

Abstract
Chronic hypoxic treatment of rats (to induce pulmonary hypertension, PHT) for 14 days increased cGMP-inhibited cAMP specific phosphodiesterase (PDE3) and cGMP binding cGMP specific phosphodiesterase (PDE5) activities in pulmonary arteries. The objective of this study was to establish the molecular basis for these changes in both animal and cell models of PHT. In this regard, RT–PCR and quantitative Western blotting analysis was applied to rat pulmonary artery homogenates and human pulmonary ‘artery' smooth muscle cell (HPASMC) lysates.
PDE3A/B gene transcript levels were increased in the main, first, intrapulmonary and resistance pulmonary arteries by chronic hypoxia. mRNA transcript and protein levels of PDE5A2 in the main and first branch pulmonary arteries were also increased by chronic hypoxia, with no effect on PDE5A1/A2 in the intra-pulmonary and resistance vessels.
The expression of PDE3A was increased in HPASMCs maintained under chronic hypoxic conditions for 14 days. This may be mediated via a protein kinase A-dependent mechanism, as treatment of cells with Br-cAMP (100 μM) mimicked chronic hypoxia in increasing PDE3A expression, while the PKA inhibitor, H8 peptide (50 μM) abolished the hypoxic-dependent increase in PDE3A transcript.
We also found that the treatment of HPASMCs with the inhibitor of κB degradation Tosyl-Leucyl-Chloro-Ketone (TLCK, 50 μM) reduced PDE5 transcript levels, suggesting a role for this transcription factor in the regulation of PDE5 gene expression.
Our results show that increased expression of PDE3 and PDE5 might explain some changes in vascular reactivity of pulmonary vessels from rats with PHT. We also report that NF-κB might regulate basal PDE5 expression.
Keywords: hypertension, pulmonary, phosphodiesterase, cAMP, cGMP, NF-κB, cardiovascular disease, genes
Introduction
The pulmonary vascular bed is a low-pressure system with a resistance approximately one-tenth of the systemic circulation. In the normal lung, pulmonary vascular tone is regulated by a balance between the effects of vasodilators/anti-proliferative agents, such as prostacyclin and isoprenaline and vasoconstrictors/co-mitogens, such as serotonin and endothelin-1 (Fishman, 1998; MacLean, 1999a; Channick & Rubin, 2000). Acute hypoxia causes pulmonary arteriolar vasoconstriction and increased pulmonary arterial pressure (Ward & Aaronson, 1999). Chronic hypoxia induces a sustained increase in pulmonary arterial pressure and pulmonary vascular smooth muscle cell proliferation and the chronic hypoxic rat is widely used as a model for the study of chronic hypoxia-induced pulmonary hypertension (PHT) (Hunter et al., 1974; Rabinovitch et al., 1979).
Phosphodiesterase (PDE) catalyses the hydrolysis of cAMP and cGMP to inactive nucleotides (Beavo, 1995) and there are at least 11 members of the PDE family (Soderling & Beavo, 2000). We have shown that cAMP and cGMP PDE activity is increased in pulmonary arteries from rats with chronic hypoxia-induced PHT (MacLean et al., 1997) and this is correlated with decreased intracellular cAMP and cGMP levels (MacLean et al., 1996).
The cGMP-inhibited cAMP PDE (PDE3) is expressed as two isoforms termed PDE3A and PDE3B. There are at least two and possibly three isoforms of PDE3A. One is exclusively membrane-bound, while another is recovered in the cytosol and microsomes. PDE3B contains an N-terminal membrane-targeting domain. The activity of PDE3 is increased in main, first branch and intrapulmonary arteries from rats maintained under chronic hypoxic conditions (MacLean et al., 1997). PDE3 inhibitors also reverse the reduced responsiveness of pulmonary vessels to isoprenaline and forskolin in rats with PHT (Wagner et al., 1997).
The cGMP binding cGMP specific PDE (PDE5) is expressed as two isoforms termed PDE5A1 and PDE5A2. PDE5 activity is increased in the first branch and intrapulmonary artery from rats maintained under chronic hypoxic conditions (MacLean et al., 1997). PDE5 activity is also elevated in an ovine model of perinatal PHT (Hanson et al., 1998) and cGMP PDE inhibitors cause vasodilation of ovine pulmonary vessels (Ziegler et al., 1995). E4021, a new PDE5 inhibitor, inhibits hypoxic-induced vasoconstriction in isolated perfused lung from rats (Cohen et al., 1996). We have also found that decreased cGMP levels contribute to pharmacological synergy resulting in enhanced pulmonary vasoconstriction (MacLean, 1999b).
Therefore, the aim of the current study was to establish the molecular mechanism underlying the hypoxic-dependent changes in PDE3 and PDE5 activity. Furthermore, others have shown that chronic hypoxia can induce activation of the nuclear factor kappaB (NF-κB) pathway (Chiarugi et al., 1999). We have therefore investigated whether the PDE3 and PDE5 genes are under the control of the NF-κB pathway in cultured human pulmonary artery smooth muscle cells (HPASMCs).
Methods
Animal studies
Male Wistar rats of 28–30 days (at start of experiment) were placed in a specially designed perspex hypobaric chamber (Royal Hallamshire Hospital, Sheffield). This was depressurised, over 2 days, to 550 mbar (equivalent PO2 of 110 mmHg). The temperature of the chamber was maintained at 21–22°C and the chamber was ventilated with air at approximately 45 1 min−1. Animals were maintained in these hypoxic/hypobaric conditions for 2 weeks. Aged-matched controls were maintained in room air under normal atmospheric pressures. The right ventricle of the heart was carefully dissected free of the septum and left ventricle and these were blotted lightly and weighed. Pulmonary hypertension was assessed by measuring the ratio of right ventricular (RV)/total ventricular (TV) weight. This is a well-established index of the degree of pulmonary hypertension in rats (Hunter et al., 1974).
Cell culture
HPASMC (Clontech) were maintained in SmGM-2 and grown to confluence. After this time, cells were maintained under normoxic and hypoxic (550 bar, 10% O2, 5% CO2, balance N2) conditions for 14 days.
Homogenate/lysate preparation
Arteries were combined and homogenized with buffer containing 0.25 M-sucrose, 10 mM-Tris/HCl, pH 7.4, 1 mM-EDTA, 0.1 mM phenylmethylsulphonyl fluoride (PMSF) and 2 mM-benzamidine. Cells were re-suspended in the same buffer and lysates prepared by passing them through a 0.24 mm gauge syringe at least five times.
PCR
The PCR reaction was carried out using cDNA prepared using reverse transcriptase. The following protocol was used for the PCR: 95°C for 5 min and 15–35 cycles of 95°C for 30 s 50°C for 30 s and 72°C for 1 min and 40 s. This was followed by a final extension of 10 min at 65°C. PCR with specific forward and reverse regular or restriction tagged primers was used to amplify PDE transcripts. The PDE3A forward primer was CTG GCC AAC CTT CAG GAA TC and the reverse primer was GCC TCT TGG TTTCCC TTT CTC. The PDE3B forward primer was AAT CTT GGT CTG CCC ATC AGT CC and the reverse primer was TTC AGT GAG GTG GTG CAT TAG CTG. The PDE5 forward primer was CGA TGC TGA TGACAG CTT GTG ATC and the reverse primer was CAAGAG CTT GCC ATT TCT GCC. The PDE3A and PDE3B primers were designed to amplify regions corresponding to 3011–3415 and 2902–3201 in human PDE3A and B respectively. The PDE5 primers were designed to amplify 2338–2637 in bovine PDE5. Results obtained for all the gene transcripts studied were obtained using RT–PCR conditions that yielded linear amplification rates.
PDE assay
The assay of PDE activity was by the two-step radiotracer method using 0.5 μM [3H] cAMP or [3H] cGMP according to Thompson & Appleman (1971).
Western blotting
Nitrocellulose membranes were blocked for 1 h at 4°C in 10 mM-phosphate buffered saline (PBS) and 0.1% (v v−1) Tween-20 containing 5% (w v−1) non-fat dried milk and 0.001% (w v−1) thimerisol. The nitrocellulose sheets were incubated overnight at 4°C in blocking solution containing anti-PDE5 antibodies (Calbiochem, U.K.). The sheets were washed with PBS and 0.1% (v v−1) Tween-20 prior to incubation with HRP-linked anti-rabbit antibodies in blocking solution for 2 h at room temperature. After washing the blots as above, the immunoreactive bands were detected using an enhanced chemiluminescence kit.
Quantification and analysis
RT–PCR and Western blotting results were quantified by densitometry using a Bio RAD imaging densitometer (Model G.S.-690) in conjunction with Molecular Analyst Software, Version 2.1 (Bio Rad laboratories (U.S.A.). Optical densities were expressed as arbitary units. Statistical comparisons were by unpaired Student t-test with significance set at P<0.05.
Results
Pulmonary hypertension
Right ventricular/total ventricular ratios were 0.202±0.013 and 0.336±0.053 for normobaric-treated versus hypobaric-treated animals (n=40).
PDE3A/B transcript levels
Two products of 405 and 300 base pairs were amplified by RT–PCR from main, first branch, intrapulmonary and small resistance vessel total RNA (Figure 1). The amplicons were not obtained when reverse transcriptase was omitted from the first strand DNA synthesis step (data not shown). The alignment of the 405 and 300 base pair products with the corresponding regions in the human PDE3A and PDE3B revealed 90 and 92% similarity in their nucleotide sequences respectively (data not shown).
Figure 1.

The detection of PDE3A/B transcripts by RT–PCR. RT–PCR with specific PDE3A/B primers using total RNA (5 μg/sample) from main, first, intrapulmonary and resistance pulmonary vessels from rats maintained under normobaric (−) and chronic hypobaric (+) conditions. RT–PCR was performed from the same RNA samples with specific G3PDH primers using total RNA (5 μg/sample) to ensure equal loading of samples. These are representative results of an experiment performed four times. Abbreviations: H=chronic hypoxia.
PDE3A/B transcript levels were elevated by chronic hypoxia in all the vessels (Figure 1). As a control for the RT–PCR, we amplified G3PDH transcript with specific primers as a method for ensuring equal amounts of total RNA had been used for the amplification of the PDE3A/B and PDE5 transcripts. Identical amounts of G3PDH transcript were amplified under normoxic and hypoxic conditions from each individual vessel (Figure 1). The PDE3A/G3PDH transcript ratio in normobaric-treated and hypobaric-treated animals respectively were: main branch, 1.03±0.02; 1.47±0.08; first branch, 1.01±0.04; 1.92±0.11; intrapulmonary, 1.03±0.05; 2.07±0.2, resistance vessels, 0.91±0.03; 1.92±0.2 (n=4, P<0.05 versus normobaric-treated animals, Student t-test). The PDE3B/G3PDH transcript ratio in normobaric-treated and hypobaric-treated animals respectively were: main branch, 1.02±0.04; 1.6±0.1; first branch, 1±0.08; 1.95±0.09; intrapulmonary, 1.01±0.06; 1.52±0.11, resistance vessels, 0.99±0.05; 1.2±0.02 (n=4, P<0.05 versus normobaric-treated animals, Student t-test).
PDE5 transcript and protein levels
PDE5A2 (Mr=93 kDa) was expressed in the main branch vessel (Figure 2a). Routinely, very low levels of PDE5A2 were found in the first branch, although this was barely detectable unless Western blots were over-exposed (Figure 2a). PDE5A1 (Mr=98 kDa) and PDE5A2 were also detected in the intrapulmonary and small resistance vessel homogenates (Figure 2a). The PDE5A1 band co-migrated with a recombinant bovine PDE5A1 standard (data not shown). Chronic hypoxia increased PDE5A2 protein levels above basal in the main branch and first branch pulmonary vessels. The per cent increase in PDE5A2 protein expression in the main branch was 54±12% (n=4, P<0.05 versus normobaric-treated animals). Chronic hypoxia did not modulate PDE5A1/A2 protein levels in the intrapulmonary and resistance vessels (Figure 2a).
Figure 2.

The detection of PDE5 transcript and protein. (a) Western blot of homogenates from main, first, intrapulmonary and resistance pulmonary vessels (10 μg protein/sample were loaded onto SDS–PAGE) with anti-PDE5 antibodies from rats maintained under normobaric (−) and chronic hypobaric (+) conditions. (b) RT–PCR with PDE5 primers using total RNA (5 μg/sample) from main and first branch vessels from rats maintained under normobaric (−) and chronic hypobaric (+) conditions. RT–PCR was performed from the same RNA samples as with specific PDE3A/B and G3PDH primers (see Figure 1). These are representative results of an experiment performed four times. Abbreviations: H=chronic hypoxia.
A single PDE5 mRNA product of 300 base pairs (25 cycles) was amplified by RT–PCR from main and first branch vessel total RNA (Figure 2b). The amplicon was not obtained when reverse transcriptase was omitted from the first strand DNA synthesis step (data not shown). The alignment of the 300 base pair product with the corresponding region in human PDE5 revealed 92% similarity in the nucleotide sequence (data not shown). Significantly, PDE5 transcript levels were increased in the main and first branch vessels by chronic hypoxia and were therefore correlated with a similar increase in PDE5 protein expression in these vessels (Figure 2b). The PDE5A/G3PDH transcript ratio in normobaric-treated and hypobaric-treated animals respectively were: main branch, 1±0.02; 1.4±0.04; first branch, 1.01±0.05; 1.65±0.05; intrapulmonary, 1.02±0.04; 1.01±0.05, resistance vessels, 0.97±0.04; 0.98±0.11 (n=4, P<0.05 versus normobaric-treated animals for main and first branch pulmonary arteries, Student t-test).
Cultured HPASMCs
We next investigated whether similar changes in the expression of PDE3 and PDE5 could be induced by chronic hypoxia in a cultured HPASMC model. PDE3A, PDE3B and PDE5 transcripts were expressed in these cells (Figure 3a). The alignment of the 405 and 300 base pair products obtained using PDE3 primers with the corresponding regions in the human PDE3A and PDE3B revealed 100% and 99% similarity in their nucleotide sequences respectively (data not shown). The alignment of the 300 base pair product from the PCR using PDE5 primers with the corresponding region in human PDE5 revealed 100% similarity in the nucleotide sequence (data not shown).
Figure 3.
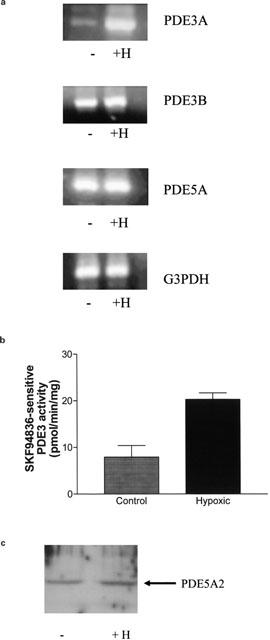
The effect of chronic hypoxia on PDE expression. (a) RT–PCR of PDE3A, PDE3B, PDE5 and G3PDH transcripts using 5 μg total RNA/sample from HPASMCs maintained under normoxic and hypoxic conditions for 14 days. (b) Histogram showing the chronic hypoxic-dependent increase in SKF94836-sensitive PDE3 activity (SKF94836, 10 μM) after 14 days. Results are means±s.d. for n=3 separate cell preparations. (c) Western blot showing the presence of PDE5A2 (93 kDa) in lysates (10 μg protein/sample were loaded onto SDS–PAGE) from cells maintained under normoxic (−) and chronic hypoxic (+) conditions for 14 days. (a) and (c) are representative results of an experiment performed at least 3–4 times. Abbreviations: H=chronic hypoxia.
PDE3A, but neither PDE5 nor PDE3B nor G3PDH transcript levels, was increased after 14 days of chronic hypoxia (Figure 3a). The PDE3A/G3PDH transcript ratios in normoxic-treated and hypoxic-treated cells respectively were: 1.01±0.06 and 2±0.1 (n=3, P<0.05 versus normoxic). For PDE3B and PDE5A, the ratios were 1.01±0.05 and 1±0.07 and 1±0.03 and 0.99±0.05 respectively (n=3–4). The increase in PDE3A transcript levels was correlated with an ∼1.5 fold increase in total cAMP PDE activity, measured at 0.5 μM cAMP. There was a 2.57 fold increase in SKF94836-sensitive PDE3A activity in response to chronic hypoxia (control versus hypoxic: 7.9±2.5 pmol min mg−1 protein versus 20.3±1.4 pmol min mg−1 protein, n=3, Figure 3b). Data showing that chronic hypoxia has no effect on PDE3B transcript levels, suggests that the increase in PDE activity can be attributed exclusively to PDE3A, since it was substantially reduced by addition of the type-selective PDE3 inhibitor, SKF94836 (10 μM) to the PDE assay. The Ki for PDE3 inhibition is approximately 2 μM (Murray et al., 1990) 10 μM SKF94836 was used to ensure complete inhibition of PDE3. It is well accepted that SKF94836 is highly selective for PDE3 inhibition at the concentration used in this study.
Western blotting with anti-PDE5 antibodies showed that the PDE5A2 isoform was expressed in cultured HPASMCs (Figure 3c). However, chronic hypoxia had no effect on protein expression (Figure 3c) or cGMP PDE activity, measured at 0.5 μM cGMP (data not shown).
We next investigated the mechanism underlying the hypoxic-dependent increase in PDE3A expression in the HPASMCs. Treatment of cultured cells with Br-cAMP (100 μM, 24 h) mimicked chronic hypoxia by inducing an increase in PDE3A transcript levels (Figure 4a). There was no significant effect of Br-cAMP on PDE3B, PDE5 or G3PDH transcript levels versus control (Figure 4a). The PDE3A/G3PDH transcript ratios in control and Br-cAMP-treated cells respectively were: 0.99±0.03 and 1.5±0.06 (n=3, P<0.05 versus control). For PDE3B and PDE5A, the ratios were 0.99±0.04 and 1±0.02 and 0.98±0.07 and 0.98±0.11 respectively (n=3). The increase in PDE3A transcript was correlated with a 1.8 fold increase in total cAMP PDE activity measured at 0.5 μM cAMP, which was completely ablated by addition of the type-selective PDE3 inhibitor, SKF94836 (10 μM) to the PDE assay. There was a 2.41 fold increase in SKF94836-sensitive PDE3A activity in response to Br-cAMP (control versus Br-cAMP-treated, 9.8±0.3pmol min mg−1 protein versus 23.6±1 pmol min mg−1, n=3, Figure 4b). The fact that Br-cAMP has no effect on PDE3B transcript levels and that SKF94836 abolishes the increase in PDE activity suggests that the increase in response to Br-cAMP can be attributed exclusively to PDE3A.
Figure 4.
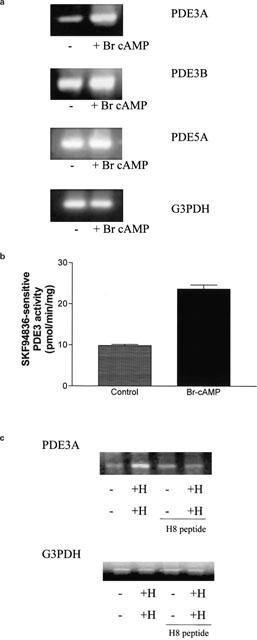
Hypoxic-induced expression of PDE3A is mediated via a cAMP-dependent mechanism. (a) RT–PCR of PDE3A, PDE3B, PDE5 and G3PDH transcripts using 5 μg total RNA/sample from HPASMCs treated with and without Br-cAMP (100 μM) for 24 h. (b) Histogram showing the increase in SKF94836-sensitive PDE3 activity (SKF94836, 10 μM) in cells treated with Br-cAMP (100 μM) for 24 h. Results are means±s.d. for n=3 separate cell preparations. (c) RT–PCR of PDE3A and G3PDH transcripts using 5 μg total RNA/sample from cells maintained under normoxic and hypoxic conditions in the presence and absence of H8 peptide (50 μM) for 14 days. (a) and (c) are representative results of an experiment performed three times. Abbreviations: H=chronic hypoxia.
Treatment with the PKA/PKG inhibitor, H8 peptide reduced the hypoxic-dependent increase in PDE3A transcript level (Figure 4c). The PDE3A/G3PDH transcript ratios were: Control, 1±0.09; Hypoxic 1.84±0.12; Control/H8 peptide, 1.11±0.15; Hypoxic/H8 peptide, 0.92±0.1 (n=3, P<0.05 for hypoxic versus normoxic-treated cells).
The effect of TLCK on PDE expression
TLCK inhibits trypsin-like serine proteinases and blocks interferon- and LPS-induced NF-κB-dependent nitric oxide synthase induction with an IC50=80 μM (Schini-Kerth et al., 1997). The treatment of HPASMCs with TLCK (100 μM, 14 days), an inhibitor of IkB degradation, substantially reduced basal PDE5 transcript levels (Figure 5a). TLCK had no effect on the chronic hypoxic-dependent increase in PDE3A transcript levels (Figure 5a). The PDE5A/G3PDH transcript ratios were Control, 1.02±0.03; Hypoxic 0.98±0.05; Control/TLCK, 0.58±0.05; Hypoxic/TLCK, 0.59±0.1 (n=3, P<0.05 for TLCK/normoxic and TLCK/hypoxic vs normoxic-treated cells). A similar TLCK-induced reduction in PDE5A2 protein expression was demonstrated in cell lysates (Figure 5b).
Figure 5.

The effect of TLCK on PDE3A and PDE5 expression. (a) RT–PCR of PDE5 and PDE3A transcripts using 5 μg total RNA/sample from HPASMCs treated with and without TLCK (100 μM, 14 days) under normoxic (−) and chronic (+) hypoxic conditions for 14 days. (b) Western blot (with anti-PDE5 antibodies) of lysates from cells treated with and without TLCK (100 μM, 14 days) under normoxic (−) and chronic (+) hypoxic conditions for 14 days. These are representative results of an experiment performed three times. The TLCK data shown for PDE5 is taken from the same gel as the experiment on PDE3A expression. Abbreviations: H=chronic hypoxia.
Discussion
Endothelial NO synthase (eNOS) is increased in both large and small pulmonary arteries from chronic hypoxic rats (Le Cras et al., 1996). However, acetylcholine-induced vasodilation is decreased in the large arteries (MacLean et al., 1995), but actually increased in the resistance arteries (MacLean & McCulloch, 1998). Consistent with this, cGMP levels are decreased in the large pulmonary arteries but unchanged in the resistance arteries (MacLean et al., 1996). Here we provide a molecular mechanism to explain these previous observations in that we have shown that PDE5 activity is increased in the large pulmonary arteries through synthesis of PDE5A2 protein, accounting for the decreased cGMP levels and subsequent decrease in acetylcholine-induced vasodilation. This might occur either via new synthesis of PDE5A2 mRNA or stabilization of pre-existing mRNA. These findings are consistent with previous reports showing that PDE5 protein expression is increased in lambs with PHT, induced by aorta-pulmonary vascular graft placement (Black et al., 2001). In our current study, PDE5 levels are unchanged in the resistance arteries. This will preserve the ability of acetylcholine to induce vasodilation, which may actually be enhanced due to increased levels of guanylyl cyclase (Li et al., 1999). This is consistent with the observations of Oka (2001) who showed that sildenafil selectively vasodilates the large pulmonary arteries but not the resistance arteries. Sildenafil also has a protective effect against the development of PHT in micc (Zhao et al., 2001). We previously demonstrated that there was an increase in total cGMP PDE activity in the main, first branch- and intra-pulmonary arteries. Further analysis indicated that calcium/calmodulin stimulated PDE was increased in the main pulmonary artery, indicating an increase in PDE1 activity and zaprinast-inhibited PDE activity was increased in the first branch and intra-pulmonary artery (MacLean et al., 1997). We chose to investigate the mechanisms behind increased PDE5 activity due to the current interest in the use of sildenafil for PHT (Sanjay et al., 2000; Ghofrani et al., 2002). There is an apparent discrepancy in the results from our previous study and the current one in that zaprinast-inhibited activity was not increased in the main pulmonary artery whilst we now show that there is an increase in PDE5A2 mRNA transcript and protein. It is possible that PDE1 and PDE5 are subject to additional regulatory factors under chronic hypoxic conditions (independent of changes in protein expression) that might explain the discrepancy. For example, phosphorylation of PDE5 by PKA markedly increases activity, but also reduces the sensitivity of PDE5 to inhibition by zaprinast (Burns et al., 1992). Therefore, it is possible that PDE5 in the main branch is refractory to inhibition by zaprinast. In this regard, Hanson et al. (1998) have shown that PDE5 is aberrantly phosphorylated in lambs with PHT.
PDE3 activity is increased in main, first branch and intrapulmonary vessels from rats maintained under chronic hypoxic conditions (MacLean et al., 1997). In light of these previous studies, the results described here suggest that this may in part be accounted for by the increase in PDE3A and PDE3B transcript levels. Therefore, the increase in PDE3A/B transcript is correlated with similar changes in activity. A hypoxic-dependent increase in PDE3A/B transcript was also detected in the resistance arteries. This was surprising because we did not detect a corresponding increase in PDE3 activity in this vessel in our previous study (MacLean et al., 1997). It is possible that the transcripts are not translated into protein in this vessel, or that PDE3 represents only a small fraction of the total cAMP hydrolysing activity.
PDE3A expression may also be increased via a cAMP-dependent mechanism in HPASMCs as evidenced by the data showing that the hypoxic-dependent increase in PDE3A expression was mimicked by treating cells with Br-cAMP and was ablated with the PKA inhibitor, H8 peptide. This data provides evidence that chronic hypoxia may induce the synthesis of PDE3A via a mechanism that might involve protein kinase A and/or CREB transcription factors. However, we cannot exclude possible effects via protein kinase G (PKG), which is also inhibited by H8 peptide. Since cAMP increased PDE3A expression, while H8 peptide reduces the hypoxic-dependent increase, we believe that there is a role for either PKA or PKG or both in the up-regulation of PDE3A. H8 peptide also inhibits myosin light chain kinase (MLCK) and PKC. However, we can exclude MLCK, because elevation in cAMP inhibits this enzyme (via PKA; Higashi et al., 1983). Under conditions of elevated cAMP, PDE3A expression is increased. Thus, if MLCK were involved, inhibition by H8 peptide should increase PDE3A expression, and not reduce it as observed here with H8 peptide. We can exclude PKC, because there is no evidence that PKA directly modulates PKC activity.
The PASMCs cells were derived from human main pulmonary arteries. There is heterogenity of PAVSMC types within the pulmonary circulation, some having a contractile phenotype and some a proliferative phenotype (Frid et al., 1997). Although the cultured cells used in this study may not be identical in phenotype to native cell phenotypes, cAMP and cGMP are anti-proliferative and also facilitate vasodilation, hence the mechanism for the preservation of cAMP and cGMP we have described is clinically relevant to both the potential vasodilator effects of sildenafil and its potential action as an anti-proliferative agent.
In contrast with main and first branch vessels, PDE5A2 expression was not modulated by chronic hypoxia in cultured human PASMCs. The reason for this difference is not known. However, one possibility may be the relative PKG content in the two systems. In this regard, it is conceivable that PDE5A2 expression may be regulated by cGMP and PKG under chronic hypoxic conditions. This would be analogous with our findings showing that cAMP/PKA may regulate PDE3A expression. Indeed, others have shown that elevated cGMP/PKG can induce PDE5 protein expression (Lin et al., 2000). It is also well established that PKG expression is markedly reduced when vascular smooth muscle cells are cultured. Thus, the inability of chronic hypoxia to modulate the expression of PDE5A2 in cultured human PASMCs maybe due to the absence of PKG.
We have also shown that treating cells with the inhibitor of IκB degradation, TLCK, reduced the basal expression of PDE5. While, TLCK does inhibit other proteases, these do not have specificity against transcription factors, such as NF-κB that could result in changes in PDE5 expression. Our finding is interesting as NF-κB is a transcription factor that regulates induction of enzymes involved in cellular stress responses (e.g. chronic hypoxia) that might affect vascular tone in PHT. For instance, NF-κB regulates expression of inducible NOS (iNOS) which in turn regulates vascular tone through the action of NO and cGMP. iNOS expression has been proposed to have a significant role in PHT (Le Cras et al., 1996). iNOS expression is also increased in the aorta of spontaneously hypertensive rats versus age-matched Wistar-Kyoto rats (Hong et al., 2000). Moreover, treatment of SHR with an NF-κB inhibitor, pyrrolidinedithiocarbamate and the inducible NOS inhibitor, aminoguanidine significantly reduced the development of hypertension and improved the diminished vascular responses to acetylcholine (Hong et al., 2000). With respect to a role for NF-κB in PHT, there is an inverse relationship between airway NO levels and NF-κB activation (Raychaudhuri et al., 1999) and NF-κB activation has been associated with the stimulated cellular oxidative stress associated with monocrotaline-induced PHT (Aziz et al., 1997). The possible inter-relationship between NF-κB and the induction of enzymes that regulate cGMP signalling (NOS and PDE5) is intriguing. Thus, the inhibitory effect of TLCK on basal PDE5 expression is important as it suggests that NF-κB inhibition might be useful in terms of suppressing PDE5 activity in PHT. This could represent an alternative approach in the treatment of PHT.
Our studies also provide a molecular mechanism to explain the beneficial effects of PDE3 and PDE5 inhibitors in reducing pulmonary vasoconstriction in models of PHT. The PDE3 inhibitor, cilostamide attenuated acute and chronic hypoxia-induced pulmonary hypertension in conscious lambs (Dukarm et al., 1998). The PDE5 inhibitor, sildenafil is a pulmonary vasodilator in conscious lambs with acute pulmonary hypertension (Weimann et al., 2000) and also prevents the development of PHT in mice exposed to hypoxia (Zhao et al., 2001), while the PDE inhibitor dipyridamole reduced PVR in newborn lambs with PHT (Dukarm et al., 1998). Indeed, the use of these PDE inhibitors may have clinical applications because dipyridamole also reduced PVR in pediatric patients with severe PHT and in some patients augmented the effects of inhaled NO (Ziegler et al., 1998). Both PDE3 and PDE5 inhibitors have been shown to amplify the pulmonary vasodilator response to inhaled prostacyclin (Schermuly et al., 1999) and such combination therapies are likely to be important approach in the future (Channick & Rubin, 2000). More recently, sildenafil has been shown to be an effective vasodilator in human patients with both primary and secondary PAH (Sanjay et al., 2000; Ghofrani et al., 2002) and is currently undergoing clinical trials for both primary and secondary PAH. Here we provide a mechanism for the effectiveness of sildenafil in PAH.
In conclusion, the results suggest that the reduced sensitivity of pulmonary arteries to certain vasodilators in rats with PHT might be due, in part, to changes in PDE3 and PDE5 expression and provide a molecular mechanism by which PDE3 and PDE5 inhibitors exert their beneficial effects. These findings are timely in the light of the recent study showing that the PDE5 inhibitor, sildenafil inhibits acute hypoxia-induced pulmonary hypertension in humans (Zhao et al., 2001) and sildenafil is now considered a potential new therapy for PHT.
Acknowledgments
We would like to thank the Universities of Glasgow and Strathclyde for the funding of a Ph.D studentship for F. Murray.
Abbreviations
- Br-cAMP
bromo-cAMP
- cAMP
adenosine 3′ 5′ cyclic monophosphate
- G3PDH
glycerol 3-phosphate dehydrogenase
- cGMP
guanosine 3′ 5′ cyclic monophosphate
- eNOS
endothelial NO synthase
- H
chronic hypoxia
- HPASMC
human pulmonary artery smooth muscle cells
- MLCK
myosin light chain kinase
- NF-κB
nuclear factor kappaB
- NO
nitric oxide
- PDE
phosphodiesterase
- PKA
protein kinase A
- PKG
protein kinase G
- PKC
protein kinase C
- PHT
pulmonary hypertension
- RV
right ventricle
- TLCK
Tosyl-Leucyl-Chloro-Ketone
- TV
total ventricle
References
- AZIZ S.M., TOBOREK M., HENNIG B., ENDEAN E., LIPKE D.W. Polyamine regulatory processes and oxidative stress in monocrotaline-treated pulmonary artery endothelial cells. Cell Biol. Int. 1997;21:801–812. doi: 10.1006/cbir.1997.0200. [DOI] [PubMed] [Google Scholar]
- BEAVO J.A. Cyclic nucleotide phosphodiesterase-functional implications of multiple isoforms. Physiol. Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- BLACK S.M., SANCHEZ L.S., MATA-GREENWOOD E., BEKKER J.M., STEINHORN R.H., FINEMAN J.R. SGC and PDE5 are elevated in lambs with increased pulmonary blood flow and pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2001;281:L1051–L1057. doi: 10.1152/ajplung.2001.281.5.L1051. [DOI] [PubMed] [Google Scholar]
- BURNS F., RODGER I.W., PYNE N.J. The catalytic sub-unit of protein kinase A triggers activation of the type V cyclic GMP-specific phosphodiesterase in guinea-pig lung. Biochem. J. 1992;283:487–491. doi: 10.1042/bj2830487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANNICK R.N., RUBIN L.J. Combination therapy for pulmonary hypertension: A glimpse into the future. Crit. Care Med. 2000;28:896–897. doi: 10.1097/00003246-200003000-00055. [DOI] [PubMed] [Google Scholar]
- CHIARUGI V., MAGNELLI L., CHIARUGI A., GALLO O. Hypoxia induces pivotal tumor angiogenesis control factors including p53, vascular endothelial growth factor and the NFkappaB-dependent inducible nitric oxide synthase and cyclooxygenase-2. J. Cancer Res. Clin. Oncol. 1999;125:525–528. doi: 10.1007/s004320050312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN A.H., HANSON K., MORRIS K., FOUTY B., MCMURTY I.F., CLARKE W., RODMAN D.M. Inhibition of cyclic 3′-5′-guanosine monophosphate-specific phosphodiesterase selectively vasodilates the pulmonary circulation in chronically hypoxic rats. J. Clin. Invest. 1996;97:172–179. doi: 10.1172/JCI118386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUKARM R.C., MORIN F.C., III, RUSSELL J.A., STEINHORN R.M. Pulmonary and systemic effects of the phosphodiesterase inhibitor dipyridamole in newborn with persistent pulmonary hypertension. Ped. Res. 1998;44:831. doi: 10.1203/00006450-199812000-00002. [DOI] [PubMed] [Google Scholar]
- FISHMAN A.L. Etiology and pathogenesis of primary pulmonary hypertension: a perspective. Chest. 1998;114 supp:242S–247S. doi: 10.1378/chest.114.3_supplement.242s. [DOI] [PubMed] [Google Scholar]
- FRID M.G., DEMPSEY E.C., DURMOWICZ A.G., STENMARK K.R. Smooth muscle cell heterogeneity in pulmonary and systemic vessels. Importance in vascular disease. Arterioscler. Thromb. Vasc. Biol. 1997;17:1203–1209. doi: 10.1161/01.atv.17.7.1203. [DOI] [PubMed] [Google Scholar]
- GHOFRANI H.A., WIEDEMANN R., SCHERMULY R.T., ROSE F., OLSCHEWSKI H., WEISSMANN N., SEEGAR W., GRIMMINGER F. Acute vasodilatory response to oral sildenafil (Viagra®) as compared to inhaled nitric oxide in pulmonary hypertension off different origin. Am. J. Crit. Care. Med. 2002;165:A573. [Google Scholar]
- HANSON K.A., ZIEGLER J.W., RYBALKIN S.D., MILLER J.W., ABMAN S.H., CLARKE W.R. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am. J. Physiol. 1998;275:L931–L941. doi: 10.1152/ajplung.1998.275.5.L931. [DOI] [PubMed] [Google Scholar]
- HIGASHI K., FUKUNAGA K., MATSUI K., MAEYAMA M., MIYAMOTO E. Purification and characterization of myosin light-chain kinase from porcine myometrium and its phosphorylation and modulation by cyclic AMP-dependent protein kinase. Biochim. Biophys. Acta. 1983;747:232–240. doi: 10.1016/0167-4838(83)90102-4. [DOI] [PubMed] [Google Scholar]
- HONG H.J., LOH S.H., YEN M.H. Suppression of the development of hypertension by the inhibitor of inducible nitric oxide synthase. Br. J. Pharmacol. 2000;131:631–637. doi: 10.1038/sj.bjp.0703603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUNTER C., BARER G.R., SHAW J.W., CLEGG E.J. Growth of the heart and lungs in hypoxic rodents: a model of human hypoxic disease. Clin. Sci. Mol. Med. 1974;46:375–391. doi: 10.1042/cs0460375. [DOI] [PubMed] [Google Scholar]
- LE CRAS T.D., XUE C., RENGASEMY A., JOHNS R.A. Chronic hypoxia upregulates endothelial and inducible NO synthase gene and protein expression in rat lung. Am. J. Physiol. 1996;270:L164–L170. doi: 10.1152/ajplung.1996.270.1.L164. [DOI] [PubMed] [Google Scholar]
- LI D., ZHOU N., JOHNS R.A. Soluble guanylate cyclase gene expression and localisation in rat lung after exposure to hypoxia. Am. J. Physiol. 1999;277:L841–L847. doi: 10.1152/ajplung.1999.277.4.L841. [DOI] [PubMed] [Google Scholar]
- LIN C., LAU A., TU R., LUE T.F. Expression of three isoforms of cGMP-binding cGMP-specific phosphodiesterase (PDE5) in human penile cavernosum. Biochem. Biophys. Res. Comm. 2000;268:628–635. doi: 10.1006/bbrc.2000.2187. [DOI] [PubMed] [Google Scholar]
- MACLEAN M.R. Endothelin and serotonin in pulmonary hypertension. J. Lab. Clin. Med. 1999a;134:105–114. doi: 10.1016/s0022-2143(99)90114-2. [DOI] [PubMed] [Google Scholar]
- MACLEAN M.R. Pulmonary hypertension, anorexigens and 5-HT: Pharmacological synergism in action. Trends Pharmacol. Sci. 1999b;20:490–495. doi: 10.1016/s0165-6147(99)01389-9. [DOI] [PubMed] [Google Scholar]
- MACLEAN M.R., JOHNSTON E.D., MCCULLOCH K.M., POOLEY L., HOUSLAY M., SWEENEY G. Phosphodiesterase isoforms in the pulmonary arterial circulation of the rat: Changes in pulmonary hypertension. J. Pharmacol. Exp. Ther. 1997;283:619–624. [PubMed] [Google Scholar]
- MACLEAN M.R., MCCULLOCH K.M. Influence of applied tension and nitric oxide on responses to endothelins in rat pulmonary resistance arteries. Br. J. Pharmacol. 1998;123:991–999. doi: 10.1038/sj.bjp.0701682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACLEAN M.R., MCCULLOCH K.M., BAIRD M. Effects of pulmonary hypertension on vasoconstrictor responses to endothelin-1 and sarafotoxin S6C and on inherent tone in rat pulmonary arteries. J. Cardiovasc. Pharmacol. 1995;26:822–830. doi: 10.1097/00005344-199511000-00020. [DOI] [PubMed] [Google Scholar]
- MACLEAN M.R., SWEENEY G., BAIRD M., MCCULLOCH K.M., HOUSLAY M., MORECROFT I. 5-Hydroxytryptamine receptors mediating vasoconstriction in pulmonary arteries from control and pulmonary hypertensive rats. Br. J. Pharmacol. 1996;119:917–930. doi: 10.1111/j.1476-5381.1996.tb15760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURRAY K.J., ENGLAND P.J., HALLAM T.J., MAGUIRE J., MOORES K., REEVES M.L., SIMPSON A.W., RINK T.J. The effects of siguazodan, a selective phosphodiesterase inhibitor, on human platelet function. Br. J. Pharmacol. 1990;99:612–616. doi: 10.1111/j.1476-5381.1990.tb12978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKA M. Phosphodiesterase 5 inhibition restores impaired Ach relaxation in hypertensive conduit pulmonary arteries. Lung Cell. Mol. Pharmacol. 2001;280:L432–L451. doi: 10.1152/ajplung.2001.280.3.L432. [DOI] [PubMed] [Google Scholar]
- RABINOVITCH M., GAMBLE W., NADAS A.S., MIETTINEN O., REID L. Rat pulmonary circulation after chronic hypoxia: haemodynamic and structural features. Am. J. Physiol. 1979;236:H818–H827. doi: 10.1152/ajpheart.1979.236.6.H818. [DOI] [PubMed] [Google Scholar]
- RAYCHAUDHURI B., DWEIK R., CONNORS M.J., BUHROW L., MALUR A., DRAZBA J., ARROLIGA A.C., ERZURUM S.C., KAVURU M.S., THOMASSEN M.J. Nitric oxide blocks nuclear factor kappaB activation in alveolar macropharges. Am. J. Respir. Cell. Mol. Biol. 1999;21:311–316. doi: 10.1165/ajrcmb.21.3.3611. [DOI] [PubMed] [Google Scholar]
- SANJAY P., WILKINSON J., GATZOULIS M.A. Sildenafil in primary pulmonary hypertension. New Eng. J. Med. 2000;343:1342–1343. doi: 10.1056/NEJM200011023431814. [DOI] [PubMed] [Google Scholar]
- SCHERMULY R.T., GHOFRANI H.A., ENKE B., WEISMMAN N., GRIMMINGER F., SEEGER W., SCHUDT C., WALMRATH D. Low-dose systemic phosphodiesterase inhibitors amplify the pulmonary vasodilatory response to inhaled prostacyclin in experimental pulmonary hypertension. Am. J. Resp. Crit. Care Med. 1999;160:1500–1506. doi: 10.1164/ajrccm.160.5.9901102. [DOI] [PubMed] [Google Scholar]
- SCHINI-KERTH V.B., BOESE M., BUSSE R., SISSLTHALER B., MULSCH A. TLCK prevents expression of iNOS in vascular smooth muscle by blocking activation of NF-κB. Athero. Thromb. Vasc. Biol. 1997;17:672–679. doi: 10.1161/01.atv.17.4.672. [DOI] [PubMed] [Google Scholar]
- SODERLING S.H., BEAVO J.A. Regulation of cAMP and cGMP: New phosphodiesterases and functions. Curr. Opin. Cell. Biol. 2000;12:174–179. doi: 10.1016/s0955-0674(99)00073-3. [DOI] [PubMed] [Google Scholar]
- THOMPSON W.J., APPLEMAN M.M. Multiple cyclic nucleotide phosphodiesterase from brain. Biochemistry. 1971;10:311–316. [PubMed] [Google Scholar]
- WAGNER R.S., SMITH C.J., TAYLOR A.M., RHOADES R.A. Phosphodiesterase inhibition improves agonist-induced relaxation of hypertensive pulmonary arteries. J. Pharmac. Exp. Ther. 1997;282:1650–1657. [PubMed] [Google Scholar]
- WARD J.P., AARONSON P.I. Mechanisms of hypoxic pulmonary vasoconstriction: can anybody be right. Respir. Physiol. 1999;115:261–271. doi: 10.1016/s0034-5687(99)00025-0. [DOI] [PubMed] [Google Scholar]
- WEIMANN J., ULLRICH R., HROMI J., FUJINO Y., CLARKE M.W., BLOCH K.D., ZAPOL W.M. Sildenafil is a pulmonary vasodilator in awake lambs with acute pulmonary hypertension. Anaesthesiology. 2000;92:1702–1712. doi: 10.1097/00000542-200006000-00030. [DOI] [PubMed] [Google Scholar]
- ZHAO L., MASON N.W., MORRELL N.W., KOJONAZAROV B., SADYKOV A., MARIPOV A., MIRRAKHIMOV M.M., ALDASHEV A., WILKINS M.R. Sildenafil inhibits hypoxia-induced pulmonary hypertension. Circulation. 2001;104:424–428. doi: 10.1161/hc2901.093117. [DOI] [PubMed] [Google Scholar]
- ZIEGLER J.W., IVY D.D., FOX J.J., KINSELLA J.P., CLARKE W.R., ABMAN S.H. Dipyridamole, a cGMP phosphodiesterase inhibitor, causes pulmonary vasodilation in the ovine fetus. Am. J. Physiol. 1995;269:H473–H479. doi: 10.1152/ajpheart.1995.269.2.H473. [DOI] [PubMed] [Google Scholar]
- ZIEGLER J.W., IVY D.D., WIGGINS J.W., KINSELLA J.P., CLARKE W.R., ABMAN S. Effects of dipyridamole and inhaled nitric oxide in pediatric patients with pulmonary hypertension. Am. J. Resp. Crit. Care Med. 1998;158:1388–1393. doi: 10.1164/ajrccm.158.5.9710117. [DOI] [PubMed] [Google Scholar]