 6 h delay almost lost this effect. This result suggests that ATA might interefere with IFN-γ-induced iNOS gene expression through uncoupling the upstream signalling cascade.
Figure 2.
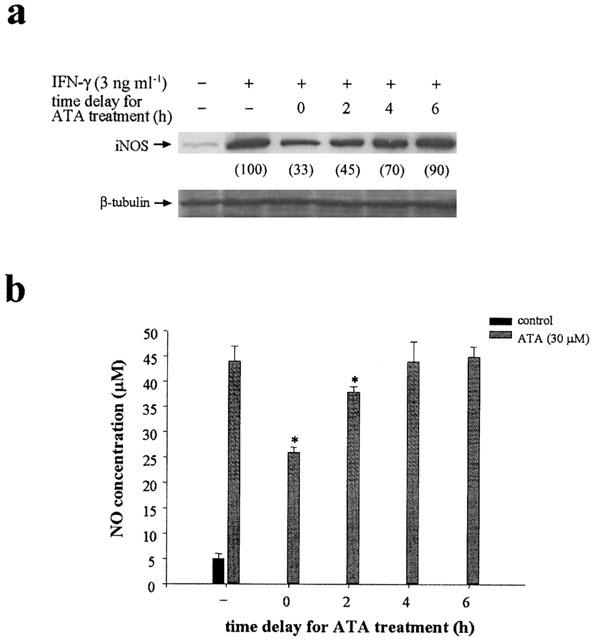
Time-dependent inhibition of IFN-γ induced iNOS expression and NO production by ATA. Cells were treated with ATA (30 μM) simultaneously as, or at different periods (2–6 h) after, IFN-γ (3 ng ml−1) stimulation. Twenty-four hours after IFN-γ stimulation, iNOS protein level (a) and NO production (b) were measured. The results shown in (a) are representative of three independent experiments. Data shown in (b) are the mean±s.e.m. from three independent experiments. *P<0.05, indicates the significant inhibition by ATA treatment.
ATA inhibits IFN-γ-induced STAT1 tyrosine phosphorylation and DNA-binding activity
Since Tyr701 phosphorylation of STAT1 following IFN-γ stimulation is associated with the priming effect on iNOS expression (Lowenstein et al., 1993; Heitmeier et al., 1999), we examined the effects of ATA on STAT1 phosphorylation and DNA binding. As shown in Figure 3a, upon treatment of RAW 264.7 macrophages with IFN-γ, a dramatic STAT1 phosphorylation at Tyr701was observed. ATA individually did not stimulate STAT1 phosphorylation, but concentration-dependently attenuated the response of IFN-γ. Consistent with this finding, nuclear level of the phosphorylated STAT1 increased with less extent in ATA treated groups (Figure 3b). In DNA binding assay, nuclear extracts of macrophages treated with IFN-γ were subjected to EMSA using SIE as a probe. As shown in Figure 3c, STAT1 binding to SIE was markedly induced after 1 h stimulation with IFN-γ, but was decreased by the pre-treatment with ATA for 20 min. This inhibitory effect of ATA also displayed a concentration-dependency within 10–100 μM.
Figure 3.
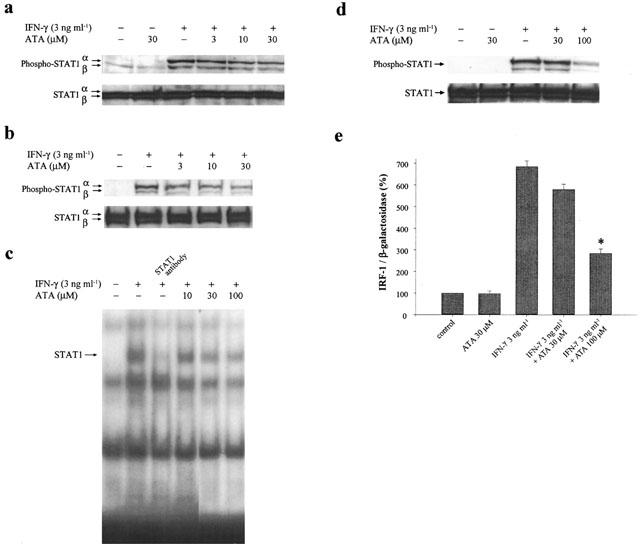
ATA inhibits IFN-γ mediated STAT1 phosphorylation, DNA binding and gene transactivation. RAW 264.7 macrophages (a, b, c) and HT-29 cells (d, e) were pretreated with ATA (3–100 μM) for 20 min, and then stimulated with 3 ng ml−1 IFN-γ for 15 min (a, b, d), 1 h (c) or 8 h (e). Total cell lysates (a, d, e) or nuclear lysates (b, c) were harvested for immunoblotting with STAT1 (a, b, d), probing with specific oligonucleotides containing binding sequences for STAT1 (c) or for luciferase assay (e). In (c), lane 3 indicated the gel shift assay, where antibody specific for STAT1 was included to detect the specific binding complex. The results in (a)–(d) are representative of three different experiments. The results in (e) are the mean±s.e.m. from three independent experiments. *P<0.05, indicates the significant inhibition by ATA treatment.
To further prove that the reduced DNA binding activity of STAT1 leads to a weak response on target gene expression, IRF-1 promoter activity was measured. In order to achieve a higher transfection efficiency, HT-29 cells instead of RAW 264.7 macrophages were used for transient transfection of IRF-1 reporter gene. In HT-29 cells, IFN-γ still kept the ability to transduce signalling from STAT1 phosphorylation to IRF-1 induction. In accordance with the effect seen in RAW264.7 macrophages, ATA at 100 μM prominently inhibited IFN-γ-elicited STAT1 phosphorylation (Figure 3d) and IRF-1 reporter activity (Figure 3e). Lower concentration of 30 μM did not alter both responses of IFN-γ in this cell type.
ATA inhibits IFN-γ-induced JAK activation
Previous studies have established the inhibitory effects of ATA on signalling protein kinases, such as IKK, ERK and p38 MAPK activation (Tsi et al., 2002), we next examined whether inhibition of IFN-γ-mediated STAT1 phosphorylation results from the action on JAK tyrosine kinase, whose activity is essential for Y701 phosphorylation and activation of latent cytoplasmic STAT1. Results of JAK1 and JAK2 kinase assays showed that the increased kinase activation by IFN-γ was reduced by ATA (3–100 μM) (Figure 4). Although Src is also an intermediate kinase to stimulate some STAT signalling (Yu et al., 1997; Chaturvedi et al., 1998), this consideration can be excluded by no effect of Src inhibitor, PP2 (up to 3 μM), to interrupt IFN-γ mediated NO response (data not shown).
Figure 4.

ATA inhibits JAK1 and JAK2 autophosphorylation. RAW 264.7 cells were pretreated with ATA at the concentrations indicated for 20 min followed by the stimulation with IFN-γ (3 ng ml−1) for 10 min. Whole cell lysates were immunoprecipitated with JAK1 or JAK2 antibody, and protein A/G beads overnight. One half of the immunoprecipitated JAK complex was subjected to SDS–PAGE, rendered for the immunoblotting, and probed with antibody specific for JAK. The other half of the immunoprecipitated JAK complex was rendered for kinase assay. After 30 min reaction at room temperature, the enzyme activity was terminated by adding SDS sample buffer and analysed by autoradiography. The results are representative of three different experiments with similar results.
ATA does not affect IFN-γ binding
Next we determined whether ATA inhibition of IFN-γ-mediated signalling results from the interference with ligand binding. Flow cytometric analysis demonstrated that there was no significant difference in the binding of FITC-IFN-γ in cells treated with 100 μM ATA or not (Figure 5).
Figure 5.
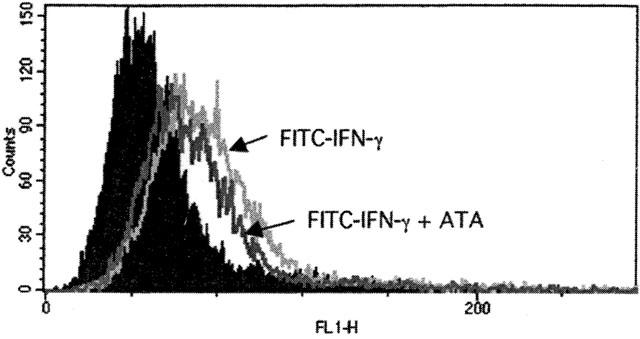
ATA does not inhibit IFN-γ binding. Cells pretreated with vehicle or ATA (100 μM) for 20 min were incubated with FITC-IFN-γ for 60 min at 37°C, followed by analysis using FACScan.
ATA inhibits cytokine-induced STATs phosphorylation in macrophages
To understand the specificity of ATA action on STAT1 signalling, we examined the signalling events of other STATs in macrophages. IL-4 and IL-10 are known to activate macrophage STAT6 and STAT3 respectively, while GM-CSF and M-CSF are known to activate macrophage STAT5 and/or STAT3 (Leonard & O'shea, 1998). The data from immunoblotting with antibodies specific for the tyrosine phosphorylated forms of STATs revealed that ATA can reduce all these four cytokines induced STAT phosphorylation at 10–100 μM (Figure 6). Significant reduction occurred at ATAT treatment with either 10 μM (for IL-4/STAT6 pathway) or 30 μM (for IL-10/STAT3, GM-CSF/STAT3 and STAT5, and M-CSF/STAT5 pathways).
Figure 6.
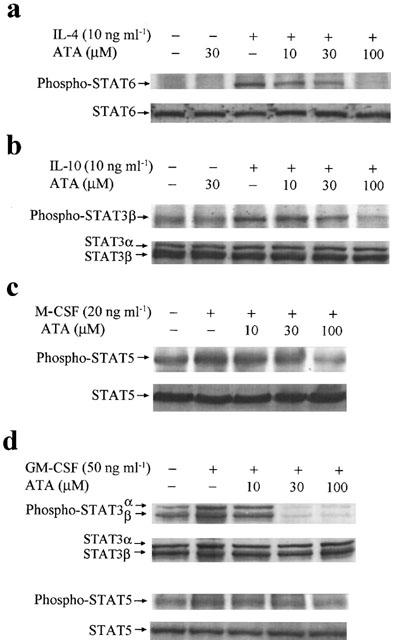
ATA inhibits STAT signalling pathways of cytokines. RAW 264.7 cells were pretreated with ATA at the concentrations indicated for 20 min, followed by the stimulation with IL-4 (10 ng ml−1), IL-10 (10 ng ml−1), M-CSF (20 ng ml−1) and GM-CSF (50 ng ml−1) for 15 min. Cell lysates were harvested for immunoblotting with STAT3, 5 or 6 antibody. The results are representative of three different experiments with similar results.
Discussion
The immunomodulatory actions of IFN-γ have been studied extensively. Macrophages, functioning as a key effect cell population in innate and adaptive immune responses, are known to be the targets for IFN-γ to accelerate Th1 cell development and to kill microbial targets. For the latter function, increases of iNOS expression and large amount of NO production are induced by IFN-γ. However, excessive NO production leads to injury of tissues adjacent to inflammatory sites, and development of chronic inflammatory process. Based on its potent bioactivity in inflammation, inhibition of iNOS production is a current strategy to develop novel anti-inflammatory drugs. In this study, we demonstrate for the first time a novel action of ATA to prevent IFN-γ-induced NO production, and this action resulting from inhibition of the upstream signallings responsible for iNOS gene regulation.
JAK-STAT1 pathway has been established to be the major signalling event for the multiple cellular functions of IFN-γ. In inducible NO production from macrophage, genetic detection of GAS, ISRE, and IRE in iNOS promoter (Lowenstein et al., 1993; Xie et al., 1993) and functional analysis (Lowenstein et al., 1993; Heitmeier et al., 1999) all indicate STAT1 as the crucial regulator for iNOS gene transcription. Measurements of iNOS mRNA level and JAK-STAT1 activity in the present study clearly indicate that ATA can reduce NO production through consequentially inhibiting the activities of JAK1 and JAK2, the tyrosine phosphorylation, nuclear translocation, and DNA binding ability of STAT1, and hence transactivation of iNOS gene. The inhibitory effect of ATA on IFN-γ is unrelated to the blockade of cytokine binding to its receptor. The concentration of ATA to achieve this signalling blockade, i.e. within 3–100 μM, is parallel to its anti-apoptotic action (Vollgraf et al., 1999; Heiduschka & Thanos, 2000; Viergutz et al., 2000) and some targets irrelevant to endonuclease inhibition (Roberts-Lewis et al., 1993; Zeevald et al., 1993; Okada and Koizumi, 1997; Tsi et al., 2002).
Besides STAT1-dependent signal cascade, we have also addressed the possible involvement of other signalling pathways in IFN-γ action. The reason for raising this concern is based on previous studies showing that some minor, but supplemented, signal pathways appear to contribute to the cell function of IFN-γ. In this regard, for example, a high concentration of IFN-γ was demonstrated to activate nuclear factor-κB (NF-κB) through tyrosine phosphorylation of IκBs in some cell types (Melillo et al., 1996; Diaz-Guerra et al., 1999), and to enhance NF-κB activation caused by TNF-α (Cheshire & Baldwin, 1997). Considering the possible involvement of NF-κB in IFN-γ-mediated iNOS gene expression, we tested NF-κB inhibitor, PDTC (Schreck et al., 1992), and performed an EMSA experiment in our system. As a result, in contrast to the effective blockade of LPS-induced NO release (Chen et al., 1998), PDTC did not alter IFN-γ-induced NO production (data not shown). Besides, we did not detect any stimulation of DNA binding to NF-κB in IFN-γ treated RAW 264.7 macrophages (data not shown). These negative results confirm previous reports showing that IFN-γ did not induce NF-κB activation (Sekine et al., 2000).
Accumulating evidence has demonstrated that MAPK signal pathways, including ERK and p38 MAPK, are crucial for iNOS gene expression (Chen et al., 1998; 1999; 2001; Kristof et al., 2001). IFN-γ was reported to activate Raf-1 (Sakatsume et al., 1998), ERK and PKC (Liu et al., 1994; Singh et al., 1996) in some cell systems, and in myocytes and microvascular endothelial cells, IFN-γ-induced iNOS gene expression requires ERK activity (Singh et al., 1996). To clear this concern of IFN-γ signalling in RAW 264.7 macrophages, we examined MAPK and PKC signalling pathways by pharmacological approaches. We found that pretreatment with 50 μM PD98059 (a MEK inhibitor), 3 μM SB203580 (a p38 MAPK inhibitor) and 1 μM Ro 31-8220 (a PKC inhibitor) did not affect IFN-γ-stimulated NO response (data not shown). By contrast, a JAK inhibitor, AG490 (30 μM) (Meydan et al., 1996), and a nonspecific tyrosine kinase inhibitor, genistein, all abrogate IFN-γ-induced NO production (data not shown). Thus, these results indicate that the activation of JAK-STAT1, but not PKC, ERK or p38 MAPK, signalling pathway is essential for iNOS induction by IFN-γ in macrophages.
Besides STAT1 is critical for IFN-γ functions in macrophages and innate immunity, IL-4, IL-10, GM-CSF and M-CSF all have been implicated as regulatory cytokines for macrophage function and immune response. For example, GM-CSF and M-CSF regulate proliferation, and cell survival during monocytic lineage development. IL-4 is a cytokine that participates in the immune response against parasitic infections (Urban et al., 1998) and in the development of allergic diseases (Henderson et al., 2000). On the contrary, IL-10 inhibits parasite killing and NO production from macrophages (Gazzinelli et al., 1992). The physiological functions of these cytokines relay on their binding to specific receptors, which leads to activation of different JAKs, and then phosphorylation of distinct tyrosine residues of the receptors. This results in the docking of distinct STATs onto the receptor through the Src homology domain, which subsequently is followed by the tyrosine phosphorylation, dimerization and nuclear translocation of STATs (Schindler & Darnell, 1995; Leonard & O'shea, 1998; Seidel et al., 2000). Although these four cytokines activate STAT3, 5, or 6 through distinct JAK members (JAK1 and JAK3 for IL-4, JAK1 and Tyk2 for IL-10, and JAK2 for GM-CSF), present data showed a common inhibition by ATA of these cytokine mediated signallings. Thus, ATA is a non-specific inhibitor to interrupt many cytokine-induced JAK-STAT signalling cascades in macrophages. In contrast to our present finding, Rui et al. (1998) observed an activation of the JAK2-STAT5 signalling pathway in Nb2 lymphoma cells by ATA alone at 250 μM. In that study, ATA also did not regulate STAT1 and STAT3. Currently, we cannot provide evidence to explain these discrepancies, but cell specificity and action relevant to concentration difference need to be considered.
In conclusion, as summarized in Figure 7, we have demonstrated that the endonuclease inhibitor ATA can effectively inhibit JAK-STAT signalling pathways in murine RAW 264.7 macrophages in response to different cytokines. This action contributes to the inhibition of iNOS gene expression and NO production caused by IFN-γ.
Figure 7.
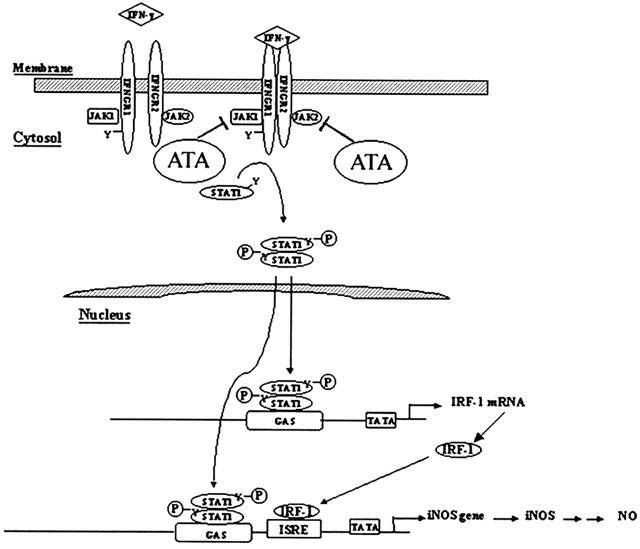
Summary of ATA inhibition of IFN-γ-mediated iNOS gene expression.
Acknowledgments
This study was supported by the grant from the National Science Council of Taiwan (NSC91-2314-B075-033).
Abbreviations
- ATA
aurintricarboxylic acid
- EMSA
electromobility gel shift assay
- ERK
extracellular signal-regulated kinase
- GAS
gamma-activated sequence
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- IFN-γ
interferon-γ
- IKK
IκB kinase
- IL-4
interleukin-4
- IL-6
interleukin-6
- IRE
IFN-γ-response elements
- ISRE
IFN-stimulated regulatory elements
- iNOS
inducible nitric oxide synthase
- IRF
interferon regulatory factor
- JAK
Janus kinases
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- M-CSF
macrophage colony-stimulating factor
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- NF-κB
nuclear factor-κB
- NO
nitric oxide
- PKC
protein kinase C
- RT–PCR
reverse transcription-polymerase chain reaction
- STAT
signal transducer and activator of transcription
References
- ANDREW D.J., HAY A.W., EVANS S.W. Aurintricarboxylic acid inhibits apoptosis and supports proliferation in a haemopoietic growth-factor dependent myeloid cell line. Immunopharmacology. 1999;41:1–10. doi: 10.1016/s0162-3109(98)00049-6. [DOI] [PubMed] [Google Scholar]
- BATISTATOU A., GREENE L.A. Aurintricarboxylic acid rescues PC12 cells and sympathetic neurons from cell death caused by nerve growth factor deprivation: correlation with suppression of endonuclease activity. J. Cell Biol. 1991;115:461–471. doi: 10.1083/jcb.115.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENCHOKROUN Y., COUPRIE J., LARSEN A.K. Aurintricarboxylic acid, a putative inhibitor of apoptosis, is a potent inhibitor of DNA topoisomerase II in vitro and in Chinese hamster fibrosarcoma cells. Biochem. Pharmacol. 1995;49:305–313. doi: 10.1016/0006-2952(94)00465-x. [DOI] [PubMed] [Google Scholar]
- BOEHM U., KLAMP T., GROOT M., HOWARD J.C. Cellular responses to interferon-γ. Annu. Rev. Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- BOWMAN T., GARCIA R., TURKSON J., JOVE R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- BROMBERG J., DARNELL J.E. The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- CATCHPOOLE D.R., STEWART B.W. Inhibition of topoisomerase II by aurintricarboxylic acid: implications for mechanisms of apoptosis. Anticancer Res. 1994;14:853–856. [PubMed] [Google Scholar]
- CHATURVEDI P., REDDY M.V., REDDY E.P. Src kinases and not JAKs activate STATs during IL-3 induced myeloid cell proliferation. Oncogene. 1998;16:1749–1758. doi: 10.1038/sj.onc.1201972. [DOI] [PubMed] [Google Scholar]
- CHEN B.C., CHEN Y.H., LIN W.W. Involvement of p38 mitogen-activated protein kinase in lipopolysaccharide-induced iNOS and COX-2 expression in J774 macrophages. Immunology. 1999;97:124–129. doi: 10.1046/j.1365-2567.1999.00747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN B.C., CHOU C.F., LIN W.W. Pyrimidinoceptor-mediated potentiation of inducible nitric-oxide synthase induction in J774 macrophages. J. Biol. Chem. 1998;273:29754–29763. doi: 10.1074/jbc.273.45.29754. [DOI] [PubMed] [Google Scholar]
- CHEN B.C., HSIEH S.L., LIN W.W. Involvement of protein kinases in the potentiation of lipopolysaccharide-induced inflammatory mediator formation by thapsigargin in peritoneal macrophages. J. Leukoc. Biol. 2001;69:280–288. [PubMed] [Google Scholar]
- CHESHIRE J.L., BALDWIN A.S., JR Synergistic activation of NF-κB by tumor necrosis factor α and γ interferon via enhanced IκBα degradation and de novo IκBβ degradation. Mol. Cell. Biol. 1997;17:6746–6754. doi: 10.1128/mcb.17.11.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIAZ-GUERRA M.J.M., CASTRILLO A., MARTIN-SANZ P., BOSCA L. Negative regulation by protein tyrosine phosphatase of IFN-γ-dependent expression of inducible nitric oxide synthase. J. Immunol. 1999;162:6776–6783. [PubMed] [Google Scholar]
- ESCARGUEIL-BLANC I., MEILHAC O., PIERAGGI M.T., ARNAL J.F., SALVAYRE R., NEGRE-SALVAYRE A. Oxidized LDLs induce massive apoptosis of cultured human endothelial cells through a calcium-dependent pathway. Prevention by aurintricarboxylic acid. Arterioscler. Thromb. Vasc. Biol. 1997;17:331–339. doi: 10.1161/01.atv.17.2.331. [DOI] [PubMed] [Google Scholar]
- GAZZINELLI R.T., OSWALD I.P., JAMES S.L., SHER A. IL-10 inhibits parasite killing and nitrogen oxide production by IFN-γ-activated macrophages. J. Immunol. 1992;148:1792–1796. [PubMed] [Google Scholar]
- HALLICK R.B., CHELM B.K., GRAY P.W., OROZCO E.M. Use of aurintricarboxylic acid as an inhibitor of nucleases during nucleic acid isolation. Nucleic Acids Res. 1977;4:3055–3064. doi: 10.1093/nar/4.9.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEIDUSCHKA P., THANOS S. Aurintricarboxylic acid promotes survival and regeneration of axotomised retinal ganglion cells in vivo. Neuropharmacology. 2000;39:889–902. doi: 10.1016/s0028-3908(99)00245-2. [DOI] [PubMed] [Google Scholar]
- HEITMEIER M.R., SCARIM A.L., CORBETT J.A. Prolonged STAT1 activation is associated with interferon-γ priming for interleukin-1-induced inducible nitric-oxide synthase expression by islets of Langerhans. J. Biol. Chem. 1999;274:29266–29273. doi: 10.1074/jbc.274.41.29266. [DOI] [PubMed] [Google Scholar]
- HENDERSON W.R., JR, CHI E.Y., MALISZEWSKI C.R. Soluble IL-4R inhibits airway inflammation following allergen challenge in a mouse model of asthma. J. Immunol. 2000;164:1086–1095. doi: 10.4049/jimmunol.164.2.1086. [DOI] [PubMed] [Google Scholar]
- HSU Y.W., CHI K.H., HUANG W.C., LIN W.W. Ceramide inhibits lipopolysaccharide-mediated nitric oxide synthase and cyclooxygenase-2 induction in macrophages: effects on protein kinases and transcription factors. J. Immunol. 2001;166:5388–5397. doi: 10.4049/jimmunol.166.9.5388. [DOI] [PubMed] [Google Scholar]
- KAMIJO R., HARADA H., MATSUYAMA T., BOSLAND M., GERECITANO J., SHAPIRO D., LE J., KOH S.I., KIMURA T., GREEN S.J., MAK T.W., TANIGUCHI T., VILCEK J. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- KRISTOF A.S., MARKS-KONCZALIK J., MOSS J. Mitogen-activated protein kinases mediate activator protein-1-dependent human inducible nitric-oxide synthase promoter activation. J. Biol. Chem. 2001;276:8445–8452. doi: 10.1074/jbc.M009563200. [DOI] [PubMed] [Google Scholar]
- LEONARD W.J., O'SHEA J.J. JAKs and STATs: biological implications. Annu. Rev. Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- LIU M.K., BROWNSEY R.W., REINER N.E. Gamma interferon induces rapid and coordinate activation of mitogen-activated protein kinase (extracellular signal-regulated kinase) and calcium-independent protein kinase C in human monocytes. Infect. Immun. 1994;62:2722–2731. doi: 10.1128/iai.62.7.2722-2731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOWENSTEIN C.J., ALLEY E.W., RAVAL P., SNOWMAN A.M., SNYDER S.H., RUSSELL S.W., MURPHY W.J. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon g and lipopolysaccharide. Proc. Natl. Acad. Sci. U.S.A. 1993;90:9730–9734. doi: 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN E., NATHAN C., XIE Q.W. Role of interferon regulatory factor 1 in inductionof nitric oxide synthase. J. Exp. Med. 1994;180:977–984. doi: 10.1084/jem.180.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELILLO G., TAYLOR L.S., BROOKS A., COX G.W., VARESIO L. Regulation of inducible nitric oxide synthase expression in IFN-gamma-treated murine macrophages cultured under hypoxic conditions. J. Immunol. 1996;157:2638–2644. [PubMed] [Google Scholar]
- MEYDAN N., GRUNBERGER T., DADI H., SHAHAR M., ARPALA E., LAPIDOT Z., LEEDER J.S., FREEDMAN M., COHEN A., GAZIT A., LEVITZKI A., ROIFMAN C.M. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645–648. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- OKADA N., KOIZUMI S. A neuroprotective compound, aurintricarboxylic acid, stimulates the tyrosine phosphorylation cascade in PC12 cells. J. Biol. Chem. 1995;270:16464–16469. doi: 10.1074/jbc.270.27.16464. [DOI] [PubMed] [Google Scholar]
- OKADA N., KOIZUMI S. Tyrosine phosphorylation of ErbB4 is stimulated by aurintricarboxylic acid in human neuroblastoma SH-SY5Y cells. Biochem. Biophys. Res. Commun. 1997;230:266–269. doi: 10.1006/bbrc.1996.5934. [DOI] [PubMed] [Google Scholar]
- RAMANA C.V., GRAMMATIKAKIS N., CHERNOV M., NGUYEN H., GOH K.C., WILLIAMS B.R.G., STARK G.R. Regulation of c-myc expression by IFN-γ through Stat1-dependent and Stat1-independent pathways. EMBO J. 2000;19:263–272. doi: 10.1093/emboj/19.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTS-LEWIS J.M., MARCY V.R., ZHAO Y., VAUGHT J.L., SIMAN R., LEWIS M.E. Aurintricarboxylic acid protects hippocampal neurons from NMDA- and ischemia-induced toxicity in vivo. J. Neurochem. 1993;61:378–381. doi: 10.1111/j.1471-4159.1993.tb03583.x. [DOI] [PubMed] [Google Scholar]
- ROSENBAUM D.M., D'AMORE J., LLENA J., RYBAK S., BALKANY A., KESSLER J.A. Pretreatment with intraventricular aurintricarboxylic acid decreases infarct size by inhibiting apoptosis following transient global ischemia in gerbils. Annal. Neurol. 1998;43:654–660. doi: 10.1002/ana.410430515. [DOI] [PubMed] [Google Scholar]
- RUI H., XU J., MEHTA S., FANG H., WILLIAMS J., DONG F., GRIMLEY P.M. Activation of Jak2-Stat5 signaling pathway in Nb2 lymphoma cells by an anti-apoptotic agent, aurintricarboxylic acid. J. Biol. Chem. 1998;273:28–32. doi: 10.1074/jbc.273.1.28. [DOI] [PubMed] [Google Scholar]
- SAKATSUME M., STANCATO L.F., DAVID M., SILVENNOINEN O., SAHARINEN P., PIERCE J., LARNER A.C., FINBLOOM D.S. Interferon γ activation of Raf-1 is Jak1-dependent and p21ras-independent. J. Biol. Chem. 1998;273:3021–3126. doi: 10.1074/jbc.273.5.3021. [DOI] [PubMed] [Google Scholar]
- SCHINDLER C., DARNELL J.E. Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu. Rev. Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- SCHRECK R., MEIER B., MANNEL D.N., DROGE W., BAEUERLE P.A. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J. Exp. Med. 1992;175:1181–1194. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEIDEL H.M., LAMB P., ROSEN J. Pharmaceutical intervention in the JAK/STT signaling pathway. Oncogene. 2000;19:2645–2656. doi: 10.1038/sj.onc.1203550. [DOI] [PubMed] [Google Scholar]
- SEKINE N., ISHIKAWA T., OKAZAKI T., HAYASHI M., WOLLHEIM C.B., FUJITA T. Synergistic activation of NF-κB and inducible isoform of nitric oxide synthase induction by interferon-γ and tumor necrosis factor-α in INS-1 cells. J. Cell. Physiol. 2000;184:46–57. doi: 10.1002/(SICI)1097-4652(200007)184:1<46::AID-JCP5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- SHUI K. Interferon-activated signal transduction to the nucleus. Curr. Opin. Cell Biol. 1994;6:253–259. doi: 10.1016/0955-0674(94)90144-9. [DOI] [PubMed] [Google Scholar]
- SIMS S.H., CHA Y., ROMINE M.F., GAO P.Q., GOTTLIEB K., DEISSEROTH A.B. A novel interferon-inducible domain: structural and functional analysis of the human interferon regulator factor 1 gene promoter. Mol. Cell. Biol. 1993;13:690–702. doi: 10.1128/mcb.13.1.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINGH K., BALLIGAND J.L., FISCHER T.A., SMITH T.W., KELLY R.A. Regulation of cytokine-inducible nitric oxide synthase in cardiac myocytes and microvascular endothelial cells. J. Biol. Chem. 1996;271:1111–1117. doi: 10.1074/jbc.271.2.1111. [DOI] [PubMed] [Google Scholar]
- STARK G.R., KERR I.M., WILLIAMS B.R.G., SILVERMAN R.H., SCHREIBER R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- TSI C.J., CHAO Y., CHEN C.W., LIN W.W. Aurintricarboxylic acid protects cell death from LPS in macrophages: decrease iNOS induction via IKK, ERK, and p38 MAPK inhibition. Mol. Pharmacol. 2002;62:90–101. doi: 10.1124/mol.62.1.90. [DOI] [PubMed] [Google Scholar]
- URBAN J.F., NOBEN-TRAUTH N., DONALDSON D.D., MADDEN K.B., MORRIS S.C., COLLINS M., FINDELMAN F.D. IL-13, IL-4Rα, and STAT6 are required for the expulsion of the gastrointestinal nematode parasite. N. brasiliensis. Immunity. 1998;8:255–264. doi: 10.1016/s1074-7613(00)80477-x. [DOI] [PubMed] [Google Scholar]
- VIERGUTZ T., LOEHRKE B., POEHLAND R., BECKER F., KANITZ W. Relationship between different stages of the corpus luteum and the expression of the peroxisome proliferator-activated receptor γ protein in bovine large lutein cells. J. Reproduct. Fertility. 2000;118:153–161. [PubMed] [Google Scholar]
- VOLLGRAF U., WEGNER M., RICHTER-LANDSBERG C. Activation of AP-1 and nuclear factor-κB transcription factors is involved in hydrogen peroxide-induced apoptotic cell death of oligodendrocytes. J. Neurochem. 1999;73:2501–2509. doi: 10.1046/j.1471-4159.1999.0732501.x. [DOI] [PubMed] [Google Scholar]
- XIE Q.W., WISHNAN R., NATHAN C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J. Exp. Med. 1993;177:1779–1784. doi: 10.1084/jem.177.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU C.L., JOVE R., BRUAKOFF S.J. Constitutive activation of the janus kinase-STAT pathway in T lymphoma overexpressing the Lck protein tyrosine kinase. J. Immunol. 1997;159:5206–5210. [PubMed] [Google Scholar]
- ZEEVALD G.D., SCHOEPP D., NICKLAS W.J. Aurintricarboxylic acid prevents NMDA-mediated excitotoxicity: evidence for its action as an NMDA receptor antagonist. J. Neurochem. 1993;61:386–389. doi: 10.1111/j.1471-4159.1993.tb03585.x. [DOI] [PubMed] [Google Scholar]