

Abstract
The mechanism(s) by which vascular endothelial growth factor (VEGF) induces endothelial nitric oxide synthase (eNOS) activation remain(s) unclear up to a certain extent. Therefore, we sought to evaluate the contribution of numerous pathways in VEGF-induced nitric oxide (NO) synthesis by measuring cGMP production. In addition, as VEGF induces the synthesis of NO and platelet-activating factor (PAF), we wanted to assess if the induction of PAF and NO is contributing to the synthesis of each other.
Herein, we show that a treatment of endothelial cells with a phospholipase C (PLC) inhibitor (U73122), a calmodulin antagonist (W-7) or with intracellular calcium chelators (EGTA/AM, BAPTA/AM) prevented VEGF-mediated eNOS Ser1177-phosphorylation and NO synthesis measured by cGMP production.
Pretreatment with phosphatidylinositol 3-kinase (PI3K) (Wortmannin, LY294002) or protein kinase C (PKC) (GF109203X, Ro318220) inhibitors attenuated eNOS Ser1177-phosphorylation mediated by VEGF, but did not alter immediate (0–10 min) cGMP synthesis induced by VEGF, but abrogated by up to 84% the delayed (10–30 min) cGMP synthesis.
Pretreatment with PAF synthesis inhibitors or with PAF receptor antagonists did not abrogate neither eNOS Ser1177-phosphorylation nor cGMP synthesis mediated by VEGF.
In conclusion, VEGF induces an immediate cGMP synthesis through the PLC-Ca2+/CaM pathway, and that the induction of delayed cGMP synthesis implies Akt and PKC activity.
Keywords: NO, VEGF, PAF, eNOS, PKC, PLC, PI3K, PLA2
Introduction
It is now well established that angiogenesis, the formation of new blood vessels from pre-existing ones, is an essential requirement for numerous physiological conditions, such as wound healing, tissular regeneration, and uterine wall thickening (Folkman & Klagsbrun, 1987; Folkman, 1991). On the other hand, angiogenesis is also involved in numerous pathologies including tumour growth, atherosclerosis, and proliferative retinopathies (Folkman & Klagsbrun, 1987; Ferrara, 1995; Moulton et al., 1999). Induction of angiogenesis consists on a multistep process involving capillary endothelial cell (EC) migration and proliferation, inflammation and subsequently the formation of a three-dimensional structure capable of carrying blood (Folkman & Klagsbrun, 1987).
As vascular endothelial growth factor (VEGF) and other growth factors can promote EC migration and proliferation in vitro and angiogenesis in vivo (Unemori et al., 1992), they are consequently considered as major candidates for the regulation of physiological and pathophysiological angiogenesis (Ferrara & Davis-Smith, 1997). However, VEGF is the only growth factor capable of promoting vascular permeability and inflammation (Connolly et al., 1989). We first showed that VEGF effect on vascular permeability is mediated through platelet-activating factor (PAF) synthesis in EC (Sirois & Edelman, 1997). Then, reported that upon Flk-1/KDR phosphorylation, VEGF leads to the activation of p38, p42/44 mitogen-activated protein kinases (MAPK), group V secreted phospholipase A2 and lyso-PAF acetyltransferase which are required for the induction of VEGF-mediated PAF synthesis (Bernatchez et al., 1999; 2001a, b). Recently, it was shown that PAF synthesis contributes to VEGF-angiogenic activity (Montrucchio et al., 2000).
However, others reported that VEGF angiogenic and inflammatory activities can be mediated through nitric oxide (NO) synthesis (Ku et al., 1993; Ziche et al., 1997; Lakshminarayanan et al., 2000; Bussolati et al., 2001; Lal et al., 2001). Despite a significant number of reports, the mechanisms by which VEGF mediates NO synthesis are not so clear and even controversial. First, it has been reported in native and transfected endothelial cells that VEGF-mediated Flk-1/KDR-autophosphorylation is leading to downstream endothelial nitric oxide synthase (eNOS) activation (He et al., 1999; Feng et al., 1999; Wu et al., 1999; Kroll & Waltenberger, 1999; Thuringer et al., 2001), and secondly, that eNOS is a Ca2+/Calmodulin (Ca2+/CaM)-dependent enzyme activated by intracellular Ca2+ release upon phospholipase C-′γ (PLC-′γ) activation (Busse & Mulsch, 1990; Brock et al., 1991; Xia et al., 1996; Wu et al., 1999). However, a recent study claimed that VEGF-mediated NO synthesis is driven through Flt-1 rather than Flk-1/KDR activation (Bussolati et al., 2001). Then, it has been shown that VEGF activates phosphatidylinositol 3-kinase (PI3K) leading to Akt phosphorylation which phosphorylates eNOS, thereby increasing eNOS enzymatic activity (Papapetropoulos et al., 1997; Dimmeler et al., 1999; Fulton et al., 1999; Michell et al., 1999). However, it was recently reported that PI3K inhibition had no or minor effect on NO release (Fleming et al., 2001; Thuringer et al., 2001). In addition, it was shown that VEGF-mediated NO production results from a bimodal system in which immediate NO synthesis is observed from an eNOS calcium-dependent activation and that delayed NO production is dependent on eNOS phosphorylation induced by intracellular mediator such as heat shock protein 90 (Hsp90) and Akt (Brouet et al., 2001). Finally, another controversial intracellular mediator associated with eNOS regulation is protein kinase C (PKC). On one side, it has been demonstrated that PKC inhibition abrogates VEGF-induced NO release (He et al., 1999), whereas another study has demonstrated that PKC activation in EC inhibits eNOS activity (Michell et al., 2001).
As VEGF induces a rapid induction of NO and PAF synthesis in EC and that the intracellular mechanisms by which VEGF induces NO synthesis are still debatable, we first sought to assess the mechanisms involved in VEGF-mediated eNOS activation. Then, we investigated the contribution of PAF in VEGF-induced NO synthesis in endothelial cells.
Methods
Cell culture
Bovine aortic endothelial cells (BAEC) were isolated from freshly harvested bovine aortas, cultured in Dulbecco's modified Eagle medium (DMEM; Life Technologies, Burlington, ON, Canada) containing 5% foetal bovine serum (Hyclone Lab., Logan, UT, U.S.A.), and antibiotics (Sigma Chem., St-Louis, MO, U.S.A.). BAEC were characterized as described previously (Sirois & Edelman, 1997; Bernatchez et al., 1999). Cells were not passaged for more than nine cycles.
Measurement of cGMP synthesis
NO synthesis from BAEC was assessed by quantifying the intracellular accumulation of cyclic guanosine monophosphate (cGMP) by using a radioimmunoassay (RIA) kit (New England Nuclear, NEN, Boston, MA, U.S.A.) (NEN). Confluent BAEC (six-well tissue culture plate) were serum-starved in DMEM for 18 h. Cells were rinsed, and a DMEM/CaCl2 (5 mM) solution added to the cells. Cells were pretreated with isobutylmethylxanthine (IBMX, 500 μM), and with inhibitors or antagonists 10 min (except for U73122 and U73343 given 60 min) prior to stimulation with phosphate-buffered saline (PBS) or agonists. The media was removed and 500 μl of ethanol (65%) was added. The cells were scraped, proteins lyophilized and diluted in RIA buffer. A predetermined amount of the antigen ScGMP-TME-[125I] was added to the cGMP produced by BAEC. ScGMP-TME-[125I] and cGMP were incubated with the antiserum complex for 18 h. The samples were centrifuged and the supernatant discarded. The amount of ScGMP-TME-[125I] in the precipitate was measured with a γ′-counter. Finally, the determination of cGMP was made by an interpolation from a standard curve.
Western blot analyses of Akt (Serine 473) and eNOS (Serine 1177) phosphorylations
Confluent BAEC (100 mm tissue culture plate) were serum-starved for 18 h in DMEM and then stimulated with agonists for various periods of time as described for cGMP synthesis±inhibitors or antagonists. Cells were lysed (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 1.2% Triton X-100, 1 mM EGTA, 1 mM, phenylmethylsulphonyl fluoride, 0.15 u ml−1 aprotinin, 10 μg ml−1 leupeptin, 1 mM NaVO3, 1 mM Microcystin LR), the plates were scraped, the lysate clarified by centrifugation, and the protein concentration of the supernatant determined by using a protein assay kit (Bio-Rad, Hercules, CA, USA). Proteins (100–150 μg) were separated on a 7.5% SDS–PAGE and transblotted onto an immunobilon-P polyvinylidene difluoride membranes (Milipore, Bedford, MA, USA). Membranes were then incubated overnight in blocking buffer containing rabbit polyclonal anti-phospho-Akt (Ser473) or anti-phospho-eNOS (Ser1177) (dilution 1 : 1000) antibodies (dilution 1 : 300) (Cell Signaling Technology Inc., Beverly, MA, U.S.A.) (Santa Cruz Biotechnologies, Inc, Santa Cruz, CA, U.S.A.). Membranes were washed with TTBS and incubated with anti-rabbit antibodies coupled to horseradish peroxidase (Santa Cruz Biotechnologies). Membranes were washed with TTBS and horseradish peroxidase was revealed by chemiluminescence. Kaleidoscope pre-stained molecular weight standards (Bio-Rad) were used as standards for SDS–PAGE.
Measurement of PAF synthesis
Platelet-activating factor synthesis in BAEC was measured by the incorporation of 3H-acetate into lyso-PAF as described previously (Sirois & Edelman, 1997; Bernatchez et al., 1999). Briefly, confluent BAEC (six-well tissue culture plates) were stimulated for 15 min in 1 ml of DMEM–HEPES (10 mM, pH 7.4), CaCl2 (5 mM), 3H-acetate (25 μCi) (NEN) plus VEGF (1 nM) (human VEGF-A165, PeproTech, Rocky Hill, NJ, U.S.A.). In various experiments, inhibitors were added 15 min prior to VEGF (1 nM) treatment. The reaction was stopped by addition of acidified methanol (50 mM acetic acid), the cells were scraped, and lipids were extracted by the Bligh and Dyer method (Bligh & Dyer, 1959) and purified by high performance liquid chromatography (HPLC). Fractions corresponding to 3H-PAF were quantified by counting radioactivity with a γ-counter. The separation and detection of 3H-PAF was confirmed by comparison with the HPLC elution pattern of a commercial 3H-PAF standard (NEN), and by the ability of PAF-containing fractions to induce platelet aggregation when compared to standard alkyl-PAF (Sirois & Edelman, 1997).
Drugs
L-NAME, U73122, U73343, GF109203X, Ro318220, W-7, A23187, EGTA/AM, BAPTA/AM, Wortmannin, LY294002, SB203580 and PD98059 were purchased from Calbiochem (La Jolla, CA, U.S.A.). BN52021 and CV3988 were purchased from Biomol Research Laboratories, Inc., (Plymouth Meeting, PA, U.S.A.). Sodium nitroprusside (SNP) and IBMX were purchased from Sigma (St-Louis, MO, U.S.A.). Alkyl-PAF was purchased from BACHEM (Torrance, CA, U.S.A.). SB203347 was donated by Dr James D. Winkler (SmithKline Beecham Pharmaceuticals; King of Prussia, PA, U.S.A.). LAU8080 was synthesized at LSU Health Sciences Center, New Orleans, LA, U.S.A.
Statistical analysis
Data are mean±s.e.mean. Statistical comparisons were made by analysis of variance followed by a Bonferroni's t-test for multiple comparison. Data were considered significantly different if values of P<0.05 were observed.
Results
VEGF effect on cGMP synthesis
The induction of NO synthesis in BAEC was quantified by measuring the intracellular cGMP accumulation using a radioimmunoassay kit. First, we determined the cGMP synthesis induced by VEGF (1 nM) at 0, 2.5, 5, 10, 15 and 20 min post-treatment. VEGF induced a time-dependent cGMP synthesis with a maximal production at 10 min (13.6 fold increase) (1.76 pmol cGMP mg−1 total protein) as compared to PBS-treated group (0.12 pmol cGMP mg−1 total protein) (Figure 1A). Then, we investigated the effect of VEGF on cGMP synthesis at various concentrations (0.01–1 nM) during a 10-min stimulation period. VEGF maximal effect was achieved at 0.1 nM with a 12.5 fold increase as compared to PBS-treated group (Figure 1B). Because of slight variation of cGMP production between experiments, data were reported as relative cGMP production (%) to baseline.
Figure 1.
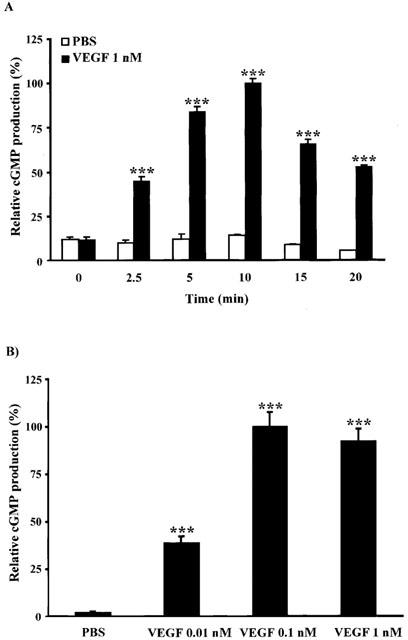
VEGF effect on cGMP production. BAEC were pretreated with (IBMX, 500 μM; 10 min) prior to VEGF stimulation in a DMEM/5 mM CaCl2 solution. Cells were scraped, proteins lyophilized and cGMP production detected by a RIA kit. (A) VEGF (1 nM) induced a time-dependent increase of cGMP production. (B) Effect of VEGF at various concentrations (0.01–1 nM) on cGMP production for a 10-min stimulation. ***P<0.001 as compared to control buffer (PBS).
Effect of eNOS and PLC pathway on VEGF-induced cGMP synthesis
First, we sought to confirm that the dose- and time-dependent effect of VEGF on cGMP synthesis was NO-dependent. Stimulation with an exogenous donor of NO (SNP, 500 μM) for 10 min induced a 14.5 fold increase in cGMP synthesis as compared to PBS-treated group (Figure 2A). Treatment with VEGF (1 nM; 10 min) induced a 13.6 fold increase which was completely blocked by a pretreatment with a competitive L-arginine analogue (L-NAME, 100 μM) (Figure 2A). Pretreatment of EC with a PLC inhibitor (U73122, 10 μM) blocked completely the VEGF-mediated cGMP synthesis, whereas its inactive analogue (U73343, 10 μM) had no effect (Figure 2A). Pretreatment with two structurally unrelated PKC inhibitors (GF109203X, 5 μM and Ro318220, 1 μM) did not prevent VEGF-induced cGMP synthesis. Pretreatment with a CaM antagonist (W-7, 250 μM) abolished the effect of VEGF on cGMP synthesis (Figure 2A).
Figure 2.

Effect of several pathway inhibitors on VEGF-induced cGMP production. BAEC were pretreated with IBMX (500 μM; 10 min), and with inhibitors or antagonists (up to 60 min) prior to VEGF stimulation in a DMEM/5 mM CaCl2 solution. cGMP production was detected by a RIA kit. Effect of VEGF (1 nM) on cGMP production upon a pretreatment with (A) eNOS inhibitor (L-NAME, 100 μM) or PLC pathway inhibitors (U73122, 10 μM; U73343, 10 μM; GF109203X, 5 μM; Ro318220, 1 μM or W-7, 250 μM). (B) cGMP synthesis upon stimulation with VEGF (1 nM) or A23187 (10 μM) on cGMP in a 5 mM CaCl2. Effect of VEGF (1 nM) on cGMP production upon a pretreatment with two chelators of calcium (EGTA/AM 100 μM or BAPTA/AM 10 μM) in a CaCl2-free solution. Effect of VEGF (1 nM) on cGMP production upon a pretreatment with (C) two PI3K inhibitors (Wortmannin, 500 nM or LY294002, 50 μM), (D) PAF mediator inhibitors (SB203580, 10 μM; PD98059, 10 μM or SB203347, 10 μM) or PAF receptor antagonists (BN52021 10 μM; LAU8080, 100 nM or CV3988, 1 μM). Effect of PAF (100 nM or 1 μM) on cGMP production. ***P<0.001 as compared to control buffer (PBS), †††P<0.001 as compared to VEGF (1 nM), *P<0.05 as compared to control buffer (PBS) in a DMEM CaCl2 free solution and †P<0.05 as compared to VEGF (1 nM) in a DMEM CaCl2 free solution.
Effect of Ca2+ chelators on VEGF-induced cGMP synthesis
We then wanted to assess the role of intracellular and extracellular Ca2+ on cGMP synthesis. A 10-min stimulation with the Ca2+ ionophore A23187 (10 μM) in presence of extracellular CaCl2 (5 mM) induced a 9.2 fold increase in cGMP synthesis (Figure 2B). The basal cGMP synthesis in an extracellular CaCl2-free DMEM solution was not different from the basal cGMP synthesis observed in the presence of CaCl2 (5 mM). However, stimulation with VEGF (1 nM) in a DMEM CaCl2-free solution induced only a 2.5 fold increase in cGMP synthesis as compared to PBS-treated group. Under the same condition (extracellular CaCl2-free solution), the addition of intracellular Ca2+ chelators such as EGTA/AM (100 μM) or BAPTA/AM (10 μM) abolished the residual cGMP synthesis mediated by a 10-min treatment with VEGF (Figure 2B).
Effect of PI3K inhibitors on VEGF-induced cGMP synthesis
Pretreatment with two unrelated PI3K inhibitors (Wortmannin, 500 nM or LY294002, 50 μM) did not prevent cGMP synthesis induced by VEGF (1 nM) for a 10-min stimulation (Figure 2C).
Effect of PAF on VEGF-induced cGMP synthesis
Selective inhibition of intracellular proteins involved in VEGF-mediated PAF synthesis, namely p38 MAPK (SB203580, 10 μM), p42/44MAPK (PD98059, 10 μM) or group V sPLA2 (SB203347, 10 μM) (Bernatchez et al., 2001a, b) did not prevent cGMP synthesis mediated by VEGF. Moreover, pretreatment with three different PAFR antagonists: extracellular PAFR antagonist (BN52021, 10 μM), intracellular PAFR antagonist (LAU8080, 100 nM) or extra- and intracellular PAF receptor antagonist (CV3988, 1 μM) (Marcheselli et al., 1990; Marcheselli & Bazan, 1994) had no inhibitory effect on cGMP synthesis mediated by VEGF (1 nM). Finally, stimulation of BAEC with exogenous PAF (100 nM and 1 μM) did not increase basal cGMP synthesis (Figure 2D).
Effect of VEGF, intracellular inhibitors, and calcium chelators on eNOS phosphorylation at Serine 1177
Treatment with VEGF (1 nM) induces a time-dependent increase in eNOS phosphorylation at Ser1177 in BAEC with a maximal phosphorylation within 10 min (Figure 3A). Treatment with VEGF (1 nM) for 10 min induced a 2.6 fold increase in eNOS phosphorylation as compared to PBS-treated group (Figure 3B). Pretreatment with L-NAME (100 μM) did not prevent VEGF-induced eNOS phosphorylation. Pretreatment with U73122 (10 μM) completely blocked not only VEGF-mediated eNOS phosphorylation but attenuated as well the basal phosphorylation level. The inactive analogue U73343 (10 μM) had only a marginal (10%) inhibitory effect on eNOS phosphorylation induced by VEGF. Pretreatment with GF109203X (5 μM) or Ro318220 (1 μM) attenuated VEGF-induced eNOS phosphorylation by 57 and 38% respectively. Finally, W-7 (250 μM) abolished both, the VEGF-induced and basal eNOS phosphorylation (Figure 3B).
Figure 3.

VEGF effect on eNOS phosphorylation at Serine 1177. Confluent BAEC were stimulated with agonists, cells were lysed and proteins were separated on a 7.5% SDS–PAGE. Western blot analysis was performed with rabbit polyclonal anti-phospho-eNOS (Ser1177) antibodies. (A) Time-dependent eNOS activation mediated by VEGF, the maximal phosphorylation was considered as control (100%). Studies (B–E) were performed with the same inhibitors described in Figure 2, and VEGF-mediated phosphorylation was considered as control (100%).
In previous figures, we showed that intracellular Ca2+ release was essential to cGMP synthesis. Herein, we observed that a 10-min stimulation with the Ca2+ ionophore A23187 (10 μM) induced a 3.3 fold increase in eNOS phosphorylation which was equivalent to the phosphorylation mediated by VEGF (1 nM) in a DMEM/5 mM CaCl2 solution (Figure 3C). In a DMEM CaCl2-free solution, there was no or marginal eNOS phosphorylation in PBS-treated group, and the absence of Ca2+ greatly attenuated (57%) the VEGF capacity to promote eNOS phosphorylation as compared to the experiments performed in presence of 5 mM CaCl2. Pretreatment with EGTA/AM (100 μM) or BAPTA/AM (10 μM) completely abrogated the residual VEGF-induced eNOS phosphorylation (Figure 3C). In addition, we observed that a 10-min pretreatment with Wortmannin (500 nM) or LY294002 (50 μM) prevented VEGF-induced eNOS phosphorylation (Figure 3D).
Effect of endogenous inhibitors of PAF synthesis and PAFR antagonists on VEGF-induced eNOS phosphorylation at Serine 1177
We used three structurally unrelated PAF synthesis inhibitors, SB203580 (10 μM), PD98059 (10 μM) or SB203347 (10 μM), which blocked respectively the activation of p38 MAPK, p42/44 MAPK and group V sPLA2 (Bernatchez et al., 2001a, b). The inhibition of these proteins did not alter eNOS phosphorylation mediated by VEGF (Figure 3E). Pretreatment with PAFR antagonists BN52021 (10 μM), LAU8080 (100 nM) or CV3988 (1 μM) did not prevent VEGF-induced eNOS phosphorylation. Finally, stimulation of BAEC with exogenous PAF (up to 1 μM) for 10 min did not increase basal eNOS phosphorylation (Figure 3E).
Effect of VEGF, intracellular inhibitors, and calcium chelators on Akt phosphorylation
Stimulation of BAEC with VEGF (1 nM) elicited a time-dependent increase in Akt phosphorylation which was nearly maximal within 5 min and maintained for an additional 5 min (Figure 4A). Then, we measured Akt phosphorylation upon a 10-min stimulation since we investigated the regulation of Akt at the time we observed maximal NO production. Treatment with VEGF (1 nM) elicited a 1.1 fold increase in Akt phosphorylation as compared to PBS-treated cells (Figure 4B). Pretreatment with L-NAME (100 μM) had no inhibitory effect on VEGF-induced Akt phosphorylation. The use of U73122 (10 μM) blocked both basal and VEGF-mediated Akt phosphorylation, whereas a pretreatment with U73343 (10 μM) had a slight effect on Akt phosphorylation induced by VEGF. Treatment with GF109203X (5 μM) or Ro318220 (1 μM) potentiated VEGF-induced Akt phosphorylation by 81 and 100% respectively. Finally, a pretreatment with W-7 (250 μM) completely blocked VEGF-mediated Akt phosphorylation (Figure 4B).
Figure 4.
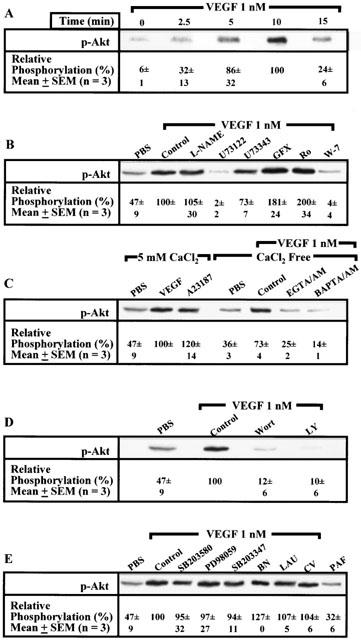
VEGF effect on Akt phosphorylation at Serine 473. Confluent BAEC were stimulated with agonists, cells were lysed and proteins separated on a 7.5% SDS–PAGE. Western blot analysis was performed with rabbit polyclonal anti-phospho-Akt (Ser473) antibodies. Studies (A–E) were performed as detailed in Figure 3.
Then, we assessed whether Akt phosphorylation is Ca2+-dependent. A 10-min stimulation with A23187 (10 μM) induced a 1.5 fold increase in Akt phosphorylation which was equivalent to the activation induced by VEGF (1 nM) as compared to PBS-treated cells. In a Ca2+-free solution, the basal Akt phosphorylation in PBS-treated cells was slightly reduced as compared to that observed in a 5 mM CaCl2 solution. In a Ca2+-free media, VEGF (1 nM) elicited a 1.1 fold increase in Akt phosphorylation as compared to PBS-treated cells, which was abolished by a pretreatment with EGTA/AM (100 μM) or BAPTA/AM (10 μM) (Figure 4C). Pretreatment with Wortmannin (500 nM) or LY294002 (50 μM) completely blocked VEGF-induced Akt phosphorylation (Figure 4D).
Effect of endogenous PAF synthesis inhibitors and PAFR antagonists on VEGF-induced Akt phosphorylation
The use of selective inhibitors capable of preventing VEGF-mediated PAF synthesis (SB203580, 10 μM; PD98059, 10 μM or SB203347, 10 μM) did not significantly attenuate Akt phosphorylation mediated by VEGF. In addition, pretreatment with unrelated PAFR antagonists (BN52021, 10 μM; LAU8080, 100 nM; CV3988, 1 μM) did not inhibit VEGF-induced Akt phosphorylation. Finally, stimulation with exogenous PAF (up to 1 μM) did not increase basal Akt phosphorylation (Figure 4E).
Effect of PI3K and PKC inhibitors on VEGF-induced delayed cGMP production
In the previous results, we showed that both PI3K and PKC inhibitors abrogated completely or partially eNOS phosphorylation but had no effect on cGMP production induced by VEGF for a 10-min stimulation. Interestingly, a recent study has demonstrated that immediate NO production was induced by intracellular calcium release in response to VEGF stimulation whereas delayed NO production was due to eNOS phosphorylation mediated by the activation of intracellular mediators such as PI3K and Hsp90 (Brouet et al., 2001). In order to measure delayed cGMP synthesis, we first sought to determine the minimal pretreatment time period required with IBMX to prevent cGMP degradation. Pretreatment with IBMX (500 μM) at 0, 2.5, 5 and 10 min prior to VEGF (1 nM) stimulation elicited a time-dependent increase in cGMP synthesis with a maximal production for a 5-min pretreatment with IBMX (Figure 5). Then, we assessed the effect of PI3K and PKC inhibitors on VEGF-induced immediate (0–10 min) and delayed (10–30 min) cGMP production. Pretreatment with two independent PI3K inhibitors (Wortmannin, 500 nM or LY294002, 50 μM) or two independent PKC inhibitors (GF109203X, 5 μM or Ro318220, 1 μM) did not attenuate immediate VEGF-mediated cGMP synthesis (0–10 min) (Figure 6; also shown in Figure 2). To evaluate the delayed cGMP synthesis, IBMX was added 5 min after VEGF stimulation and the experiment was stopped at 30 min. In the same condition (IBMX was added 5 min after VEGF stimulation), we quantified the cGMP synthesis from 5 to 10 min (data not shown) and subtracted this value from the total delayed cGMP synthesis. Therefore, in Figure 6 we present the delayed cGMP synthesis observed between 10 and 30 min upon VEGF stimulation. We observed that PI3K inhibitors (Wortmannin, 500 nM or LY294002, 50 μM) and PKC inhibitors (GF109203X, 5 μM or Ro318220, 1 μM) attenuated VEGF-induced delayed cGMP synthesis by 52, 62, 80 and 84% respectively (Figure 6).
Figure 5.
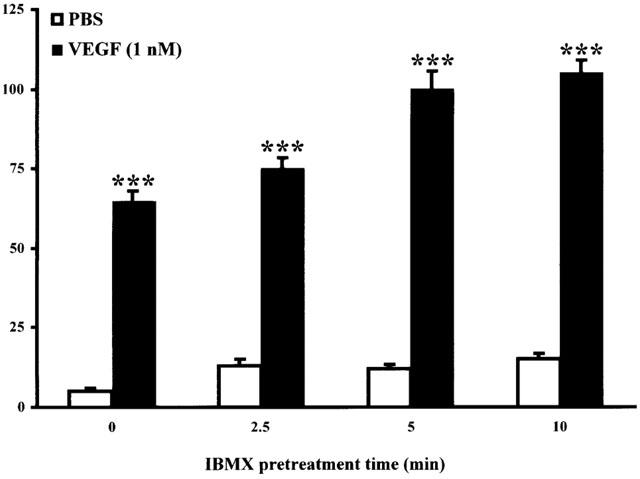
Effect of IBMX pretreatment on VEGF-induced immediate cGMP synthesis. Confluent BAEC were incubated 10 min in a DMEM 5 mM CaCl2 solution. IBMX (500 μM) was added for different periods of time (0, 2.5, 5, 10 min) prior to a 10-min stimulation with VEGF (1 nM). Cells were scraped, proteins lyophilized and cGMP production detected by a RIA kit. ***P<0.001 as compared to control buffer (PBS).
Figure 6.
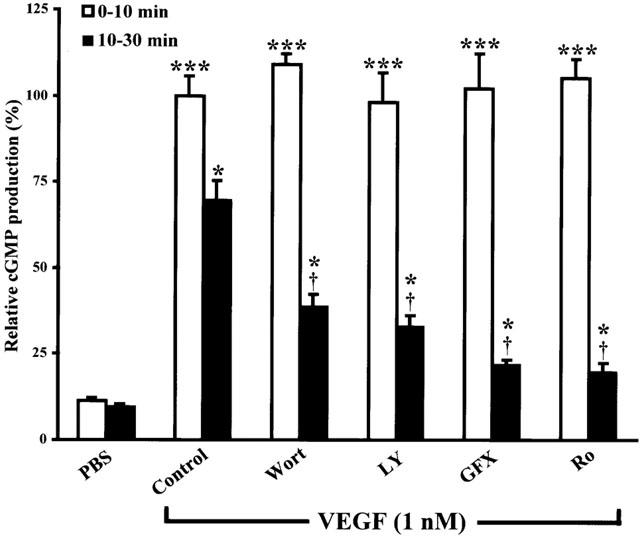
Effect of PI3K and PKC inhibitors on VEGF-induced immediate and delayed cGMP synthesis. Immediate cGMP synthesis was measured as described in Figure 2. Delayed cGMP synthesis was performed using confluent BAEC incubated 10 min with PKC inhibitors (GF109203X, 5 μM or Ro318220, 1 μM) or PI3K inhibitors (Wortmannin, 500 nM or LY294002, 50 μM) in a DMEM 5 mM CaCl2 solution prior to VEGF (1 nM) stimulation. IBMX (500 μM) was added at 5 min after VEGF stimulation and the experiment was stopped 30 min after VEGF stimulation. In another set of experiments and under the same conditions (IBMX added 5 min after VEGF stimulation), we quantified cGMP synthesis produced between 5 and 10 min (data not shown) and subtracted this amount from total delayed cGMP synthesis. Therefore, in Figure 6 (black columns) we show delayed cGMP synthesis observed between 10 and 30 min after VEGF stimulation. ***P<0.001 as compared to control buffer (PBS) for immediate cGMP synthesis; *P<0.05 as compared to control buffer (PBS) for delayed cGMP synthesis and †P<0.05 as compared to VEGF (1 nM) for delayed cGMP.
Effect of VEGF-induced NO synthesis on PAF synthesis
As VEGF induces NO and PAF synthesis in EC, we wanted to assess if VEGF-mediated PAF synthesis was regulated by the induction of NO synthesis. Pretreatment with L-NAME (100 μM), or a 15-min stimulation with SNP (500 μM), in absence of VEGF, did not alter the basal level of PAF synthesis. Treatment with VEGF (1 nM) for 15 min induced a 6.2 fold increase of PAF synthesis as compared to PBS-treated cells. Pretreatment with L-NAME (100 μM) did not prevent VEGF-induced PAF synthesis. Finally, a combined treatment of BAEC with VEGF (1 nM) plus SNP (500 μM) did not alter VEGF-mediated PAF synthesis (Figure 7).
Figure 7.
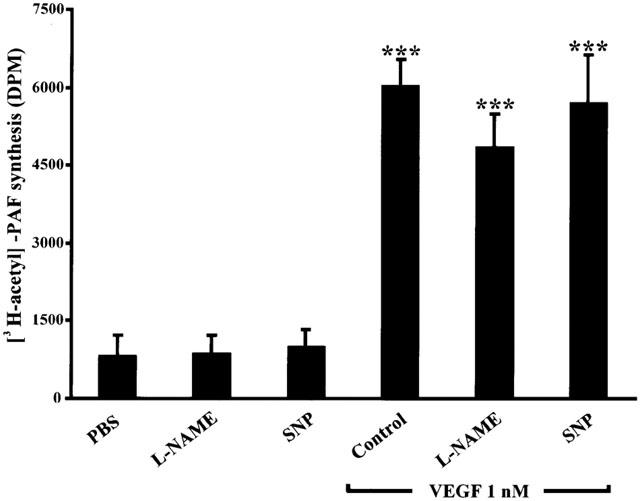
Effect of VEGF-induced nitric oxide on PAF synthesis. BAEC were grown to confluence. The media was replaced by 1 ml of HBSS-HEPES (10 mM)+CaCl2 (5 mM). Inhibitors were added 15 min prior to stimulation. VEGF (1 nM) was added to stimulate BAEC: alone as a control, with L-NAME (100 μM) pretreatment or in addition with the exogenous NO donor SNP (500 μM). L-NAME (100 μM) and SNP (500 μM) were added without VEGF to evaluate their effect on basal PAF synthesis. ***P<0.001 as compared to control buffer (PBS).
Discussion
There have been several studies investigating the mechanisms by which agonists are mediating eNOS activation. However, these reports did not highlight an exclusive pathway shared by various agonists leading to eNOS activation. In addition, there are some discrepancies regarding the mechanisms involved in eNOS activation and NO production mediated by VEGF, peculiarly for those involving the contribution of Akt and/or PKC as intracellular mediators. Furthermore, a recent study has demonstrated that immediate NO production was induced by intracellular calcium release in response to VEGF, and that delayed NO production was mediated by eNOS phosphorylation which results from the activation of intracellular mediators such as PI3K and Hsp90 (Brouet et al., 2001). Herein, we investigated the contribution of Akt, PKC, PLC and calcium on eNOS activation by assessing its phosphorylation at Ser1177, and by measuring immediate and delayed cGMP synthesis. In addition, as VEGF induces the synthesis of NO and PAF in endothelial cells and that both mediators might play key roles in the inflammatory and angiogenic activities mediated by VEGF (Ku et al., 1993; Sirois & Edelman, 1997; Ziche et al., 1997; Lakshminarayanan et al., 2000; Montrucchio et al., 2000; Bussolati et al., 2001; Lal et al., 2001), we then wanted to assess if the synthesis of PAF and NO was affecting the synthesis of each other.
In the current study, we measured NO synthesis by quantifying cGMP accumulation as previously described (Ishii et al., 1991; Ziche et al., 1997; He et al., 1999; Kroll & Waltenberger, 1999; Fleming et al., 2001). First, we confirmed that cGMP production was NO-dependent by treating the endothelial cells with an exogenous NO donor (SNP) which induced cGMP production and by the blockade of cGMP production with L-NAME (Figure 2A), which is in agreement with previous reports (Ziche et al., 1997; He et al., 1999; Kroll & Waltenberger, 1999). Treatment with L-NAME did not attenuate VEGF-induced eNOS Ser1177-phosphorylation as L-NAME is a L-arginine competitive analogue and therefore interfering downstream to eNOS phosphorylation (Figure 3B).
Ca2+/CaM and eNOS activation
It is well known that eNOS can be regulated through intracellular calcium release induced by various agonists such as bradykinin, VEGF and histamine (Govers & Rabelink, 2001). The mechanism by which VEGF releases intracellular calcium in EC is consequent to PLC activation. On one hand, PLC has the ability to induce DAG synthesis and IP3 which promotes the release of intracellular calcium from the endoplasmic reticulum (Brock et al., 1991). Herein, we showed that the inhibition of PLC with U73122 abolished eNOS Ser1177 and Akt phosphorylation, and cGMP synthesis mediated by VEGF. Therefore, confirming the crucial role played by PLC on eNOS activation. The contribution of intracellular calcium elevation on eNOS Ser1177-phosphorylation and immediate NO/cGMP synthesis was furthermore supported by the fact that calcium ionophore A23187 was as potent as VEGF to mediate eNOS activation and nearly equivalent at mediating cGMP synthesis (Figures 2B and 3C). Moreover, in a calcium-free media, VEGF had the ability to increase both eNOS Ser1177-phosphorylation and cGMP synthesis but at a lower extent to that observed in a 5 mM CaCl2-DMEM solution. These data illustrate that intracellular calcium release upon PLC activation is sufficient to mediate eNOS activation and cGMP production, which is potentiated by an extracellular calcium entry into endothelial cells (Figures 2B and 3C).
Immediate cGMP synthesis is Akt-independent
In our study, we observed that the blockade of PLC activation, Ca2+/CaM complex formation, and that the use of intracellular calcium chelators abrogated Akt phosphorylation mediated by VEGF. In addition, a treatment with a calcium ionophore induced Akt activation. These data suggest that Akt phosphorylation is Ca2+/CaM complex-dependent. As previously reported (Papapetropoulos et al., 1997), we observed that Akt phosphorylation is also mediated by PI3K activation since VEGF-mediated Akt phosphorylation was abrogated by pretreatment either with Wortmannin or LY294002. Therefore, our data imply that Akt phosphorylation requires intracellular calcium elevation, consequent Ca2+/CaM complex formation, and PI3K activation, since the blockade of one or the other inhibits Akt activation. Our hypothesis is supported by recent studies in which it was shown that Akt phosphorylation can be induced through the activation of Ca2+/CaM-dependent protein kinase kinase (CaM-KK), and that a treatment with a CaM antagonist prevented insulin-induced Akt phosphorylation (Tokumitsu & Soderling, 1996; Yano et al., 1998; Yang et al., 2000).
More interestingly, we observed that the blockade of Ca2+/CaM-dependent Akt phosphorylation abrogated immediate cGMP production, whereas the blockade of PI3K-dependent Akt phosphorylation did not prevent immediate cGMP production. In the previous section, we defined that Ca2+/CaM complex formation was essential for eNOS activation and cGMP production. Consequently, our data suggest that immediate NO/cGMP synthesis requires eNOS-Ca2+/CaM complex interaction as defined previously (Busse & Mulsch, 1990), and that Akt activation is not necessary for the initial induction of NO/cGMP production.
Delayed cGMP synthesis in PKC- and Akt-dependent
Aside the synthesis of IP3 and intracellular calcium release, PLC promotes as well PKC activation through DAG synthesis. PKC is a calcium-dependent enzyme whose role in eNOS activation has been thoroughly assessed. However, its contribution remains controversial. Indeed, a previous study showed that treatment with a PKC inhibitor completely blocked cGMP synthesis induced by VEGF (He et al., 1999), whereas others (Michell et al., 2001) showed that PKC phosphorylates eNOS at Thr495 and dephosphorylates eNOS at Ser1177 thereby showing an inhibitory action of PKC on eNOS activation. In our study, we found that despite the fact that two unrelated PKC inhibitors (GF109203X or Ro318220) attenuated eNOS Ser1177-phosphorylation by 57 and 38% respectively they had no inhibitory effect on immediate cGMP synthesis (Figure 2B). Interestingly, a recent study has demonstrated that immediate NO production can be induced exclusively by calcium release upon stimulation with VEGF and that eNOS phosphorylation mediated by VEGF leads to a delayed positive effect on NO synthesis (Brouet et al., 2001). The exact mechanism responsible for an increase of eNOS enzymatic activity is unclear, although the late phosphorylation at Ser1177 is thought to potentiate eNOS sensitivity for Ca2+/CaM complex (Dimmeler et al., 1999; Fulton et al., 1999; Gratton et al., 2000; McCabe et al., 2000). Therefore, we quantified the delayed cGMP synthesis (10–30 min) upon VEGF stimulation and we found that the use of two PKC inhibitors (GF109203X or Ro318220) almost completely blocked VEGF-induced delayed cGMP synthesis (80 and 84% respectively). This latter result confirms a positive role played by PKC on eNOS regulation which is in agreement with two recent studies (He et al., 1999; Thuringer et al., 2001).
It has previously been shown with specific PI3K inhibitors that blockade of Akt activation reduces partially the VEGF-mediated cGMP production (Papapetropoulos et al., 1997). Then, it has been demonstrated that activated Akt possesses the ability to phosphorylate eNOS at Ser1177, thereby increasing eNOS enzymatic activity (Dimmeler et al., 1999; Fulton et al., 1999; Michell et al., 1999). However, two recent reports have suggested that the inhibition of PI3K with specific inhibitors had no or slight inhibitory effect on NO release (Fleming et al., 2001; Thuringer et al., 2001). In our study, we found that despite the fact that two independent unrelated PI3K inhibitors (Wortmannin or LY294002) prevented eNOS Ser1177-phosphorylation induced by VEGF, they had no effect on VEGF-induced immediate NO synthesis (Figure 2D). However, pretreatment with these two inhibitors attenuated by 52 and 62% respectively the delayed cGMP synthesis induced by VEGF (Figure 6). This latter result is in agreement with a recent study (Brouet et al., 2001) which demonstrated the positive effect of Akt on delayed NO production induced by VEGF. It is suggested that delayed Akt effect on eNOS is attributable to eNOS phosphorylation at Ser1177 which reduces CaM dissociation from the activated eNOS when intracellular calcium returns to its basal level (Dimmeler et al., 1999; Fulton et al., 1999; McCabe et al., 2000).
The inhibition of delayed cGMP synthesis mediated by PKC inhibitors was greater than the one mediated by PI3K inhibitors, even though eNOS Ser1177-phosphorylation blockade was more efficient with the PI3K inhibitors. This is in agreement with a recent study which demonstrated that Ro318220 had a greater inhibitory effect than Wortmannin on VEGF-induced NO synthesis (Thuringer et al., 2001). Moreover, a recent study has suggested that temporal sequence of events leading to eNOS activation by VEGF implies KDR-PLC-Ca2+-PKC respectively (Wu et al., 1999). Accordingly, we hypothesize that PKC action on eNOS may occur first and then facilitate Akt action on eNOS. This might explain the greater cGMP synthesis inhibition observed with the PKC inhibitors even though eNOS phosphorylation blockade was more important with PI3K inhibitors.
Absence of crosstalks between PAF and NO synthesis mediated by VEGF
In the current study we observed that VEGF-mediated cGMP production is independent from PAF synthesis. The absence of PAF contribution on NO synthesis was not due to a lack of biological activity of PAF and/or its receptor, as we observed by Western blot analysis and confocal microscopy the expression of PAF receptors, that PAF was able to promote endothelial cell migration, and that a preatreatment with PAF receptor antagonists inhibited VEGF-chemotactic activity (data not shown). Finally, we observed that NO synthesis does not contribute to promote VEGF-mediated PAF synthesis (Figure 7). Therefore, our data suggest that VEGF-induced NO and PAF synthesis are mediated by independent pathways (Figure 8).
Figure 8.
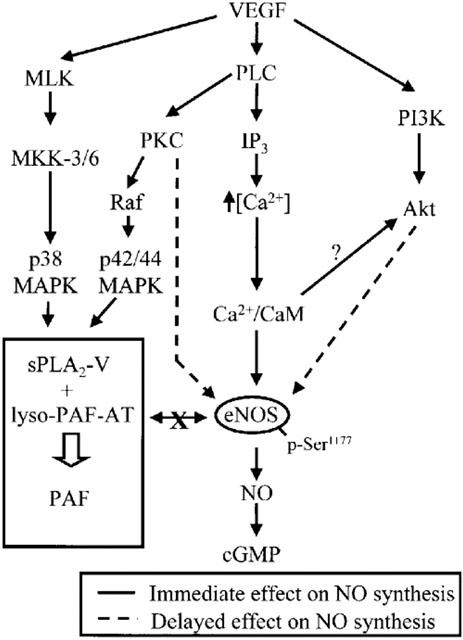
Proposed intracellular pathways for the induction of NO and PAF synthesis by VEGF in endothelial cells. VEGF induces immediate NO synthesis through the PLC-Ca2+/CaM pathway and the induction of delayed NO synthesis implies Akt and PKC activition. In addition, NO and PAF synthesis mediated by VEGF involves two independent pathways.
In conclusion, VEGF induces an immediate NO/cGMP synthesis which is independent from Akt activation but rather driven through PLC activation which is leading to Ca2+/CaM complex formation and eNOS activation. However, VEGF induces as well a delayed NO/cGMP synthesis which implies the activation of PKC and Akt. In addition, we showed that NO and PAF synthesis mediated by VEGF involves two independent intracellular pathways.
Acknowledgments
This study was supported by grants from the Canadian Institutes of Health Research (MOP-43919) and from the Heart and Stroke Foundation of Québec to Dr Sirois. Mr Bernatchez is recipient of a studentship from the CIHR and Dr Sirois was recipient of a scholarship from the Heart and Stroke Foundation of Canada, and is currently recipient of a scholarship from the Canadian Institutes of Health Research.
Abbreviations
- BAEC
bovine aortic endothelial cells
- CaM
calmodulin
- cGMP
cyclic guanosine monophosphate
- DMEM
Dulbecco's modified Eagle medium
- EC
endothelial cells
- eNOS
endothelial nitric oxide synthase
- DAG
diacylglycerol
- Hsp90
heat shock protein, IBMX, isobutylmethylxanthine
- IP3
inositol triphosphate
- L-NAME
NG-nitro-L-arginine methyl ester
- MAPK
mitogen-activated protein kinase
- NO
nitric oxide
- PAF
platelet activating factor
- PAFR
platelet activating factor receptor
- PI3K
phosphatidylinositol 3-kinase
- PKC
protein kinase C
- PLA2
phospholipase A2
- PLC
phospholipase C
- RIA
radioimmunoassay
- SNP
sodium nitroprusside
- VEGF
vascular endothelial growth factor
References
- BERNATCHEZ P.N., ALLEN B.G., GÉLINAS D.S., GUILLEMETTE G., SIROIS M.G. Regulation of VEGF-induced endothelial cell PAF synthesis: role of p42/44 MAPK, p38 MAPK and PI3K pathways. Br. J. Pharmacol. 2001b;134:1253–1262. doi: 10.1038/sj.bjp.0704367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERNATCHEZ P.N., SOKER S., SIROIS M.G. Vascular endothelial growth factor effect on endothelial cell proliferation, migration, and platelet-activating factor synthesis is Flk-1-dependent. J. Biol. Chem. 1999;274:31047–31054. doi: 10.1074/jbc.274.43.31047. [DOI] [PubMed] [Google Scholar]
- BERNATCHEZ P.N., WINSTEAD M.V., DENNIS E.A., SIROIS M.G. VEGF stimulation of endothelial cell PAF synthesis is mediated by group V 14 kDa secretory phospholipase A2. Br. J. Pharmacol. 2001a;134:197–205. doi: 10.1038/sj.bjp.0704215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLIGH E.G., DYER W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- BROCK T.A., DVORAK H.F., SENGER D.R. Tumor-secreted vascular permeability factor increases cytosolic Ca2+ and von Willebrand factor release in human endothelial cells. Am. J. Pathol. 1991;138:213–221. [PMC free article] [PubMed] [Google Scholar]
- BROUET A., SONVEAUX P., DESSY C., BALLIGAND J-L., FERON O. Hsp90 ensures the transition from the early Ca2+-dependent to the late phosphorylation-dependent activation of the endothelial nitric-oxide synthase in vascular endothelial growth factor-exposed endothelial cells. J. Biol. Chem. 2001;276:32663–32669. doi: 10.1074/jbc.M101371200. [DOI] [PubMed] [Google Scholar]
- BUSSE R., MULSCH A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated calmodulin. FEBS Lett. 1990;265:133–136. doi: 10.1016/0014-5793(90)80902-u. [DOI] [PubMed] [Google Scholar]
- BUSSOLATI B., DUNK C., GROHMAN M., KONTOS C.D., MASON J., AHMED A. Vascular endothelial growth factor receptor-1 modulates vascular growth factor-mediated angiogenesis via nitric oxide. Am. J. Physiol. 2001;159:993–1008. doi: 10.1016/S0002-9440(10)61775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONNOLLY D.T., HEUVELMAN D.M., NELSON R., OLANDER J.V., EPPLEY B.L., DELFINO J.J., SIEGEL N.R., LIMGRUBER R.M., FEDER J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J. Clin. Invest. 1989;84:1470–1478. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIMMELER S., FLEMING I., FISSLTHALER B., HERMANN C., BUSSE R., ZEIHER A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- FENG Y., VENEMA V.J., VENEMA R.C., TSAI N., CALDWELL R.B. VEGF induces nuclear translocation of Flk-1/KDR, endothelial nitric oxide synthase, and caveolin-1 in vascular endothelial cells. Biochem. Biophys. Res. Commun. 1999;256:192–197. doi: 10.1006/bbrc.1998.9790. [DOI] [PubMed] [Google Scholar]
- FERRARA N. Vascular endothelial growth factor. The trigger for neovascularization in the eye. Lab. Invest. 1995;72:615–618. [PubMed] [Google Scholar]
- FERRARA N., DAVIS-SMITH T. The biology of vascular endothelial growth factor. Endo. Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- FLEMING I., FISSLTHALER B., DIMMELER S., KEMP B.E., BUSSE R. Phosphorylation of Thr495 regulates Ca2+/Calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001;88:e68–e75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- FOLKMAN J. What is the evidence that tumors are angiogenesis-dependent. J. Natl. Cancer Inst. 1991;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- FOLKMAN J., KLAGSBRUN M. Angiogenic factors. Science. 1987;235:442–447. doi: 10.1126/science.2432664. [DOI] [PubMed] [Google Scholar]
- FULTON D., GRATTON J-P., MCCABE T.J., FONTANA J., FUJIO Y., WALSH K., FRANKE T.F., PAPAPETROPOULOS A., SESSA W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOVERS R., RABELINK T.J. Cellular regulation of endothelial nitric oxide synthase. Am. J. Physiol. 2001;280:F193–F206. doi: 10.1152/ajprenal.2001.280.2.F193. [DOI] [PubMed] [Google Scholar]
- GRATTON J-P., FONTANA J., O'CONNOR D.S., GARCÍA-CARDEÑA G., MCCABE T.J., SESSA W.C. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and Caveolin-1 complex in vitro. J. Biol. Chem. 2000;275:22268–22272. doi: 10.1074/jbc.M001644200. [DOI] [PubMed] [Google Scholar]
- HE H., VENEMA V.J., GU X., VENEMA R.C., MARRERO M.B., CALDWELL R.B. Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through Flk-1/KDR activation of c-Src. J. Biol. Chem. 1999;274:25130–25135. doi: 10.1074/jbc.274.35.25130. [DOI] [PubMed] [Google Scholar]
- ISHII K., CHANG B., KERWIN J.F., JR, WAGENAAR F.L., HUANG Z.J., MURAD F. Formation of endothelium-derived relaxing factor in porcine kidney epithelial LLC-PK1 cells: an intra- and intercellular messenger for activation of soluble guanylate cyclase. J. Pharmacol. Exp. Ther. 1991;256:38–43. [PubMed] [Google Scholar]
- KROLL J., WALTENBERGER J. A novel function of VEGF receptor-2 (KDR): rapid release of nitric oxide in response to VEGF-A stimulation of endothelial cells. Biochem. Biophys. Res. Commun. 1999;265:636–639. doi: 10.1006/bbrc.1999.1729. [DOI] [PubMed] [Google Scholar]
- KU D.D., ZALESKI J.K., LIU S., BROCK T.A. Vascular endothelial growth factor induces EDRF-dependent relaxation in coronary arteries. Am. J. Physiol. 1993;265:H586–H592. doi: 10.1152/ajpheart.1993.265.2.H586. [DOI] [PubMed] [Google Scholar]
- LAKSHMINARAYANAN S., ANTONETTI D.A., GARDNER T.W., TARBELL J.M. Effect of VEGF on retinal microvascular endothelial hydraulic conductivity: The role of NO. Invest. Ophthalmol. Vis. Sci. 2000;41:4256–4261. [PubMed] [Google Scholar]
- LAL B.K., VARMA S., PAPPAS P.J., HOBSON R.W., II, DURAN W.N. VEGF increases permeability of the endothelial cell monolayer by activation of PKB/Akt, endothelial nitric oxide, and MAP kinase pathways. Microvasc. Res. 2001;62:252–262. doi: 10.1006/mvre.2001.2338. [DOI] [PubMed] [Google Scholar]
- MARCHESELLI V.L., BAZAN N.G. Platelet-activating factor is a messager in the electroconvulsive shock-induced transcriptional activation of c-fos and zif-268 in hippocampus. J. Neurosci. Res. 1994;37:54–61. doi: 10.1002/jnr.490370108. [DOI] [PubMed] [Google Scholar]
- MARCHESELLI V.L., ROSSOWSKA M.J., DOMINGO M.T., BRAQUET P., BAZAN N.G. Distinct platelet-activating factor binding sites in synaptic endings and in intracellular membranes of rat cerebral cortex. J. Biol. Chem. 1990;265:9140–9145. [PubMed] [Google Scholar]
- MCCABE T.J., FULTON D., ROMAN L.J., SESSA W.C. Enhanced electron flux and reduced calmodulin dissociation may explain “calcium-independent” eNOS activation by phosphorylation. J. Biol. Chem. 2000;275:6123–6128. doi: 10.1074/jbc.275.9.6123. [DOI] [PubMed] [Google Scholar]
- MICHELL B.J., CHEN Z-P., TIGANIS T., STAPLETON D., KATSIS F., POWER D.A., SIM A.T., KEMP B.E. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J. Biol. Chem. 2001;276:17625–17628. doi: 10.1074/jbc.C100122200. [DOI] [PubMed] [Google Scholar]
- MICHELL B.J., GRIFFITHS J.E., MITCHELHILL K.I., RODRIGUEZ-CRESPO I., TIGANIS T., BOZINOVSKI S., ORTIZ DE MONTELLANO P.R., KEMP B.E., PEARSON R.B. The Akt kinase signals directly to endothelial nitric oxide synthase. Cur. Biol. 1999;9:845–848. doi: 10.1016/s0960-9822(99)80371-6. [DOI] [PubMed] [Google Scholar]
- MONTRUCCHIO G., LUPIA E., BATTAGLIA E., DEL SORBO L., BOCCELLINO M., EMANUELLI G., CAMUSSI G. Platelet-activating factor enhances vascular endothelial growth factor-induced endothelial cell motility and neoangiogenesis in a murine matrigel model. Arterioscler. Thromb. Vasc. Biol. 2000;20:80–88. doi: 10.1161/01.atv.20.1.80. [DOI] [PubMed] [Google Scholar]
- MOULTON K.S., HELLER E., KONERDING M.A., FLYNN E., PALINSKI W., FOLKMAN J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. 1999;99:1726–1732. doi: 10.1161/01.cir.99.13.1726. [DOI] [PubMed] [Google Scholar]
- PAPAPETROPOULOS A., GARCÍA-CARDEÑA G., MADRI J.A., SESSA WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIROIS M.G., EDELMAN E.R. VEGF effect on vascular permeability is mediated by synthesis of platelet-activating factor. Am. J. Physiol. 1997;272:H2746–H2756. doi: 10.1152/ajpheart.1997.272.6.H2746. [DOI] [PubMed] [Google Scholar]
- THURINGER D., MAULON L., FRELIN C. Rapid transactivation of the VEGF receptor KDR/Flk-1 by the bradykinin B2 receptor contributes to eNOS activation in cardiac capillary endothelial cells. J. Biol. Chem. 2001;277:2028–2032. doi: 10.1074/jbc.M109493200. [DOI] [PubMed] [Google Scholar]
- TOKUMITSU H., SODERLING T.R. Requirements for calcium and calmodulin in the calmodulin kinase activation cascade. J. Biol. Chem. 1996;271:5617–5622. doi: 10.1074/jbc.271.10.5617. [DOI] [PubMed] [Google Scholar]
- UNEMORI E.N., FERRARA N., BAUER E.A., AMENTO E.P. Vascular endothelial growth factor induces interstitial collagenase expression in human endothelial cells. J. Cell. Physiol. 1992;153:557–562. doi: 10.1002/jcp.1041530317. [DOI] [PubMed] [Google Scholar]
- WU H.M., YUAN Y., ZAWIEJA D.C., TINSLEY J., GRANGER H.J. Role of phospholipase C, protein kinase C, and calcium in VEGF-induced venular hyperpermeability. Am. J. Physiol. 1999;276:H535–H542. doi: 10.1152/ajpheart.1999.276.2.H535. [DOI] [PubMed] [Google Scholar]
- XIA P., AIELLO L.P., ISHII H., JIANG Z.Y., PARK D.J., ROBINSON G.S., TAKAGI H., NEWSOME W.P., JIROUSEK M.R., KING G.L. Characterization of vascular endothelial growth factor's effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J. Clin. Invest. 1996;98:2018–2026. doi: 10.1172/JCI119006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG C., WATSON R.T., ELMENDORF J.S., SACKS D.B., PESSIN J.E. Calmodulin antagonists inhibit insulin-stimulated GLUT4 (Glucose Transporter 4) translocation by preventing the formation of phosphatidyinositol 3,4,5- trisphosphate in 3T3L1 adipocytes. Mol. Endocrinol. 2000;14:317–326. doi: 10.1210/mend.14.2.0425. [DOI] [PubMed] [Google Scholar]
- YANO S., TOKUMITSU H., SODERLING T.R. Calcium promotes cell survival through CaM-K Kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- ZICHE M., MORBIDELLI L., CHOUDHURI R., ZHANG H-T., DONNINI S., GRANGER H.J., BICKNELL R. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J. Clin. Invest. 1997;99:2625–2634. doi: 10.1172/JCI119451. [DOI] [PMC free article] [PubMed] [Google Scholar]