

Abstract
Levobupivacaine and ropivacaine are the pure S(−) enantiomers of N-butyl- and N-propyl-2′,6′-pipecoloxylidide, developed as less cardiotoxic alternatives to bupivacaine. In the present study, we have analysed the effects of levobupivacaine, ropivacaine and bupivacaine on HERG channels stably expressed in CHO cells.
The three drugs blocked HERG channels in a concentration-, time- and state-dependent manner. Block measured at the end of 5 s pulses to −10 mV induced by 20 μM bupivacaine (52.7±2.0%, n=15) and ropivacaine (55.5±2.7%, n=13) was similar (P>0.05) and both lower than that induced by levobupivacaine (67.5±4.2%, n=11) (P<0.05).
Dextrobupivacaine (20 μM) was less potent (47.2±5.2%, n=10) than levobupivacaine (P<0.05), indicating stereoselective HERG channel block.
Block induced by the three local anaesthetics exhibited a steep voltage dependence in the range of channel activation. In all cases, block measured at the maximum peak current at a test potential of 0 mV after promoting recovery from inactivation (I→O) was lower than that observed at the end of 5-s pulses (I+O).
Levobupivacaine, ropivacaine and bupivacaine accelerated HERG inactivation kinetics, slowed the recovery from inactivation and shifted the inactivation curve towards more negative membrane potentials. The three local anaesthetics induced a rapid time-dependent decline after using a protocol that quickly activates HERG channels.
All these results suggest that: (1) these drugs bind to the open and the inactivated states of HERG channels, (2) they stabilize HERG channels in the inactivated state, and (3) block induced by bupivacaine enantiomers is stereoselective.
Keywords: Bupivacaine, HERG, ropivacaine, levobupivacaine, stereoselectivity
Introduction
Bupivacaine is an amide-type local anaesthetic widely used for regional anaesthesia (Strichartz, 1987) that exhibits a high cardiotoxicity, likely due to its ability to depress the intracardiac conduction velocity and cardiac contractility (Kotelko et al., 1984). These bupivacaine effects have been related to its blocking properties of Na+ and Ca2+ channels (Clarkson & Hondeghem, 1985; Sanchez-Chapula, 1988). Moreover, several cases of long QT (LQT) syndrome, associated to polymorphic ventricular arrhythmias (torsades de pointes) have been reported in animal models (Wheeler et al., 1988; Solomon et al., 1990) and in humans after its accidental intravascular injection (Scott et al., 1989). Although drug-induced LQT could result from the inhibition of any voltage-gated K+ current that contributes to ventricular repolarization, almost all known drugs exhibiting this effect block IKr (Roden et al., 1996; Keating & Sanguinetti, 1996; Tamargo, 2000) that is generated by the activation of HERG potassium channels (Sanguinetti & Jurkiewicz, 1990; Sanguinetti et al., 1995). In fact, it is widely accepted that acquired LQT syndrome is due to an excessive HERG channels block (Mitcheson et al., 2000). Thus, sensitivity of HERG K+ channels to drugs has become a major issue in the development of new and safer drugs (Roden et al., 1996).
Bupivacaine induced cardiotoxicity has been mostly related to the effects of its R(+) enantiomer (Luduena et al., 1972; Aberg, 1972), which exhibits a higher potency for blocking cardiac Na+ and hKv1.5 channels (Valenzuela et al., 1995a, b; Nau et al., 2000). Levobupivacaine and ropivacaine have been recently developed as less cardiotoxic alternatives to racemic bupivacaine (Akerman et al., 1988; Scott et al., 1989; Foster & Markham, 2000). Both local anaesthetics are the pure S(−) enantiomers of N-butyl-pipecoloxylidide and N-propyl-pipecoloxylidide; i.e. they only differ in the length of the N-substituent that is a butyl and a propyl group for levobupivacaine and ropivacaine, respectively. Both are weak bases (pKa=8.1) and thus, they are predominantly charged at the physiological pH (Strichartz, 1987).
The effects of racemic bupivacaine on HERG channels have been previously studied both in Xenopus oocytes and after transient transfection in CHO cells (Lipka et al., 1998; González et al., 2001b). Potency of bupivacaine-induced block of HERG channels was higher in CHO cells, likely due to the different expression system used in both studies. The effects of levobupivacaine and ropivacaine on HERG channels are unknown at the present time. Therefore, the purpose of the present study was to analyse and compare the electrophysiological effects of levobupivacaine and ropivacaine on HERG channels stably expressed in CHO cells with those induced by racemic bupivacaine. The effects of racemic bupivacaine have been analysed again because of the different method of transfection used (transiently or stably transfected CHO cells). Preliminary results of the present study have been published in abstract form (González et al., 2002).
Methods
Cell culture
CHO cells stably transfected with the gene encoding HERG channels (a gift of Drs Nattel, Hébert and Weerapura) were cultured at 37°C in Ham's F12 medium supplemented with geneticine (600 μg ml−1), penicillin-streptomycin (800 UI and 200 μg ml−1, respectively) and bovine serum 10%, in a 5% CO2 atmosphere. Cultures were passaged every 3–5 days by use of a brief trypsin treatment. Before experimental use, the cells were removed from the dish with a rubber policeman, a procedure that left the majority of the cells intact. The cell suspension was stored at room temperature (21–23°C) and used within 12 h in all the experiments reported.
Drugs
Levobupivacaine, dextrobupivacaine and ropivacaine (a gift from Chiroscience, Cambridge, UK) and racemic bupivacaine (Sigma Chemical, St. Louis, MO, USA) were dissolved in distilled deionized water to yield stock solutions of 1 mM from which further dilutions were made.
Electrophysiological recording
The intracellular pipette-filling solution contained (in mM): K-aspartate 80, KCl 50, phosphocreatine 3, KH2PO4 10, MgATP 3, HEPES-K 10, and EGTA 5 and was adjusted to pH 7.25 with KOH. The bath solution contained (in mM): NaCl 130, KCl 4, CaCl2 1.8, MgCl2 1, HEPES-Na 10, and glucose 10, and was adjusted to pH 7.4 with NaOH. HERG currents were recorded at room temperature (21–23°C) using the whole-cell patch-clamp technique (Hamill et al., 1981) with an Axopatch 200A patch-clamp amplifier (Axon Instruments, Foster City, CA, U.S.A.). Micropipettes were pulled from borosilicate glass capillary tubes (GD-1; Narishige, Tokyo, Japan) on a programmable horizontal puller (Sutter Instrument Co., San Rafael, CA, U.S.A.) and heat-polished with a microforge (Narishige). Micropipettes resistance was 1–3 MΩ. Mean uncompensated access resistance was 2.3±0.4 Mω, and cell capacitance was 11.5±0.7 pF (n=10). Thus, no significant voltage errors (<5 mV) were expected with the electrodes used. HERG currents were filtered at 100 Hz and sampled at 200 Hz. Cells were held at −80 mV. After control data were obtained, bath perfusion was switched to drug-containing solution. The effects of drug infusion were monitored with test pulses to −10 mV applied every 30 s until steady state was obtained. Steady-state current-voltage relationships (IV) were obtained by averaging the current over a small window (2–5 ms) at the end of 5-s depolarizing pulses. Between −80 and −50 mV only passive linear leak was observed and least squares fit to these data were used for passive leak correction. Deactivating tail currents were recorded at −60 mV. The activation curves were obtained from the tail current amplitude measured just after the capacitive transient. Other pulse protocols are described in the Results section. Command potentials, data acquisition and measurements were done using the CLAMPFIT program of PCLAMP 6.0.1, Origin 6.0.1 (Microcal Software, Northampton, MA, U.S.A.) and by custom-made analysis programs. Deactivation was fitted to a biexponential process:
where τ1 and τ2 are the system time constants, A1 and A2 are the amplitudes of each component of the exponential, and C is the baseline value. Half-maximal voltages (Eh) and slope factors (s) of activation and inactivation were determined by fitting data with a Boltzmann equation: y=1/[1+exp(−(E−Eh/s)]. The curve-fitting procedure used a non-linear least-squares (Gauss-Newton) algorithm. Goodness of fit was judged by comparing χ2 values statistically (F test) and by inspection for systematic non-random trends in the difference plot.
Drug-induced block was measured at the end of depolarizing pulses of 5 s in duration from −80 mV to −10 mV, unless indicated otherwise. The degree of inhibition obtained for each drug concentration of levobupivacaine, ropivacaine or bupivacaine [f=(1−IDrug/IControl)×100] was used to calculate the EC50 and nH values from the fitting of these values to a Hill equation of the form:
Voltage dependence of block was determined as follows: leak-corrected tail current in the presence of drug was normalized to matching control to yield the fractional block at each voltage [f=1−(IDrug/IControl)]. The voltage dependence of block was fitted to a Boltzmann function based on the Woodhull model:
where z, F, R and T have their usual meaning, δ represents the fractional electrical distance, i.e., the fraction of the transmembrane electrical field sensed by a single charge at the receptor site and KD* represents the apparent dissociation constant at the reference potential (0 mV) (Woodhull, 1973).
Statistical methods
Results are expressed as mean±s.e.mean. Direct comparisons between mean values in control conditions and in the presence of drug for a single variable were performed by paired Student's t-test. Differences were considered significant if P value was less than 0.05. Comparisons between the three groups were performed by a one-way analysis of variance (ANOVA), with a posterior Newman-Keuls test if P<0.05.
Results
Concentration-dependence of HERG block induced by levobupivacaine, ropivacaine and bupivacaine
Figure 1 shows original HERG current records obtained after applying 5 s depolarizing pulses from a holding potential of −80 mV up to +50 mV in 10-mV steps in the absence and in the presence of 20 μM levobupivacaine, ropivacaine and bupivacaine. Tail currents were elicited by repolarization of the membrane potential to −60 mV after each voltage step. Depolarizations to values more positive than −50 mV elicited an outward current followed by a hooked deactivating tail current. The maximum outward current was observed at ≈−10 mV (−6.7±1.2 mV, n=33) and it decreased at more positive membrane potentials. Thus, steady-state drug-induced block was measured at the end of 5 s depolarizing pulses to −10 mV. The degree of block induced by 20 μM levobupivacaine (67.5±4.2%, n=11) was higher than that induced by 20 μM ropivacaine (55.5±2.7%, n=13; P<0.05) or 20 μM bupivacaine (52.7±2.0%, n=15; P<0.05). Levobupivacaine, ropivacaine and bupivacaine blocked HERG tail curents by 55.1±3.3% (n=11), 45.6±3.0% (n=11) and 51.6±2.1% (n=15), respectively, with only significant differences between levobupivacaine and ropivacaine (P<0.05). Block induced by bupivacaine was similar when measured both at −10 mV and at the maximum tail current (P>0.05). However, levobupivacaine and ropivacaine induced a higher degree of block at −10 mV than at the tail current (P<0.05), suggesting that both local anaesthetics bind preferentially during the depolarizing step (i.e., during the activation and inactivation of HERG channels) (Table 1).
Figure 1.
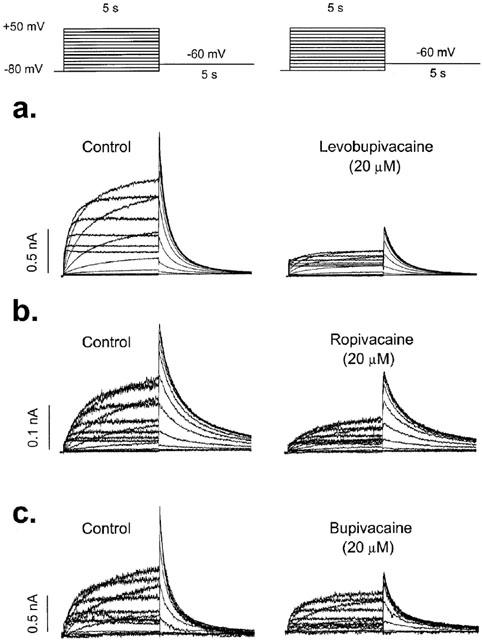
Original records obtained upon depolarization from a holding potential of −80 mV to +50 mV in 10 mV steps and upon repolarization to −60 mV. Current records obtained in the absence and in the presence of 20 μM levobupivacaine (a), ropivacaine (b) and bupivacaine (c).
Table 1.
Degree of inhibition induced by 20 μM levobupivacaine, ropivacaine and bupivacaine at the end of 5 s pulses to −10 mV and at the tail HERG currents

Figure 2 shows the concentration-response curves for the blocking effects of HERG channels induced by levobupivacaine, ropivacaine and bupivacaine calculated in a range of concentrations between 2 μM and 500 μM. Suppression of current at the end of 5 s depolarizations to −10 mV was used as an index of steady-state inhibition (f=[1−(IDrug/IControl)]×100). Non-linear least-squares fits of the concentration-response equation (see Materials) to the individual data points yielded apparent EC50's for levobupivacaine, ropivacaine and bupivacaine of 10.2±1.2 μM (n=27), 20.3±2.5 μM (n=27), and 18.0±1.1 μM (n=33) with significant differences between levobupivacaine and ropivacaine (P<0.001) and also between levobupivacaine and bupivacaine (P<0.001). The Hill coefficients obtained by this fitting procedure were 1.02±0.14, 0.81±0.13 and 1.16±0.13, for levobupivacaine, ropivacaine and bupivacaine, respectively. These nH values close to unity suggest that binding of one drug molecule/channel is sufficient to block potassium permeation.
Figure 2.
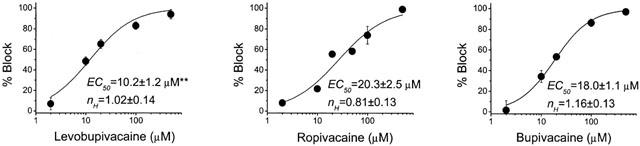
Concentration dependence of levobupivacaine-, ropivacaine- and bupivacaine-induced block of HERG channels. Reduction of current (relative to control) at the end of 5-s depolarizing steps from −80 mV to −10 mV was used as index of block. The continuous line represents the fit of the experimental data to a Hill equation. Each point represents the mean±s.e.mean of 3–16 experiments. **P<0.001. The r2 values for these fits were 0.99199, 0.97676 and 0.99794 for levobupivacaine, ropivacaine and bupivacaine respectively.
Stereoselective block of HERG channels induced by bupivacaine enantiomers
As shown in Figures 1 and 2, levobupivacaine was more potent than bupivacaine in blocking HERG channels. Since bupivacaine is the mixture of two enantiomers S(−) bupivacaine (levobupivacaine) and R(+)bupivacaine (dextrobupivacaine), these results would suggest that block of HERG channels by bupivacaine enantiomers is stereoselective. In order to analyse this hypothesis, the effects of dextropuvicaine (20 μM) on HERG channels were studied. Dextrobupivacaine did block HERG channels by 43.7±5.8% (n=8; P<0.01 vs levobupivacaine) and 33.3±6.0% (n=8; P<0.01 vs levobupivacaine) when measured at the end of 5 s depolarizing pulses to −10 mV and at the maximum amplitude of the tail currents recorded at −60 mV. Thus, HERG channels block by bupivacaine is stereoselective, levobupivacaine being ≈2 fold more potent than dextrobupivacaine.
Voltage-dependent block of HERG channels induced by levobupivacaine, ropivacaine and bupivacaine
Figure 3a shows the IV relationships obtained by plotting the amplitude of the HERG current at the end of 5 s pulses vs membrane potential in the absence and in the presence of 20 μM levobupivacaine (left panel), ropivacaine (middle panel) and bupivacaine (right panel). Under control conditions, the IV relationship exhibits the characteristic bell-shape that increases from −40 mV to ≈−10 mV and, due to the fast C-type inactivation of HERG channels, it decreased with further depolarization (Spector et al., 1996; Smith et al., 1996). The three drugs decreased the amplitude of the current at all membrane potentials tested. Figure 3b shows the activation curves obtained under control conditions and in the presence of 20 μM of levobupivacaine, ropivacaine or bupivacaine. The activation curves obtained in the presence of drug were normalized to match those obtained in its absence (dashed lines). As it can be observed, levobupivacaine, ropivacaine or bupivacaine did not modify the midpoint or the slope factors of the activation curves. Figure 3c shows the relationships between the relative tail current in the presence of the drugs and the membrane potential, superimposed with the activation curve of the channels obtained under control conditions (Eh and s values of the current were −9.9±1.1 mV and −10.0±0.3 mV, n=48, respectively). In the presence of the three local anaesthetics the relative current steeply decreased in the membrane potential range coinciding with the activation of HERG channels, indicating that levobupivacaine, ropivacaine and bupivacaine inhibited HERG current in a voltage-dependent manner. Between 0 mV and +50 mV, a slight increase in block was observed that was quantified based on the Woodhull model. The solid line represents the fit to a Boltzmann equation, from which δ values of 0.10±0.01 (n=9), 0.10±0.02 (n=6) and 0.11±0.02 (n=8) for levobupivacaine, ropivacaine and bupivacaine were obtained.
Figure 3.
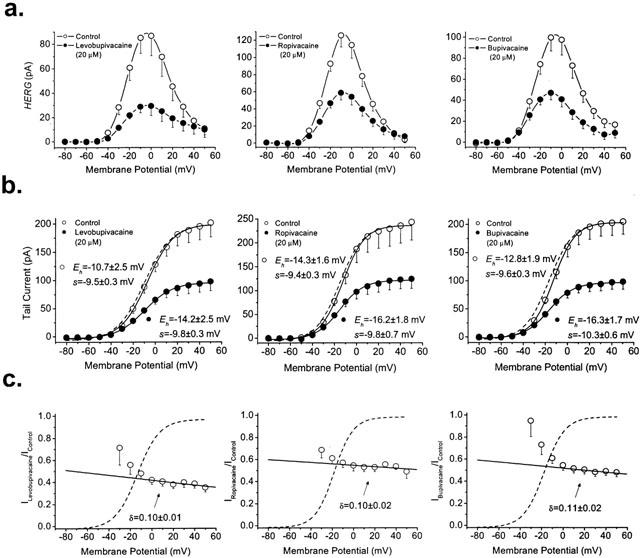
Voltage-dependent effects of levobupivacaine, ropivacaine and bupivacaine induced block of HERG channels. (a) IV relationships (5 s isochronal) of HERG channels obtained in the absence and in the presence of 20 μM of each drug. (b) Activation curves of HERG channels obtained under control conditions and in the presence of levobupivacaine, ropivacaine or bupivacaine. Dotted lines reflect the normalized activation curves obtained in the presence of drug and matching control values. Note that none of the three drugs shifted the activation curve. (c) Relative current vs membrane potential. Block increases in the range of membrane potentials that coincides with the activation of HERG channels. At membrane potentials positive to 0 mV block slightly increased consistent with δ values of ≈0.10 for the three drugs. Dashed lines represent the activation curves obtained under control conditions. Each point represents the mean±s.e.mean of 6–9 experiments. The r2 values for the Boltzmann fits of the activation curves were always >0.99. In the case of the Boltzmann fits to obtain the δ values, r2 were 0.8548, 0.7868 and 0.8152 for levobupivacaine, ropivacaine and bupivacaine, respectively.
Time dependent block of HERG channels
In order to explore the effects of levobupivacaine, ropivacaine and bupivacaine on the inactivation kinetics of HERG channels, the membrane potential was held at −80 mV and a three pulse protocol was applied, consisting in a 2 s depolarizing prepulse to +40 mV that fully activates HERG channels followed by a 20 ms hyperpolarizing pulse to −120 mV, which promotes the recovery from inactivation. Then, a 20 ms test pulse to different voltages between 0 mV and +50 mV was applied (Figure 4). Levobupivacaine, ropivacaine and bupivacaine accelerated the inactivation when measured at 0 mV decreasing the time constant of this process from 11.1±1.4 ms to 7.9±1.2 ms (n=4, P<0.01), from 14.9±2.1 to 11.5±1.2 ms (n=7, P<0.05) and from 14.9±1.9 to 10.7±1.0 ms (n=5, P<0.05), respectively. Block induced by levobupivacaine, ropivacaine and bupivacaine was measured at the maximum peak current at membrane potentials between 0 and +50 mV after a 20 ms hyperpolarizing pulse to −120 mV, which promotes the I→O transition. This block was significantly lower than that measured at the end of 5 s depolarizing pulses (Figure 1) to the same test potential (Table 2).
Figure 4.
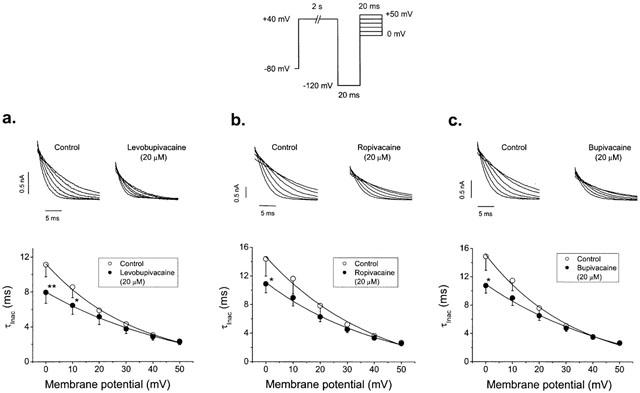
Effects of 20 μM levobupivacaine (a), ropivacaine (b) and bupivacaine (c) on the inactivation kinetics of HERG. Current records were obtained by using the protocol shown in the top of the figure. Top panels show current records of test pulses obtained in the absence and in the presence of 20 μM of each drug. Bottom panels show plots of the time constant of inactivation at different membrane potentials in the absence and in the presence of each drug. Each point represents the mean±s.e.mean of 4–5 experiments. *P<0.05 vs control conditions.
Table 2.
Degree of block of HERG channels induced by 20 μM levobupivacaine, ropivacaine and bupivacaine at the end of 5 s pulses to 0 mV and after a pulse to −120 mV

In order to further study the possible time-dependent block induced by these drugs, the voltage pulse protocol shown in Figure 5 was used. Because activation of HERG channels is strongly voltage dependent, from a holding potential of −80 mV HERG channels were rapidly activated by a 5-ms step to +180 mV followed by a 200 ms step to 0 mV, before returning to −60 mV and then to the holding potential (Figure 5a). By using this pulse protocol, the control current amplitude was nearly constant during the step to 0 mV. In the presence of 100 μM levobupivacaine, ropivacaine or bupivacaine, following the step to 0 mV, HERG current exponentially declined to a new steady state level (Figure 5a). In order to quantify this time dependency of block, we represented the relative current (IDrug/IControl) during the 200 ms pulse to 0 mV (Figure 5b). These current traces were fitted by a monoexponential function (solid lines in Figure 5b), and the time constants (τB) for levobupivacaine, ropivacaine and bupivacaine averaged 29.9±5.1 ms (n=4), 36.8±6.0 ms (n=4; P>0.05) and 36.1±3.8 ms (n=4; P>0.05), respectively. These results are consistent with binding of these drugs to activated channels with rapid kinetics, and explain why time-dependent block was not observed during the application of 5 s depolarizing pulses to 0 mV. This pulse protocol also allowed us to measure the recovery kinetics from inactivation by fitting the hook of the tail current at −60 mV (Figure 5c). As it can be observed, 100 μM levobupivacaine, ropivacaine and bupivacaine slowed the recovery process from 6.9±0.3 to 9.4±1.3 ms (n=5, P<0.01), from 7.1±0.4 to 9.8±0.3 ms (n=4, P<0.01) and from 6.4±0.9 to 10.0±2.5 ms (n=3, P<0.01), respectively.
Figure 5.
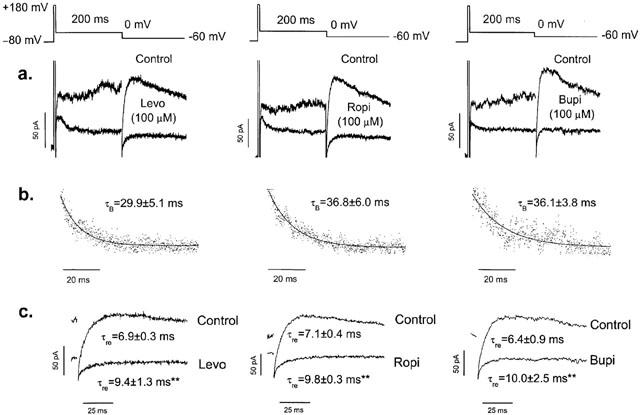
Time-dependent block of levobupivacaine, ropivacaine and bupivacaine after a fast activation of HERG channels, using the pulse protocol shown at the top. (a) original current records obtained in the absence and in the presence of 100 μM levobupivacaine, ropivacaine and bupivacaine. (b) relative current (IDrug/IControl) during the step to 0 mV together with the monoexponential fit to obtain the time constant (τB) for the development of block. (c) tail current records under control conditions and in the presence of drug by using this protocol. Note that the recovery from inactivation is slower in the presence of drug. **P<0.01 vs recovery from inactivation under control conditions. Numerical data in panels (b) and (c) represent the mean±s.e.mean of 4–5 experiments. The r2 values for the monoexponential fits shown in panel (b) were 0.81923, 0.88711 and 0.78239 for levobupivacaine, ropivacaine and bupivacaine, respectively.
Time-dependent block was also observed at the tail currents in the presence of 20 μM levobupivacaine and bupivacaine but not in the presence of ropivacaine when recorded at −60 mV after a 5 s depolarizing pulse to 0 mV (Figure 6). In the absence of drug, HERG tail currents followed an exponential decay that was fitted to a biexponential equation with fast (τf) and slow (τs) time constants. Levobupivacaine (20 μM) slowed the τf from 278.3±31.4 to 537.6±62.9 ms (n=9; P<0.001) and the τs from 1345.7±134.5 to 1706.8±169.7 ms (n=9; P<0.05). Bupivacaine (20 μM) also slowed the deactivation process by increasing τf from 283.7±11.5 to 462.2±59.7 ms (n=12; P<0.01) and the τs from 1579.5±114.1 to 2060.9±250.7 ms (n=12; P<0.05). However, as it can be observed in Figure 6b, ropivacaine (20 μM) did not modify the kinetics of the deactivation process. In the absence of drug, τf and τs averaged 460.8±64.2 and 1975.1±293.4 ms (n=10) values, and in the presence of ropivacaine, τf and τs averaged 436.8±47.0 and 2123.1±356.1 ms, respectively (n=10, P>0.05).
Figure 6.
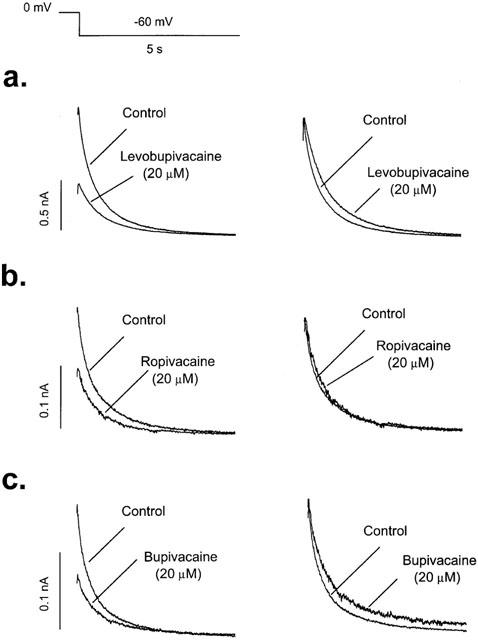
Time-dependent effects of 20 μM levobupivacaine (a), ropivacaine (b) and bupivacaine (c) on HERG deactivation. Tail currents were recorded at −60 mV after a 5-s depolarizing pulse to 0 mV. Left panels show superimposed current traces obtained in the absence and in the presence of each drug. Right panels show both current traces normalized to match control values. Note that levobupivacaine and bupivacaine, but not ropivacaine, slow the deactivation kinetics.
Effects of levobupivacaine, ropivacaine and bupivacaine on HERG inactivation availability
In order to analyse the effects of levobupivacaine, ropivacaine and bupivacaine on the inactivation availability, a three-pulse voltage clamp protocol was used (Figure 7a) (Spector et al., 1996; Smith et al., 1996; Johnson et al., 1999). Figure 7 shows the effects of 20 μM levobupivacaine, ropivacaine and bupivacaine on the voltage dependence of channel open-inactivated distribution (‘inactivation availability'). The dynamic nature of HERG currents precludes direct measurements of ‘steady state' inactivation, but the data shown approximate a voltage dependence of the distribution of channels between open and inactivated states. The 1 s step to +30 mV fully activates and inactivates the channels. The second step to the test potential (that ranged between −120 mV and +10 mV) allows some fraction of the channels to recover from inactivation. The instantaneous current after the third step to +30 mV allows measurements of the fraction of channels that have recovered from inactivation during the preceding test step. Left panels in Figure 7 show a plot of the current measured at +30 mV as a function of voltage. As it can be observed, the current falls off at negative potentials because the channels also begin to deactivate during the 20 ms pulse. We corrected the current magnitude for the amount of deactivation at each voltage by fitting the deactivating tail currents with an exponential function and then back-extrapolated this fit to the beginning of the hyperpolarizing pulse, which permitted us to estimate the fraction of channels deactivating during the 20-ms pulse and then increased the outward current accordingly (Smith et al., 1996). Right panels of Figure 7 show the Boltzmann fit of the corrected data in the absence and in the presence of the three local anaesthetics. Levobupivacaine, ropivacaine and bupivacaine at 20 μM shifted the voltage dependence of channel availability towards more negative potentials (determined by a Boltzmann fit) by 15.1±1.8 mV (n=4, P<0.05), 11.0±2.8 mV (n=5, P<0.05) and 12.0±2.6 mV (n=4, P<0.05), respectively, whereas none of them modified the slope factor. These results indicate that at any given membrane potential, HERG channels were more likely to be inactivated in the presence of levobupivacaine, ropivacaine or bupivacaine than under control conditions.
Figure 7.
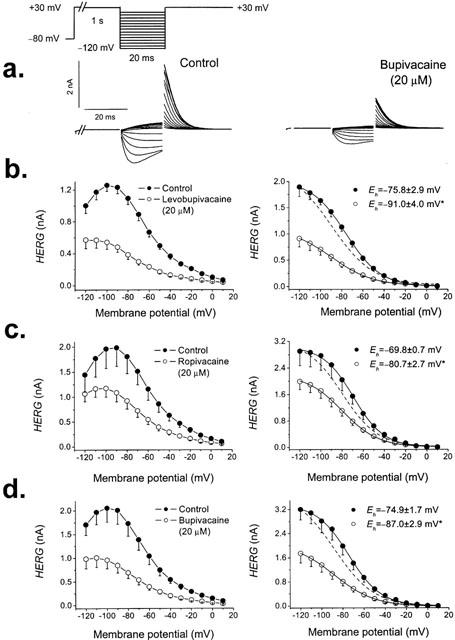
Apparent voltage dependence of channel availability. The pulse protocol used to obtain each data point is shown in the top of the Figure. (a) original traces obtained after applying such pulse protocol in the absence and in the presence of 20 μM bupivacaine. The peak current observed immediately after stepping to +30 mV was then plotted vs the test potential. Currents were recorded in the absence and in the presence of 20 μM of levobupivacaine (b), ropivacaine (c) and bupivacaine (d). Currents decreased at negative potentials due to deactivation during the 20 ms pulse. Left panels show non-corrected data and right panels show corrected data for deactivation (see Results). Each point represents the mean±s.e.mean of 4–5 experiments. The dashed lines represent the normalized fits to matching control. *P<0.05.
Discussion
In the present study, the effects of levobupivacaine, ropivacaine and bupivacaine on HERG channels stably expressed on CHO cells have been analysed. Our main findings were: (1) levobupivacaine, ropivacaine and bupivacaine blocked the HERG channels in a concentration-, time- and state-dependent manner, (2) levobupivacaine was ≈2 fold more potent that ropivacaine or bupivacaine in blocking these channels and (3) block of HERG channels by bupivacaine enantiomers was stereoselective, levobupivacaine being ≈2 fold more potent than dextrobupivacaine.
Effects of levobupivacaine, ropivacaine and bupivacaine on HERG channels
Levobupivacaine, ropivacaine and bupivacaine blocked HERG channels in a concentration-, time- and state-dependent manner. Block induced by these local anaesthetics was voltage-dependent, in such a way that block steeply increased in the range of potentials coinciding with the range of activation of the channels. At membrane potentials positive to 0 mV, block induced by these drugs increased with a shallower slope, consistent with δ values ≈0.10. At physiological pH, levobupivacaine, ropivacaine and bupivacaine are predominantly positively charged (pKa=8.1) and this voltage dependence of block can be interpreted as the consequence of the effects of the transmembrane electrical field on the interaction between the cationic form of the drugs and their receptor site at the channel. However, the relative voltage insensitivity of the inhibition at potentials more depolarized than 0 mV is difficult to reconcile with a simple electrostatic interaction, which predicts a continuous increase in the inhibition with progressive depolarization. Block induced by the three local anaesthetics under study increases over the range of voltages where the channels activate, suggesting that their binding may derive a significant fraction of its voltage sensitivity through coupling to channel gating. Unfortunately, at depolarized voltages the open and inactivated conformations of HERG channels are in rapid equilibrium, making it difficult to unequivocally identify the state(s) with which these three drugs interact.
Whereas bupivacaine induced a similar inhibition of the HERG current when measured at the end of depolarizing pulses to −10 mV and at the maximum tail currents, levobupivacaine and ropivacaine inhibited this current to a higher extent when measured at the end of depolarizing steps to −10 mV than at the maximum tail current. HERG channels inactivate faster than activate and thus, during depolarization, the amplitude of the current is reduced and, on repolarization, channels close through the open state, resulting in tail currents with higher amplitude (Spector et al., 1996; Smith et al., 1996). Therefore, these results may suggest that levobupivacaine and ropivacaine exhibit a lower affinity for the open than for the inactivated or the closed states of HERG channels. In agreement with these results, block induced by the three local anaesthetics studied, when measured at the maximum peak current of a test pulse to 0 mV applied after a hyperpolarizing pulse to −120 mV (that promotes the I→O transition), was significantly lower than that observed at the same voltage at the end of a 5 s depolarizing pulse. Moreover, levobupivacaine, ropivacaine and bupivacaine accelerated the inactivation process, slowed down the recovery from inactivation and shifted the inactivation availability curve without modifying the midpoint of the activation curve, thus suggesting a higher affinity for the inactivated state of HERG channels.
Levobupivacaine, ropivacaine and bupivacaine induced block was also time-dependent, being evident after a prepulse to +180 mV, suggesting a rapid drug binding (τB averaging ≈30 ms for the three drugs) to activated HERG channels, as previously described for cocaine (Zhang et al., 2001). Also, levobupivacaine and bupivacaine, but not ropivacaine, slowed the deactivation process of HERG channels, consistent with an open channel mechanism by which these drugs prevent channel closing by a ‘foot in the door' mechanism, similar to what has been proposed for quaternary ammonium compounds in potassium channels (Armstrong, 1971). The different effects on the deactivation kinetics observed with ropivacaine could be attributed to a different association or dissociation kinetics of this drug to its receptor site at the channel level. Another possibility could be that the n-butyl substituent present in levobupivacaine and bupivacaine, but not the shorter n-propyl of ropivacaine, can prevent the closure of the activation gate (‘foot in the door' mechanism). In fact, another new local anaesthetic, IQB-9302, blocks HERG channels to a similar extent than bupivacaine but, similarly to ropivacaine, it does not slow the deactivation process (González et al., 2001a, b). IQB-9302 has a similar chemical structure than bupivacaine, with the exception of the N-substituent, which is a cyclopropylmethyl instead of a butyl group. The length of the cyclopropylmethyl and the propyl groups are close (5.2 vs 5.9 Å, respectively) and shorter than the butyl group present in levobupivacaine and bupivacaine (7.2 Å), suggesting that a minimum length in the N-substituent is required to prevent the closure of the activation gate. All these results may suggest that these three local anaesthetics bind to both, open and inactivated states of HERG channels. Moreover, from the results on the inactivation kinetics and the recovery kinetics of inactivation, we can conclude that levobupivacaine, ropivacaine and bupivacaine stabilize HERG channels in the inactivated state.
Stereoselective block of HERG channels and structure-activity relationship
Levobupivacaine resulted to be more potent than bupivacaine, suggesting that block induced by the enantiomers of bupivacaine of HERG channels could be stereoselective. In fact, dextrobupivacaine (R(+)bupivacaine) resulted to be a less potent inhibitor of HERG current than levobupivacaine (S(−)bupivacaine). To our knowledge, this is the first evidence of drug stereoselective block of HERG channels. These findings suggest specified drug-channel interactions, since both enantiomers have identical biophysical properties, but differ in their three-dimensional structure (Franks & Lieb, 1991). It has been reported that bupivacaine, ropivacaine and IQB-9302 block cardiac hKv1.5 channels in a stereoselective manner (Valenzuela et al., 1995a; 1997; Longobardo et al., 1998; González et al., 2001a). Interestingly, in hKv1.5 channels, the R(+) enantiomers of these drugs were more potent than the S(−) enantiomers. Molecular determinants of stereoselective bupivacaine block of hKv1.5 channels are related to drug interactions with the amino acids T505, L508 and V512 located at the S6 segment (Franqueza et al., 1997). HERG channels have a phenylalanine (F656) at the V512 equivalent position that has been involved in binding of drugs with very different chemical structures, such as methanesulphonanilides, terfenadine and cisapride and, together with Y652 are unique to eag/erg K+ channels (Mitcheson et al., 2000). These structural differences could explain the opposite stereoselective effects of bupivacaine enantiomers observed in HERG and Kv1.5 channels.
The results shown in the present paper provide support for the fact that levobupivacaine exhibits a higher potency to block HERG channels than ropivacaine and bupivacaine. The only structural difference between these drugs is the N-substituent and/or the presence of another enantiomer (as in the case of racemic bupivacaine, which is the mixture of S(−)bupivacaine and R(+)bupivacaine). Thus, it seems that potency of block of HERG channels by bupivacaine-type local anaesthetics is related to the length of their N-substituent, as in the case of hKv1.5 channels (Valenzuela et al., 1995a; Longobardo et al., 1998; González et al., 2001b).
Practical implications of this study
Ventricular arrhythmias, including torsades de pointes, have been observed after accidental intravascular administration of bupivacaine to humans (Scott et al., 1989). Similar arrhythmias are commonly reported in patients with congenital or acquired long QT syndrome (Tamargo, 2000). It has been reported that block of cardiac Na+ and hKv1.5 channels induced by bupivacaine is stereoselective with the R(+) enantiomer being more potent that the S(−) one (Valenzuela et al., 1995a, b). Similar results have been observed with ropivacaine enantiomers on hKv1.5 channels (Valenzuela et al., 1997; Longobardo et al., 1998). Most adverse effects induced by bupivacaine have been attributed to dextrobupivacaine (Luduena et al., 1972; Aberg, 1972). Therefore, levobupivacaine and ropivacaine, the pure S(−) enantiomers of N-butyl- and N-propyl-pipecoloxylidide, have been developed as safer alternatives to racemic bupivacaine. The present study shows that levobupivacaine, ropivacaine and bupivacaine block HERG channels in a time-, state- and concentration-dependent manner at relevant clinical concentrations, and that levobupivacaine is the most potent local anaesthetic to block HERG channels, out of the three tested. These results could explain recent studies in which the incidence of ventricular arrhythmias was similar with these three anaesthetics (Groban et al., 2000). However, caution should be exerted before extrapolating the present results to the clinics, since many factors (temperature, β subunits, etc.) are not similar under these experimental conditions.
Acknowledgments
This study has been supported by CICYT SAF99-0069 and FIS 01/1130 Grants. The authors want to express their thanks to Drs Nattel, Hébert and Weerapura for providing us with the cell line expressing HERG channels, to Drs J.P. Johnson and F. Pérez-Vizcaíno for their valuable comments on the manuscript and to Guadalupe Pablo for her excellent technical assistance.
Abbreviations
- δ
fractional electrical distance
- EGTA
ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid
- IKur
ultrarapid delayed rectifier potassium current
- IV
Current-voltage relationship
References
- ABERG G. Toxicological and local anaesthetic effects of optically active isomers of two local anaesthetic compounds. Acta Pharmacol. Toxicol. 1972;31:273–286. [PubMed] [Google Scholar]
- AKERMAN B., HELLBERG I.B., TROSSVIK C. Primary evaluation of the local anaesthetic properties of the amino amide agent ropivacaine (LEA 103) Acta Anaesthesiol. Scand. 1988;32:571–578. doi: 10.1111/j.1399-6576.1988.tb02788.x. [DOI] [PubMed] [Google Scholar]
- ARMSTRONG C.M. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARKSON C.W., HONDEGHEM L.M. Mechanism for bupivacaine depression of cardiac conduction: fast block of sodium channels during the action potential with slow recovery from block during diastole. Anesthesiology. 1985;62:396–405. [PubMed] [Google Scholar]
- FOSTER R.H., MARKHAM A. Levobupivacaine: A review of its pharmacology and use as a local anaesthetic. Drugs. 2000;59:551–579. doi: 10.2165/00003495-200059030-00013. [DOI] [PubMed] [Google Scholar]
- FRANKS N.P., LIEB W.R. Stereospecific effects of inhalational general anesthetic optical isomers on nerve ion channels. Science. 1991;254:427–430. doi: 10.1126/science.1925602. [DOI] [PubMed] [Google Scholar]
- FRANQUEZA L., LONGOBARDO M., VICENTE J., DELPÓN E., TAMKUN M.M., TAMARGO J., SNYDERS D.J., VALENZUELA C. Molecular determinants of stereoselective bupivacaine block of hKv1.5 channels. Circ. Res. 1997;81:1053–1064. doi: 10.1161/01.res.81.6.1053. [DOI] [PubMed] [Google Scholar]
- GONZÁLEZ T., ARIAS C., MORENO I., CABALLERO R., DELPÓN E., TAMARGO J., VALENZUELA C. Comparative effects of bupivacaine, levobupivacaine, and ropivacaine on HERG channels stably expressed in CHO cells. Biophys. J. 2002;82:581a. doi: 10.1038/sj.bjp.0704978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GONZÁLEZ T., LONGOBARDO M., CABALLERO R., DELPÓN E., SINISTERRA J.V., TAMARGO J., VALENZUELA C. Stereoselective effects of the enantiomers of a new local anaesthetic, IQB-9302, on a human cardiac potassium channel (Kv1.5) Br. J. Pharmacol. 2001a;132:385–392. doi: 10.1038/sj.bjp.0703844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GONZÁLEZ T., LONGOBARDO M., CABALLERO R., DELPÓN E., TAMARGO J., VALENZUELA C. Effects of bupivacaine and a novel local anaesthetic, IQB-9302, on human cardiac K+ channels. J. Pharmacol. Exp. Ther. 2001b;296:573–583. [PubMed] [Google Scholar]
- GROBAN L., DEAL D.D., VERNON J.C., JAMES R.L., BUTTERWORTH J. Ventricular arrhythmias with or without programmed electrical stimulation after incremental overdosage with lidocaine, bupivacaine, levobupivacaine, and ropivacaine. Anesth. Analg. 2000;91:1103–1111. doi: 10.1097/00000539-200011000-00011. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- JOHNSON J.P., BALSER J.R., BENNETT P.B. Enhancement of HERG K+ currents by Cd2+ destabilization of the inactivated state. Biophys J. 1999;77:2534–2541. doi: 10.1016/s0006-3495(99)77088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEATING M.T., SANGUINETTI M.C. Molecular genetic insights into cardiovascular disease. Science. 1996;272:681–685. doi: 10.1126/science.272.5262.681. [DOI] [PubMed] [Google Scholar]
- KOTELKO D.M., SHNIDER S.M., DAILEY P.A., BRIZGYS R.V., LEVINSON G., SHAPIRO W.A., KOIKE M., ROSEN M.A. Bupivacaine-induced cardiac arrhythmias in sheep. Anesthesiology. 1984;60:10–18. doi: 10.1097/00000542-198401000-00004. [DOI] [PubMed] [Google Scholar]
- LIPKA L.J., JIANG M., TSENG G.N. Differential effects of bupivacaine on cardiac K channels: role of channel inactivation and subunit composition in drug-channel interaction. J. Cardiovasc. Electrophysiol. 1998;9:727–742. doi: 10.1111/j.1540-8167.1998.tb00960.x. [DOI] [PubMed] [Google Scholar]
- LONGOBARDO M., DELPÓN E., CABALLERO R., TAMARGO J., VALENZUELA C. Structural determinants of potency and steroselective block of hKv1.5 channels induced by local anesthetics. Mol. Pharmacol. 1998;54:162–169. doi: 10.1124/mol.54.1.162. [DOI] [PubMed] [Google Scholar]
- LUDUENA F., BOGADO E., TULLAR B. Optical isomers of mepivacaine and bupivacaine. Arch. Int. Pharmacodyn. Ther. 1972;200:359–369. [PubMed] [Google Scholar]
- MITCHESON J.S., CHEN J., LIN M., CULBERSON C., SANGUINETTI M.C. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U.S.A. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAU C., WANG S.Y., STRICHARTZ G.R., WANG G.K. Block of human heart hH1 sodium channels by the enantiomers of bupivacaine. Anesthesiology. 2000;93:1022–1033. doi: 10.1097/00000542-200010000-00026. [DOI] [PubMed] [Google Scholar]
- RODEN D.M., LAZZARA R., ROSEN M., SCHWARTZ P.J., TOWBIN J., VINCENT G.M. Multiple mechanisms in the long-QT syndrome. Current knowledge, gaps, and future directions. The SADS Foundation Task Force on LQTS. Circulation. 1996;94:1996–2012. doi: 10.1161/01.cir.94.8.1996. [DOI] [PubMed] [Google Scholar]
- SANCHEZ-CHAPULA J. Effects of bupivacaine on membrane currents of guinea-pig ventricular myocytes. Eur. J. Pharmacol. 1988;156:303–308. doi: 10.1016/0014-2999(88)90274-9. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JIANG C., CURRAN M.E., KEATING M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JURKIEWICZ N.K. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCOTT D.B., LEE A., FAGAN D., BOWLER G.M., BLOOMFIELD P., LUNDH R. Acute toxicity of ropivacaine compared with that of bupivacaine. Anesth. Analg. 1989;69:563–569. [PubMed] [Google Scholar]
- SMITH P.L., BAUKROWITZ T., YELLEN G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- SOLOMON D., BUNEGIN L., ALBIN M. The effect of magnesium sulfate administration on cerebral and cardiac toxicity of bupivacaine in dogs. Anesthesiology. 1990;72:341–346. doi: 10.1097/00000542-199002000-00021. [DOI] [PubMed] [Google Scholar]
- SPECTOR P.S., CURRAN M.E., ZOU A., KEATING M.T., SANGUINETTI M.C. Fast inactivation causes rectification of the IKr channel. J. Gen. Physiol. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRICHARTZ G.R. Local Anesthetics 1987Berlin, Springer-Verlag; >1st Ed [Google Scholar]
- TAMARGO J. Drug-induced torsade de pointes: from molecular biology to bedside. Jpn. J. Pharmacol. 2000;83:1–19. doi: 10.1254/jjp.83.1. [DOI] [PubMed] [Google Scholar]
- VALENZUELA C., DELPÓN E., FRANQUEZA L., GAY P., SNYDERS D.J., TAMARGO J. Effects of ropivacaine on a potassium channel (hKv1.5) cloned from human ventricle. Anesthesiology. 1997;86:718–728. doi: 10.1097/00000542-199703000-00025. [DOI] [PubMed] [Google Scholar]
- VALENZUELA C., DELPÓN E., TAMKUN M.M., TAMARGO J., SNYDERS D.J. Stereoselective block of a human cardiac potassium channel (Kv1.5) by bupivacaine enantiomers. Biophys. J. 1995a;69:418–427. doi: 10.1016/S0006-3495(95)79914-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VALENZUELA C., SNYDERS D.J., BENNETT P.B., TAMARGO J., HONDEGHEM L.M. Stereoselective block of cardiac sodium channels by bupivacaine in guinea pig ventricular myocytes. Circulation. 1995b;92:3014–3024. doi: 10.1161/01.cir.92.10.3014. [DOI] [PubMed] [Google Scholar]
- WHEELER D.M., BRADLEY E.L., WOODS W.T. The electrophysiologic actions of lidocaine and bupivacaine in the isolated, perfused canine heart. Anesthesiology. 1988;68:201–212. doi: 10.1097/00000542-198802000-00005. [DOI] [PubMed] [Google Scholar]
- WOODHULL A.M. Ionic blockade of sodium channels in nerve. J. Gen. Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG S., RAJAMANI S., CHEN Y., GONG Q., RONG Y., ZHOU Z., RUOHO A., JANUARY C.T. Cocaine blocks HERG, but not KvLQT1+minK, potassium channels. Mol. Pharmacol. 2001;59:1069–1076. doi: 10.1124/mol.59.5.1069. [DOI] [PubMed] [Google Scholar]