

Abstract
KCNQ K+ channels are thought to underlie the M current of neurons. To probe if the KCNQ2 and KCNQ3 subtypes underlie the M current of rat superior cervical ganglia (SCG) neurons and of hippocampus, we raised specific antibodies against them and also used the cysteine-alkylating agent N-ethylmaleimide (NEM) as an additional probe of subunit composition.
Tested on tsA-201 (tsA) cells transfected with cloned KCNQ1-5 subunits, our antibodies showed high affinity and selectivity for the appropriate subtype. The antibodies immunostained SCG neurons and hippocampal sections at levels similar to those for channels expressed in tsA cells, indicating that KCNQ2 and KCNQ3 are present in SCG and hippocampal neurons. Some hippocampal regions contained only KCNQ2 or KCNQ3 subunits, suggesting the presence of M currents produced by channels other than KCNQ2/3 heteromultimers.
We found that NEM augmented M currents in SCG neurons and KCNQ2/3 currents in tsA cells via strong voltage-independent and modest voltage-dependent actions. Expression of individual KCNQ subunits in tsA cells revealed voltage-independent augmentation of KCNQ2, but not KCNQ1 nor KCNQ3, currents by NEM indicating that this action on SCG M currents likely localizes to KCNQ2. Much of the voltage-independent action is lost after the C242T mutation in KCNQ2.
The correspondence of NEM effects on expressed KCNQ2/3 and SCG M currents, along with the antibody labelling, provide further evidence that KCNQ2 and KCNQ3 subunits strongly contribute to the M current of neurons. The site of NEM action may be important for treatment of diseases caused by under-expression of these channels.
Keywords: potassium channel, antibodies, M current, KCNQ channel, cysteine, N-ethylmaleimide
Introduction
M current is a non-inactivating, neuronal K+ current with slow kinetics that is strongly modulated by the actions of G protein-coupled receptors. Wide interest in M current stems from its prevalence in the nervous system, its importance in controlling neuronal excitability (Jones et al., 1995; Wang & McKinnon, 1995) and its role in some forms of congenital epilepsy (Lerche et al., 2001). Muscarinic inhibition occurs via the M1 subtype of muscarinic acetylcholine receptor (Bernheim et al., 1992; Hamilton et al., 1997), pertussis toxin-insensitive G proteins of the Gq/11 class (Delmas et al., 1998; Haley et al., 1998) and an unidentified diffusible cytoplasmic messenger (Selyanko et al., 1992). M currents have also been characterized in hippocampal neurons (Halliwell and Adams, 1982). As in sympathetic neurons, hippocampal M currents are suppressed by muscarinic stimulation (reviewed in Brown et al., 1990), but there, another augmenting mode of modulation has also been described involving somatostatin receptors and possible 2nd-messenger actions of phospholipase A2 and arachadonic acid (Schweitzer et al., 1990; 1993).
The molecular correlate of M-type channels eluded researchers until recently. In the past 3 years, four members of the KCNQ family of K+ channel genes, consisting of KCNQ2-5 (Biervert et al., 1998; Charlier et al., 1998; Kubisch et al., 1999; Lerche et al., 2000; Singh et al., 1998) have emerged as candidates to underlie the M current of different neuronal cell types. These genes were identified from mutations that cause epileptic syndromes in humans, presumably resulting from neuronal hyper-excitability. Interestingly, mutations in the first member of this channel family, KCNQ1 (formerly called KvLQT1), that greatly lower the activity of the cardiac IKs current, produce a hyper-excited state of cardiac myocytes as well, leading to a form of long QT syndrome (Shalaby et al., 1997; Wang et al., 1996). Which of the KCNQ subunits make the classic M current of neurons? Heterologous expression of KCNQ2 and KCNQ3 subunits yields K+-selective channels that have important features of M current, including slow activation kinetics, a more negative threshold for activation relative to other K+ channels (Wang et al., 1998), and sensitivity to stimulation of muscarinic receptors (Selyanko et al., 2000; Shapiro et al., 2000). However, expression of KCNQ1 (Selyanko et al., 2000), KCNQ4 (Kubisch et al., 1999) and KCNQ5 (Lerche et al., 2000) also yield slowly-gated K+ currents that are suppressed by muscarinic agonists (Schroeder et al., 2000; Selyanko et al., 2000). Thus, M current could be formed by assembly of several different subunits of the KCNQ family, necessitating further characterization of the subunits that make M-type channels.
The M current has been most studied in neurons from the rat superior cervical ganglion (SCG), and in those cells, mRNA for KCNQ2, KCNQ3 (Wang et al., 1998) and KCNQ5 (Schroeder et al., 2000) has been detected. As heteromeric KCNQ2/3 channels and SCG M current have similar sensitivity to blockade by tetraethylammonium (TEA) ions and the compounds linopirdine and XE991, the McKinnon lab proposed that these heteromultimers underlie the M current of SCG neurons (Wang et al., 1998). Here, we focus on KCNQ2 and KCNQ3 and use antibodies specific to them, and additional pharmacology, to localize those subunits to these cells. Our results indicate that SCG neurons, as well as some hippocampal regions, express KCNQ2 and KCNQ3 subunits. Interestingly, other hippocampal regions express only one of these subunits, suggesting that in some regions of the hippocampus, M currents are made by channels other than KCNQ2/3 heteromultimers. Additionally, we demonstrate a potentially important mode of subunit-specific M current modulation.
Methods
Cells and expression of KCNQ2/3 channels
Plasmids containing human KCNQ1, human KCNQ2, rat KCNQ3, human KCNQ4 and human KCNQ5 (Genbank accessions NM000218, AF110020, AF091247, AF105202 and AF249278, respectively) were kindly given to us by Michael Sanguinetti (KCNQ1, University of Utah, Salt Lake City, UT, U.S.A.), David McKinnon (KCNQ2, KCNQ3, SUNY, Stony Brook, N.Y., U.S.A.), Thomas Jentsch (KCNQ4, Zentrum fur Molekulare Neurobiologie, Hamburg, Germany) and Klaus Steinmayer (KCNQ5, Aventis Pharm., Frankfurt Am Main, Germany). A plasmid containing the mouse M1 muscarinic receptor was given to us by Neil Nathanson (University of Washington). Human tsA-201 (tsA) cells are a simian virus 40 (SV40) T-antigen-expressing derivative of the human embryonic kidney cell line 293 (HEK293). Cells were grown in tissue culture dishes (Falcon) in tsA growth medium (DMEM or DMEM/F-12 nutrient mixture+10% heat-inactivated foetal bovine serum+0.2% penicillin/streptomycin) in a humidified incubator at 37°C (5% CO2) and passaged about every 5 days after exposure to Ca2+-free saline for 2 min. Plasmids were transfected as follows: DNA (approx. 2 μg total) was combined with 10 μl of Superfect transfection reagent (Qiagen) and 100 μl serum/antibiotic-free DMEM medium. After a 10 min wait for complexes to form, this was mixed with 600 μl tsA growth medium and incubated with tsA cells, grown to about 50% confluency, for 2 h which were then returned to the incubator. The next day, cells were plated onto poly-L-lysine coated coverslip chips and used within 3 days for electrophysiological experiments. As a marker for successfully transfected cells, 0.2 μg DNA encoding green fluorescent protein was co-transfected with channel and receptor DNA. Using this protocol, we found that >95% of green fluorescing cells express M-like currents in control experiments.
Mutagenesis
Amplicons for KCNQ2 subunit mutants T2 and C242T were produced by polymerase chain reaction (PCR) as follows. One nanogram of human KCNQ2 template, 60 pmol of dNTPs (Life Technologies, Grand Island, NY, U.S.A.), 4.0 U of Pfu polymerase (Stratagene, La Jolla, CA, U.S.A.), and 25 pmol of each primer (Life Technologies) were combined in a final reaction volume of 100 μl. PCR was performed in a DNA thermal cycler (model 480; Perkin-Elmer, Foster City, CA, U.S.A.) for 28 cycles with annealing temperatures appropriate for the oligonucleotide sequences. PCR products were ethanol-precipitated, pelleted, washed in 70% ethanol, vacuum-dried, resuspended in 10 mM Tris and 0.1 mM EDTA, pH 8.0 (T1/10E) buffer, and gel-purified on a 1% Seaplaque agarose gel (FMC Bioproducts, Rockland, ME, U.S.A.). Amplicons detected by ethidium bromide staining were excised and purified with 0.22 μm Micropure separators (Amicon, Beverly, MA, U.S.A.). Eluted DNA was extracted with phenol-chloroform and precipitated with ethanol. To make the chimeric KCNQ3/KCNQ2 channel, we first inserted an XhoI restriction site into KCNQ3 using overlap extension PCR (Ho et al., 1989). The inserted XhoI site was used to cut out a fragment of DNA that was replaced by the corresponding piece of DNA from KCNQ2 that had a naturally occurring XhoI site.
Purified PCR products or cut DNA fragments and vector pcDNA3 (Invitrogen, Carlsbad, CA, U.S.A.) were digested with the appropriate restriction endonucleases (New England Biolabs, Beverly, MA, U.S.A.), phenol-chloroform extracted, ethanol precipitated, pelleted, washed in 70% ethanol, vacuum-dried, and resuspended in T1/10E. The vector was dephosphorylated with shrimp alkaline phosphatase (Boehringer Mannheim, Indianapolis, IN, U.S.A.) and gel-purified as above. Inserts and vector were ligated with T4 DNA ligase (New England Biolabs) overnight at 16°C. Competent XL-1 Blue Escherichia coli (Stratagene) were transformed and plated on LB-amp-tet agar plates. Plasmid DNA was isolated from broth cultures started from amp-tet-resistant bacterial colonies (Qiagen), and the plasmid DNA was screened by appropriate restriction endonuclease (New England Biolabs) digestion. Mutations were confirmed by automated DNA sequencing (Applied Biosystems, Perkin-Elmer).
Generation of antibodies
Rabbit polyclonal antibodies were raised to the amino termini of KCNQ2 and KCNQ3 and the carboxy terminus of KCNQ2. Briefly, three glutathione S-transferase fusion proteins were constructed containing the first 70 amino acids of KCNQ2, the first 71 amino acids of KCNQ3, and residues 655–810 of the KCNQ2 carboxy terminus, respectively. These regions were chosen based on their lack of similarity to other KCNQ family members. The respective sequences were amplified using PCR and primers incorporating BamHI and EcoRI restriction sites on the 5′ and 3′ end of amplicons, respectively. After digestion with BamHI and EcoRI, amplicons were subcloned into pGEX-3X (Pharmacia). Fusion proteins were expressed and purified and antibodies raised and characterized as previously described (Twitchell et al., 1997).
Immunocytochemistry
TsA cells and SCG cultures Cells were grown on poly-lysine-coated coverslips and fixed in 4% paraformaldehyde, washed twice with 100 mM sodium phosphate (PB, pH 7.4), three times with PB+150 mM NaCl (PBS), and blocked with 5% goat serum and 0.1% saponin in PBS (PBS+GS). Cells were incubated overnight with primary antibody diluted in PBS+GS at 4°C. The following day, cells were washed five times with PBS then incubated with secondary antibody in PBS+GS for 45 min. Rabbit antibodies were detected with Texas Red anti rabbit (1 : 100, Jackson Immunoresearch) and mouse antibody detected with FITC anti-mouse (1 : 150, Jackson Immunoresearch). Cells were then washed three times with PBS, twice with PB, and three times with water. Air-dried coverslips were mounted on slides with a drop of Vectashield (Vector Laboratories).
Hippocampal sections
Adult rats were anaesthetized with nembutal and intracardially perfused with a solution of 4% paraformaldehyde in PB. The brains were removed and postfixed for 2 h. The tissue was then sunk in successive solutions of 10% and 30% (w/v) sucrose in PB at 4°C over 72 h. Sagittal sections (40 μm) of the brain were cut on a sliding microtome and placed in PB until the tissue was processed for immunocytochemistry.
Free-floating sections
Brain sections from rats were processed as free-floating sections and were rinsed in 0.1 M Tris buffer (TB), pH 7.4, for 15 min, in 0.1 M Tris buffered saline (TBS), pH 7.4, for 15 min, blocked using 2% avidin in TBS for 30 min, rinsed in TBS for 30 min, blocked using 2% biotin for 30 min, and finally rinsed in TBS for 30 min. The tissue sections were then incubated in purified anti-KCNQ2 (diluted 1 : 200) or anti-KCNQ3 serum (diluted 1 : 400) for 36 h at 4°C. All antibodies were diluted in a solution containing 0.10% Triton X-100 and 1% NGS in 0.1 M TBS. Control sections were incubated in pre-immune rabbit serum, in antibody preincubated with 3 μg/ml of the antigen to which the antibody was raised against, or in solution lacking the primary antibody. In all three instances, no specific staining was observed. Tissue sections were rinsed in TBS for 60 min and incubated in biotinylated goat anti-rabbit IgG diluted 1 : 300 for 1 h at 37°C. The tissue sections were rinsed with TBS for 60 min and incubated in Avidin D-fluorescein diluted 1 : 300 for 1 h at 37°C. The sections were rinsed in TBS for 10 min, rinsed in TB for 20 min, and then mounted on gelatin-coated slides, coverslipped with Vectashield and sealed with nail polish.
Confocal microscopy
Stained cells and sections were viewed using excitation and emission filters appropriate for FITC and Texas Red with a BioRad MRC600 confocal microscope at the W.M. Keck Imaging Facility at the University of Washington, or with an Olympus FV-500 confocal microscope at the Optical Imaging Core Facility at the University of Texas Health Science Center. Images were typically collected as single images, 3–5 μm above the surface of the coverslip. When comparisons were being made between blocking, preimmune serum, and immune serum, images were always collected and processed with identical gain settings. Post-acquisition processing was done in Photoshop (Adobe), Canvas (Denaba), and PowerPoint (Microsoft).
SCG sympathetic neuron cultures
SCG neurons were taken from 3- to 5-week-old male rats (Sprague-Dawley) and cultured for 1 day. Rats were anaesthetized with CO2 and decapitated. Neurons were dissociated using methods of Bernheim et al. (1991) and suspended twice in DMEM+10% heat-inactivated calf serum. Cells were plated on 4×4 mm glass coverslips (coated with poly-L-lysine) and incubated at 37°C (5% CO2). Fresh culture medium containing nerve growth factor (50 ng/ml) was added to the cells 3 h after plating.
Current recording and analysis
The whole-cell configuration of the patch-clamp technique was used to voltage clamp and dialyze cells at room temperature (22–25°C). Electrodes were pulled from glass hematocrit tubes (VWR Scientific Corp., Seattle, WA, U.S.A.) and fire-polished. They had resistances of 1–3 MΩ when measured in Ringer and filled with internal solution. Membrane current was measured under whole-cell clamp with pipette and membrane capacitance cancellation, sampled at 5 ms and filtered at 200–500 Hz. The whole-cell access resistance was 3–10 MΩ. Junction potentials have been corrected by −2 mV. Cells were placed in a 100 μl chamber through which solution flowed at 1–2 ml min−1. Inflow to the chamber was by gravity from several reservoirs, selectable by activation of solenoid valves. Bath solution exchange was complete by <30 s.
For most experiments, KCNQ2/3 currents from tsA cells were studied by holding the cell at −20 mV and applying a 500–650 ms hyperpolarizing step to −60 mV, followed by a 650 ms pulse back to the holding potential, every 4–5 s. The amplitude of the current was usually defined as the outward current at the holding potential sensitive to block by 50 μM XE991. Like HEK293 cells (Yu and Kerchner, 1998), tsA cells have small endogenous voltage-gated K+ currents. These currents have an onset rate more than 10 fold faster than KCNQ2/3 currents, show little observable tail current (deactivation) at −60 mV, and are just starting to activate at −20 or 0 mV (data not shown). Thus, the KCNQ2/3 current was easily distinguished from the endogenous current. In some experiments that did not use XE991 and had little leak or endogenous current, the KCNQ2/3 current amplitude was defined by the holding current. In the few cells with significant endogenous current or significant leak current, the amplitude of the KCNQ2/3 current was taken as the difference between the holding current and a point 20–25 ms after the start of the activating step back to the holding potential of −20 mV (which is enough time for activation of the endogenous current). Cells exhibited variable ‘run-down' in the amplitude of KCNQ currents, and currents usually stabilized within several minutes of whole-cell dialysis. Cells in which the ‘run-down' was excessive were not studied.
M-type currents in SCG cells were studied by holding the membrane potential at −25 mV and applying a 500 ms hyperpolarizing pulse to −60 mV every 4 s. M current amplitude was measured at −60 mV from the decaying time course of deactivating current as the difference between the average of a 10 ms segment taken 20–30 ms into the hyperpolarizing step and the average during the last 50 ms of that step. All results are reported as mean±s.e.mean.
Electrophysiology solutions and materials
The external Ringer solution used to record KCNQ currents in tsA cells contained (mM): NaCl 160, KCl 2.5, CaCl2 2, MgCl2 1, HEPES 10, pH adjusted to 7.4 with NaOH. For recordings from SCG neurons, the CaCl2 concentration was 5 mM and 8 mM glucose was added. The pipette solution contained (mM): KCl 175, MgCl2 5, HEPES 5, 0.1 1,2-bis(2-aminophenoxy)ethane N,N,N′,N′-tetraacetic acid (BAPTA), Na2ATP 3, NaGTP 0.1, pH titrated to 7.4 with KOH.
Reagents were obtained as follows: BAPTA (Molecular Probes); ATP and GTP (Pharmacia LKB Biotechnology); DMEM, DMEM/F-12 mixture, foetal bovine serum, horse serum, nerve growth factor, Penicillin/Streptomycin (Gibco); oxotremoreine-M, RBI; N-ethylmaleimide (NEM) (Sigma); XE991 was kindly given to us by Michael E. Schnee (Dupont Pharmaceuticals).
Results
Antibodies specifically label KCNQ2 and KCNQ3 subunits
To determine the localization of KCNQ2 and KCNQ3 subunits in neurons, we raised rabbit antisera against fusion proteins containing sequences corresponding to both the amino- and carboxy-termini of KCNQ2 and the amino-terminal of KCNQ3. We will call these n-Q2, c-Q2 and n-Q3 antibodies, respectively. We tested the efficacy of the antibodies by performing immunocytochemistry on tsA cells transiently transfected with the cDNA coding for either KCNQ2 or KCNQ3 subunits. Previous work indicates that transfection and expression of KCNQ2 and KCNQ3 subunits in several mammalian cell lines, including tsA cells, recapitulates M-like currents with many of the biophysical, pharmacological and modulatory characteristics of M current in SCG neurons (Selyanko et al., 2000; Shapiro et al., 2000). TsA cells were fixed and immunostained with the rabbit antibodies 2–4 days after transfection. The cDNA coding for green fluorescent protein was included in the transfections as a reporter for successfully transfected cells. We found that the antibodies were efficacious, and specific for the individual subunits they were designed to detect. Figure 1A shows strong labelling of tsA cells transfected with either the KCNQ2 or KCNQ3 constructs. These confocal images, all from the same group of transfected cells, show immunostaining by either the c-Q2 antibody (top left), n-Q2 (bottom left), or n-Q3 (bottom right). Controls for specificity of the antibody labelling included the occlusion of staining by pre-incubation of the antibodies with the immunizing peptide (Figure 1A top middle) and the lack of reactivity of the pre-immune serum when applied to this same group of transfected cells (Figure 1A top right). To determine the specificity of our antibodies among the KCNQ subtypes, we transfected cells with KCNQ1-5 individually and tested for immunostaining by the three antibodies. Figure 1B shows the result for the c-Q2 antibodies. Cells transfected with KCNQ2 were labelled strongly, but not those transfected with KCNQ1, KCNQ3, KCNQ4 and KCNQ5. The n-Q2 and n-Q3 antibodies showed similar specificity (data not shown). In addition, only cells that fluoresced green from co-expressed GFP displayed KCNQ2 or KCNQ3 immunoreactivity.
Figure 1.
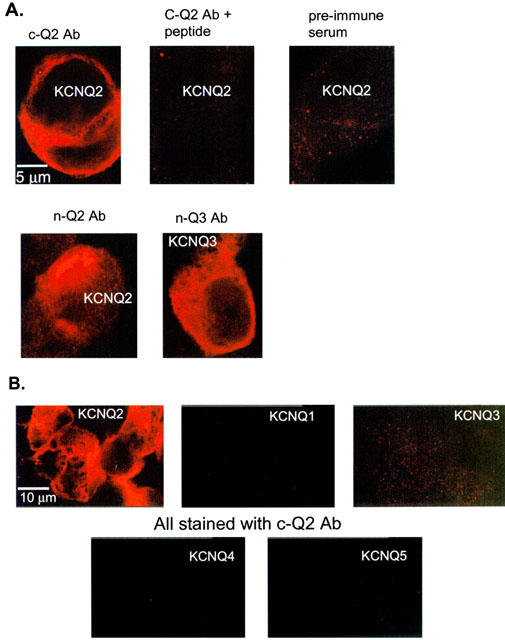
Immunostaining of KCNQ2 and KCNQ3 channels heterologously expressed in tsA cells. Confocal images of transiently transfected tsA cells labelled with antibodies to the amino-terminal of KCNQ2 (n-Q2), the carboxy-terminal of KCNQ2 (c-Q2), and the amino-terminal of KCNQ3 (n-Q3). The constructs transfected into the tsA cells are labelled in white on the appropriate panels. (A) Images of cells transfected with KCNQ2 top, bottom left) of KCNQ3 (bottom right) and immunostained with n-Q2 (bottom left) c-Q2 (top left), c-Q2 plus 1 μg/ml immunizing protein (top centre), or pre-immune serum (top right). The indicated scale bar applies to all panels. The gain settings used to acquire and process these images were identical. (B) Images of cells transfected individually with KCNQ1-5 and immunostained with the c-Q2 antibody. The indicated scale bar applies to all panels. For (A) and (B), the serum dilution was 1 : 1000.
Localization of KCNQ2 and KCNQ3 in SCG neurons and hippocampal sections using antibodies
To determine if SCG neurons express KCNQ2 and KCNQ3, we used our n-Q2, c-Q2 and n-Q3 antibodies to immunostain cultured rat SCG neurons (Figure 2). We found that the n-Q3 antibody stained the SCG cells strongly when used at similar concentrations to those used in the experiments on expressed KCNQ3 channels in tsA cells. As with tsA cells, the labelling of SCG cells was efficacious (middle row), blocked by the immunizing peptide (bottom left), and the pre-immune antibody did not label the cells (bottom right). Interestingly the n-Q3 antibody stained both the cell bodies and neural processes, suggesting that M-type channels localize to both regions of SCG cells. The c-Q2 antibody also strongly labelled SCG neurons (top right), and the labelling was blocked by the immunizing peptide (data not shown). The n-Q2 antibody labelled SCG cells as well, but less strongly than the c-Q2 antibody (top left). Perhaps the presumed association of KCNQ3 with KCNQ2 into channel tetramers interferes with access of antibodies to the amino terminus of KCNQ2. In conclusion, these data suggest that KCNQ2 and KCNQ3 have a strong presence in the SCG neurons that have been widely used in studies of M current.
Figure 2.
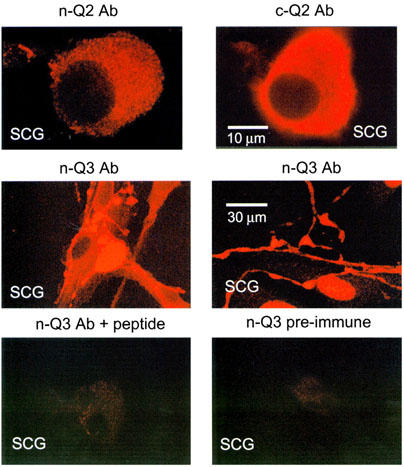
SCG neurons express both KCNQ2 and KCNQ3. SCG cultures were prepared and immunostained with the KCNQ2 and KCNQ3 antibodies after 1–2 days in culture. The n-Q3 (centre row) antibodies strongly labelled the SCG neurons both at the soma and on neural processes. The staining as blocked by the immunizing protein used to make n-Q3 and the pre-immune antibody did not label the cells (bottom row, similar results for c-Q2 not shown). The cells were more weakly labelled by the n-Q2 antibody (top left) than by the c-Q2 antibody. The scale bar in the top right panel applies to the top row, and that in the middle right panel applies to the other panels.
Immunostaining was then performed in the hippocampus (Figure 3) with the anti-KCNQ2 and anti-KCNQ3 antibodies to determine the localization of KCNQ2 and KCNQ3 in that brain region. The n-Q2 antibody stained the granule cell layer and the mossy fibers in the central hilus of the dentate gyrus extensively, and the outer molecular layer and outer hilus less well (Figure 3A). There was little staining in the inner molecular layer. The staining was blocked when the antibody is pre-adsorbed with the immunizing peptide (Inset). In regions CA3 and CA2, there is intense staining of the mossy fibers and their terminals (Figure 3B). The pyramidal neurons and an occasional interneuron are stained to a lesser extent. In the CA1 region pyramidal cells, their apical dendrites, and an occasional interneuron are moderately labelled (Figure 3C). In contrast, staining with the n-Q3 antibody in the dentate region was mainly in interneurons and astrocytes (Figure 3D). There is no staining apparent in the mossy fibers. The specific staining was abolished when the antibody was pre-adsorbed with the immunizing peptide (Inset). In the CA3 and CA2 regions, there is light staining of pyramidal cells and several interneurons and more intense staining of astrocytes (Figure 3E). In the CA1 region (Figure 3F), there is intense staining of scattered interneurons in addition to astrocyte staining. Pyramidal neurons are less intensely stained. Lower levels of staining are apparent in the stratum lacunosum-moleculare. The neuropil of the stratum oriens and stratum radiatum is lightly stained. Collectively these findings indicate that there is a specific and distinct pattern of expression of KCNQ2 and KCNQ3 subunits in the hippocampus. Moreover, in addition to KCNQ2/3 heteromultimers, they also express either as homomeric channels or as heteromeric channels with subunits other than KCNQ2 and KCNQ3.
Figure 3.
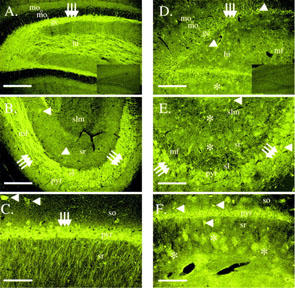
KCNQ2 and KCNQ3 are labelled in the rat hippocampal formation. (A) Dentate gyrus stained with the n-Q2 antibody demonstrating intense staining of the granule cell layer and mossy fibers. The outer molecular layer is lightly stained while staining is absent from the inner molecular layer. moo, outer molecular layer, moi, inner molecular layer, gc, granule cell layer, hi, hilus. Arrows indicate granule cells. Scale bar is 300 μm. The inset shows labelling from the n-Q2 antibody pre-adsorbed with the immunizing peptide. (B) Regions CA3 and CA2 stained with the n-Q2 antibody demonstrating intense staining of the mossy fibers and their terminals and, to a lesser extent, pyramidal cells and interneurons. mf, mossy fibers, pyr, pyramidal cell layer, sl, stratum lucidum, sr, stratum radiatum, slm, stratum lacunosum-moleculare. Arrows indicate pyramidal cells. Arrowheads identify interneurons. Scale bar is 300 μm. (C) Region CA1. Pyramidal cells, their apical dendrites, and an occasional interneuron are moderately to strongly labelled. Scale bar corresponds to 120 μm. so, stratum oriens. Arrows identify pyramidal cells. Arrowheads identify interneurons. (D, E, F) Micrographs of KCNQ3 immunostaining in the rat hippocampal formation with the n-Q3 antibody. (D) Dentate region illustrating staining of astrocytes and interneurons in this region. Block of this labelling with the immunizing peptide is shown in the inset. Scale bar is 250 μm. (E) Region CA3. Moderate staining of astrocytes and scattered interneurons is evident. Pyramidal cells are diffusely labelled. Mossy fibers (stratum lucidum) do not appear to be stained. so, stratum oriens, pyr, pyramidal cell layer, sl, stratum lucidum, sr, stratum radiatum. Arrows indicate pyramidal cells. Arrowheads identify interneurons. Asterisks denote astrocytes. Scale bar is 120 μm. (F) Region CA1. Intense staining of scattered interneurons is evident. Pyramidal neurons and a population of astrocytes are less intensely stained. Lower levels of staining are apparent in the stratum lacunosum-moleculare. The neuropil of the stratum oriens and stratum radiatum is lightly stained. so, stratum oriens, pyr, pyramidal cell layer, sr, stratum radiatum, slm, stratum lacunosum-moleculare. Arrows indicate pyramidal cells. Arrowheads identify interneurons. Asterisks denote astrocytes. Scale bar is 250 μm.
NEM enhances currents of both SCG M current and KCNQ2/3 currents in tsA cells via two distinct mechanisms
We had previously observed that the cysteine-alkylating agent N-ethylmaleimide (NEM) increases the amplitude of heteromeric KCNQ2/3 currents expressed in tsA cells (Shapiro et al., 2000). We sought to understand the underlying molecular and biophysical mechanisms of the NEM-induced augmentation of the current, and to use NEM action as another test of the correspondence of M-type channels to KCNQ2/3 heteromultimers. Since M-type currents arise from voltage-gated channels, an augmentation of the current at a particular voltage could arise from two effects: (1) a general increase in the amplitude of the current that is the same at all voltages, or (2) a shift of the voltage dependence of the conductance such that it activates at less depolarized potentials. To determine possible mechanisms of NEM current enhancement and to test the specificity of NEM action, we applied NEM to various combinations of expressed KCNQ subunits.
To ascertain the action of NEM on the voltage dependence of KCNQ2/3 currents in tsA cells, we applied a family of voltage steps from a holding potential of −80 mV, and measured the amplitude of the tail current at −60 mV following each test current (Figure 4A,B). We found that bath application of NEM (50 μM, 2 min) augmented the KCNQ2/3 currents via both a general increase in the amplitude of the current at all potentials, and a shift of the voltage dependence of the conductance to less depolarized potentials. Fitting the tail current amplitudes before and after NEM treatment to Boltzmann equations shows that NEM shifted the voltage dependence of activation by −13 mV (Figure 4B). We scaled up the control data (dotted curve) to show more clearly that both a general increase in amplitude and a shift in the voltage dependence are required to explain the actions of NEM on the KCNQ2/3 conductance. The mean effect of NEM on the voltage necessary for half-maximal activation (V1/2) was −11±4.0 mV (n=5) and the mean increase of the amplitude at −20 mV was 2.0±0.3 fold (n=6).
Figure 4.
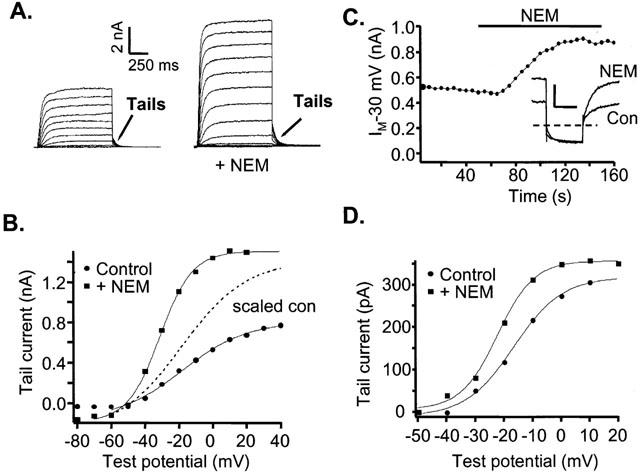
NEM enhances amplitude and shifts the voltage dependence of homomeric KCNQ2/3 currents in tsA cells and M current in SCG neurons. (A) Currents in tsA cells expressing KCNQ2/3 subunits. Families of currents before (left) and after NEM (50 μM, 2 min) treatment (right), elicited by voltage steps in the range of −80 to +40 mV in 10 mV increments. (B) Activation curve for KCNQ2/3 channels expressed in tsA cells subunits before and after NEM application. Test potential steps range from −80 mV to +40 mV in 10 mV increments, followed by a step to −60 mV where tail-current amplitudes were measured. Tail currents were fitted by a single exponential, extrapolated to the beginning of the tail potential step, and the fitted amplitudes were plotted vs test potential. The data were fitted by Boltzmann equations of the form Itail=Imax/(1+exp(V1/2-V)/k), where Itail is the fitted tail current amplitude, Imax is the maximum current, V1/2 is the voltage at which the conductance is half-activated, and k is the slope factor. For the control curve, V1/2 and k were −18 and 19 mV, and for the NEM curve, they were −31 and 10 mV. The dotted curve (scaled con) is the fitted Boltzmann relation of the control data, scaled up so that the Imax is equal to that of the post-NEM curve. (C) Amplitudes of M currents in SCG neurons voltage-clamped to −30 mV and stepped repeatedly to −60 mV every 5 s. Filled circles represent the average current at −25 mV for the first 300 ms of a given sweep. Inset shows the current traces both before (con) and after NEM (NEM) treatment. Scale bars 500 pA, 300 ms. (D) Activation curve of M current in SCG neurons, obtained as for KCNQ2/3 currents in tsA cells, before and after application of NEM. Voltage steps range from −50 mV to +20 mV in 10 mV increments. The tail current potential was −60 mV. The data were fitted by Boltzmann equations as in (B). For the control curve, V1/2 and k were −16 and 8.5 mV, and for the NEM curve, they were −22 and 6.4 mV.
We then tested the actions of NEM on M current in SCG neurons. If the channel responsible for SCG M current consists of heteromultimers of KCNQ2 and KCNQ3 subunits, then M current in SCG cells should have similar sensitivity to NEM. To observe the effect of NEM on current amplitude, we used our usual M current protocol on SCG cells in whole-cell clamp and recorded M currents elicited from repeated voltage pulses. Bath-application of NEM caused a large increase in M-current amplitude (Figure 4C). To separate the possible effects of NEM on the amplitude and the voltage dependence of M current, we used a tail current protocol similar to that used above in tsA cells. Plotted in Figure 4D are the amplitudes of the tail currents vs test potential, fitted by Boltzmann functions. NEM shifted the voltage dependence of M current slightly, by −6.0±1.9 mV (n=4, this shift was not significant at the P<0.05 level) and increased the amplitude of the current by 2.8±0.6 fold (n=5). Thus, the actions of NEM on heteromeric KCNQ2/3 currents expressed in tsA cells and M current in SCG neurons are similar.
NEM enhances current amplitude in KCNQ2 but not KCNQ3 or KCNQ1 homomultimers expressed in tsA cells
We next wanted to localize the region responsible for the dual effects of NEM, first to the individual subunit, and then to specific residues on this subunit. This information may help us both identify the specific KCNQ subunits in SCG neurons as well as provide information on important structural elements involved in regulating current amplitudes from these channels. For these experiments, we expressed KCNQ1, KCNQ2, and KCNQ3 subunits individually in tsA cells and tested the effects of NEM application. We first tested the subunit specificity of the voltage-independent increase in current amplitude. NEM profoundly enhanced the current amplitude from KCNQ2 homomultimers to levels 5 fold greater than the control current (5.0±1.1 fold, n=4, Figure 5A). This effect was even greater than the 2 fold increase seen for expressed KCNQ2/3 heteromultimers in tsA cells. Conversely, neither KCNQ1 nor KCNQ3 homomultimers showed a significant increase in current amplitude after application of NEM at voltages where the channels were maximally activated (1.0±0.1 fold, n=6, and 0.9±0.1 fold, n=6 respectively) (Figure 5B,C). For the experiments on KCNQ2 and KCNQ3, we used the far greater sensitivity of KCNQ2 vs KCNQ3 channels to TEA blockade to verify the expression of the appropriate homomeric channel type. Thus, application of 1 mM TEA substantially blocked currents in cells expressing KCNQ2 channels, but did not affect cells expressing KCNQ3 channels, in accord with expectations from previous work (Hadley et al., 2000; Shapiro et al., 2000; Wang et al., 1998). We also co-transfected these cells with M1 muscarinic receptors and confirmed that the currents were suppressed by application of the muscarinic agonist oxotremorine (oxo-M) (Shapiro et al., 2000; Selyanko et al., 2000).
Figure 5.
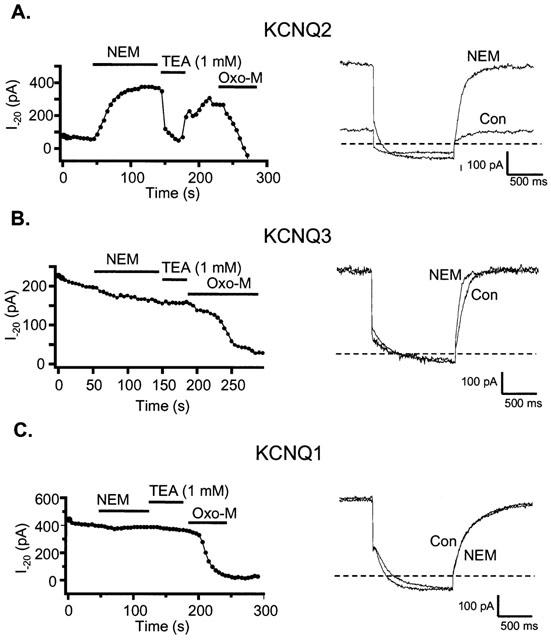
Augmentation of current by NEM is specific to the KCNQ2 subunit. TsA cells individually transfected with KCNQ2 (A), KCNQ3 (B) or KCNQ1 (C) subunits were voltage-clamped to −20 mV and stepped repeatedly to −70 mV every 5 s. Plotted as filled circles are the mean current amplitudes at −20 mV for the first 300 ms of each sweep. NEM (50 μM), TEA (1 mM) or oxo-M (10 μM) were bath-applied during the times indicated by the bars. The right hand panels show the current traces both before (con) and after NEM (NEM) treatment.
We next asked if there was subunit specificity to the shift in voltage dependence of KCNQ2/3 currents caused by NEM. We used a similar voltage pulse protocol as that in Figure 4 to analyse the shift in V1/2. Plotted in Figure 6 are the tail current amplitudes vs test potential for KCNQ2 or KCNQ3 homomultimers, fitted to Boltzmann equations. In addition to the large augmentation in current amplitude at all voltages, we found that NEM shifted the activation of KCNQ2 homomultimers by −16±4.5 mV (Figure 6A,C). The control Boltzmann curve is scaled up to display this shift more clearly (Figure 6A). Although KCNQ3 homomultimers did not show a voltage-independent enhancement of current amplitude, there was a −6.3±0.7 mV shift in the V1/2 (Figure 6B). The voltage-shift data are summarized in Figure 6C. Also included are the mean shifts in V1/2 for KCNQ2/3 heteromultimers and for SCG M current. The results are qualitatively similar for all these groups. Thus, we conclude that, whereas residues in KCNQ2, but not in KCNQ3, are responsible for the large voltage-independent enhancement of M-type current amplitude by NEM, residues important for the shift in the voltage dependence of M current activation are likely found on both subunits.
Figure 6.
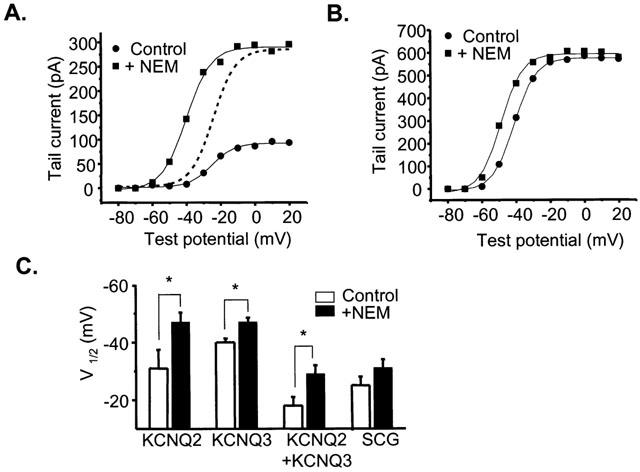
NEM-induces a hyperpolarizing shift in the V1/2 of activation of KCNQ channels. (A) Activation curve for tsA cells expressing KCNQ2 subunits before and after application of NEM. Voltage steps range from −80 mV to +40 mV in 10 mV increments. The tail current potential was −70 mV. The data were fitted by Boltzmann equations (solid lines) of the form Itail=Imax/(1+exp(V1/2-V)/k), where Itail is the fitted tail current amplitude, Imax is the maximum current, V1/2 is the voltage at which the conductance is half-activated, and k is the slope factor. The dotted curve is the fitted Boltzmann relation of the control data, scaled up so that the Imax is equal to that of the post-NEM curve. (B) Activation curve for tsA cells expressing KCNQ3 subunits before and after the application of NEM. Voltage steps range from −80 mV to +40 mV in 10 mV increments. The tail current potential was −70 mV. These data were also fitted by Boltzmann equations (solid lines) (C) Mean V1/2 of various KCNQ expressing cells as well as SCG neurons both before (open) and after NEM (filled) (*significant difference, Student's paired t-test, P⩽0.05).
Residue C242 of KCNQ2 may mediate part of the effect of NEM on this channel
NEM alkylates cysteine residues. Since it crosses the membrane rapidly, the site of action cannot be localized to one side of the membrane or another. Alignment of the KCNQ2 and KCNQ3 amino acid sequences reveals that five cysteine residues are conserved between the subunits whereas KCNQ2 has 11 cysteine residues not found on KCNQ3. We sought to identify the specific residues that mediate the augmentation by NEM. Because the voltage-independent effect of NEM on KCNQ2 currents was so large, we focused on this effect, rather than on the shift in voltage dependence. Our strategy relied on site-directed mutagenesis, based on systematic elimination of candidate cysteines (Figure 7A). Mutant KCNQ2 subunits were expressed individually in tsA cells and tested for NEM augmentation.
Figure 7.
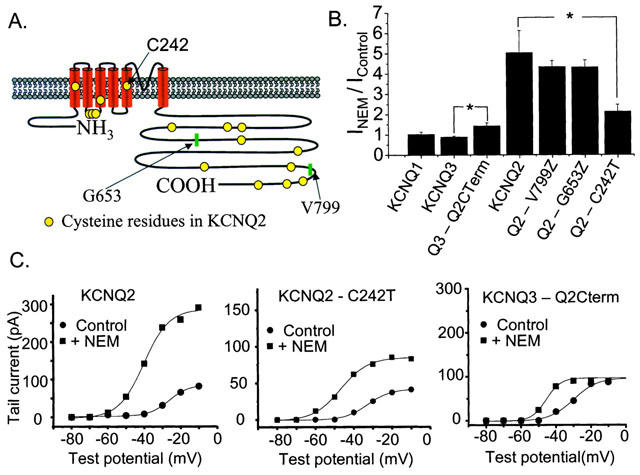
Investigation of possible regions on KCNQ2 responsible for NEM action. (A) Diagram of the location of cysteine residues on the KCNQ2 subunit. (B) Mean NEM-induced augmentation of current amplitudes for homomeric channels made up of the construct indicated below the column (*represents significant difference, Student's t-test, P⩽0.05). (C) Activation curves for tsA cells expressing KCNQ2 (left), KCNQ2-C242T (middle), or the KCNQ3/2 chimera (right) before and after the application of NEM. Voltage steps range from −80 mV to 0 mV in 10 mV increments. The tail current potential was −70 mV.
We first truncated the KCNQ2 subunit at V799, eliminating three cysteines in the most distal portion of the carboxy-terminus from the subunit. NEM application still caused a large increase in current amplitude (4.3±0.3 fold, n=3) similar to wild type KCNQ2 (Figure 7B). We next truncated the KCNQ2 subunit at G653, thereby removing four additional cysteines. Again, this mutant showed a large enhancement of current amplitude after application of NEM (4.3±0.4 fold, n=3) (Figure 7B). Truncation upstream of G653 resulted in a loss of channel expression.
To determine if the cytoplasmic carboxy-terminal cysteines of KCNQ2 that we were unable to remove by truncation are the site of action of NEM, we made a chimeric protein that replaced the carboxy-terminus of KCNQ3 with that of KCNQ2 (KCNQ3–Q2Cterm). We reasoned that if the cysteines in the KCNQ2 carboxy-terminus were responsible for the actions of NEM then we would expect that this KCNQ3/2 chimera would confer the amplitude-enhancing actions of NEM to the previously insensitive KCNQ3. Tested on the chimeric subunit, NEM increased the current amplitude slightly at −20 mV (1.4±0.2 fold, n=3) unlike the case for wild type KCNQ3 (Figure 7B). However, we found that this resulted solely from a shift of the voltage dependence of the KCNQ3/2 chimera to slightly more depolarized potentials, relative to that of wild type KCNQ3. Thus, the increase in amplitude at −20 mV of the KCNQ3/2 chimeric currents was the result of the leftward shift in voltage dependence of activation caused by NEM and not the conferring of the voltage-independent enhancement onto KCNQ3 by the KCNQ2 carboxy-terminal. The activation data for a cell expressing the KCNQ3/2 chimera are plotted in Figure 7C (right panel), before and after NEM treatment. The activation curve is shifted to more depolarized voltages in comparison to that of wild type KCNQ3, such that −20 mV is no longer at the peak of the activation curve. Thus, the leftward shift in activation caused by NEM results in an enhancement of current amplitude at −20 mV and no voltage-independent augmentation of the current, such as that seen for wild type KCNQ2, is required to account for it. Thus, we conclude that the distal seven cysteines in the cytoplasmic carboxy-terminal portion of KCNQ2 do not mediate the voltage-independent enhancement of current caused by NEM.
We next mutated the cysteine residue at position 242 of KCNQ2, in the S5 region of the channel, to a threonine. For KCNQ2-C242T, application of NEM increased the current to a significantly smaller extent than in wild type KCNQ2. Plotted in Figure 7C (middle) are the activation data from one such experiment, as well as data from a cell expressing wild type KCNQ2 (left). These data are summarized in Figure 7B. NEM caused only a doubling (2.1±0.3 fold, n=5) of the current amplitude for cells expressing the C242T mutant compared to the 5 fold increase in current amplitude for cells expressing wild type KCNQ2. These data indicate that C242 of KCNQ2 is likely responsible for much of the voltage-independent enhancement of current amplitude caused by NEM.
Discussion
This work provides further evidence that KCNQ2 and KCNQ3 subunits are present in SCG neurons, using two different approaches. The first approach is immunochemical. Using antibodies raised against regions of KCNQ2 and KCNQ3 subunits, we showed that both are strongly expressed in SCG cells. The second approach is pharmacological. We first explored the biophysical and molecular basis of the augmentation of currents from cloned KCNQ channels by the sulfhydryl-alkylating reagent NEM. We then tested M current in SCG neurons for NEM action and show that the effect on endogenous M current is very similar to that on expressed KCNQ2/3 heteromultimers. Both types of experiments indicate that KCNQ2 and KCNQ3 subunits must make at least a strong contribution to the M current of sympathetic neurons.
We also tested for the presence of KCNQ2 and KCNQ3 in sections from the hippocampal formation and find the presence there of both types of subunits. Interestingly, the staining patterns indicate that these two subunits are expressed in some hippocampal regions independently of each other. In the dentate, we saw strong labelling of KCNQ2 in the mossy fibers in the dentate hilus and in granule cells, as did Cooper et al. (2001). In area CA3, the strongest labelling was in the mossy fibers of the stratum lucidum and weaker labelling in the interneurons of the stratum radiatum and the pyramidal cell layer, similar to the earlier study (Cooper et al., 2001). Unlike that study, we also found strong KCNQ2 localization in the pyramidal cell layer of CA1. In general, our data suggest a role for KCNQ2 in modulating transmitter release from mossy fibers and a role in modulating the excitability of pyramidal cells and some interneurons. We also found a strong presence of KCNQ3 subunits in both CA1 and CA3. The most intense staining was of astrocytes, interneurons in CA1, and pyramidal cells in CA3. Interestingly, the mossy fibers of the stratum lucidum of CA3 showed very little staining for KCNQ3, even though strong labelling was seen there for KCNQ2. Although expression of KCNQ2 as homomultimers seems low in all cells studied thus far (Jentsch, 2000), these data may predict the unexpected findings of a large population of homomeric KCNQ2 channels. Alternatively, the mossy fiber KCNQ2 channels may exist as heteromultimers with KCNQ5 or other channels. The intense labelling of hippocampal astrocytes for KCNQ3 is provocative. Their function in those cells is unknown, but may be related to regulation of astrocyte homeostasis or indirectly, to regulation of neuronal excitability. Although some of the protein expression that we see could be non-functional, the data suggest that KCNQ2 and KCNQ3 subunits could express as homomultimers.
Recently, it has been suggested that the newest member of the KCNQ family to be cloned, KCNQ5, also contributes to M current in the SCG (Schroeder et al., 2000). We did not test for the presence of KCNQ5 in these cells. However, a major rationale for the suggestion of Wang et al. (1998) that KCNQ2/3 heteromultimers make SCG M current comes from the very similar sensitivity to TEA blockade. KCNQ2 is the only subunit among KCNQ1-5 that has a tyrosine at the position analogous to Y449 in Shaker K+ channels, the pore residue that confers high sensitivity to external TEA block (MacKinnon & Yellen, 1990). KCNQ2 currents are thus highly TEA sensitive, KCNQ3 currents very TEA insensitive, and KCNQ2/3 currents of intermediate sensitivity, quantitatively in accord with the TEA sensitivity of SCG M current (Hadley et al., 2000; Shapiro et al., 2000; Wang et al., 1998). KCNQ5 has a threonine in this position, and, like KCNQ3, is quite TEA insensitive (Schroeder et al., 2000). These authors also show that KCNQ5 does not assemble with KCNQ2 in oocytes, making KCNQ2/KCNQ5 heteromultimers (which would also have the corresponding TEA sensitivity to M current) in SCG cells unlikely. In addition, KCNQ5 has a valine at the 276 position corresponding to C242 and KCNQ2, and should thus be only weakly sensitive to NEM augmentation. Thus, it will be interesting to see how KCNQ5 can make a strong contribution to SCG M current, given this subunit-specific sensitivity to TEA blockade and NEM action.
We find that NEM enhances KCNQ currents and SCG M current via two distinct mechanisms. One voltage independent mechanism results in a powerful increase in the amplitude of the currents. We attribute this effect to KCNQ2 and not to KCNQ3, since homomeric expression of KCNQ1-3 subunits shows that only KCNQ2 currents demonstrate this type of augmentation of the current. Using site-directed mutagenesis, we find that this effect is strongly reduced when C242 in S5 of KCNQ2 is mutated. Since this NEM action does not involve a shift in the voltage dependence of gating and is far too rapid to be the result of an increase in the number of channels, it must result from an increase in the maximal open probability (Po,max) of the channel. The available single-channel data indicate that Po,max of KCNQ2 homomultimers is ∼0.25 and that of KCNQ2/3 heteromultimers ∼0.36 (Selyanko et al., 2001). Thus, an increase by NEM of Po,max for these channels to near unity could account for the large voltage-independent increase in whole-cell current amplitude.
Mutation of C242 may well reduce the NEM augmentation of the current by eliminating a cysteine modified by NEM; however, we cannot exclude the possibility that C242T channels have a larger Popen,max than wild type KCNQ2 channels, such that an increase of Popen,max to unity now only produces a 2 fold, rather than a 5 fold, increase in the current. How would alkylation of C242 in the S5 region affect Po,max? Much molecular and physiological data indicate that voltage sensing in voltage-gated channels occurs largely in the fourth transmembrane domain (S4) (reviewed in Hille, 2001), whereas other molecular and, now, structural data (Doyle et al., 1998) indicate that the channel gate involves movement of S5 and the sixth transmembrane domain (S6). Thus, there must be some intramolecular coupling mechanism that links voltage-induced conformational changes in S4 to those in S5 and S6 that constitute gating and mutations of S5 in Shaker K+ channels produce large changes in the stability of the open state (Kanevsky et al., 1999; Shieh et al., 1997). These authors found that altering residues in S5 caused changes in gating kinetics, an effect of NEM action that we did not observe. However, the introduction of bulky aromatic or aliphatic side chains in S5 (Kanevsky & Aldrich, 1999) and alkylation of C242 in KCNQ2 both strongly bias the respective channels towards opening. It will be important to examine the effects of NEM on KCNQ channels in patches, and see the effects of action of NEM directly at the single-channel level. NEM is also known to inhibit the action of PTX-sensitive (Gi/o) G proteins (Jakobs et al., 1982; Milligan, 1988; Shapiro et al., 1994). If these G proteins were tonically inhibiting M-type currents then part or all of NEM augmentation of the currents could be due to relief of tonic G protein inhibition. We view this as unlikely however, since G protein actions on M-type channels have been shown to be wholly PTX-insensitive (Brown et al., 2000).
The second mechanism of action of NEM involves a shift in the voltage dependence of activation. Since this action is common to homomeric KCNQ2 and KCNQ3, and to heteromeric KCNQ2/3 channels, as well as SCG M current, the site of action of NEM producing the shift in gating should be on both subunits. This shift in gating will also act to increase M-type channel activity, especially at resting potentials typical of neurons. Interestingly, retigabine, a drug now in clinical trials as an anticonvulsant, also increases M-type currents by shifting their voltage dependence of activation (Main et al., 2000; Tatulian et al., 2001; Wickenden et al., 2000). In the case of retigabine, the action is strongest on KCNQ3 subunits and weakest on KCNQ1 subunits (Tatulian et al., 2001). It will be interesting to determine whether the effects of this drug are additive to those of NEM and if they act at a common site on the channel proteins. The actions of NEM described in this work, via both voltage-dependent and voltage-independent mechanisms, were quite similar between heteromeric KCNQ2/3 currents and M current of SCG neurons. Thus, we add the common action of NEM as additional evidence that heteromeric KCNQ2/3 channels underlie neuronal M current.
Loss-of-function mutations of KCNQ2 and KCNQ3 are involved in inherited epileptic disorders (Biervert et al., 1998; Charlier et al., 1998; Singh et al., 1998). Although these mutations frequently cause loss of channel function in expressed homomeric KCNQ channels, co-expression of the mutant subunit with wild type subunits results in only a 20–30% reduction in current amplitude when compared to experiments using an equivalent amount of wild type RNA (Schroeder et al., 1998). Although interpretations of such experiments are difficult, they suggest that the epileptic syndromes resulting from mutation of either KCNQ2 or KCNQ3 follow relatively minor perturbations in KCNQ current density. Thus, pharmacological agents that enhance M current may be valuable therapeutic tools. NEM would not be a candidate because of its widespread deleterious actions on cellular proteins, but understanding the mechanism/locus by which NEM enhances M current may be important in designing or identifying agents which will specifically enhance M current.
Acknowledgments
We thank Drs Nikita Gamper and Jeffry Isaacson for comments on the manuscript. This work was supported by NIH grants NS08174 (B. Hille), DA00286 (K. Mackie), DA11322 (K. Mackie), DA07278 (J. Roche) and NS43394 (M.S. Shapiro).
Abbreviations
- BAPTA
0.1 1,2-bis(2-aminophenoxy)ethane N,N,N′,N′-tetraacetic acid
- NEM
N-ethylmaleimide
- SCG
superior cervical ganglia
References
- BERNHEIM L., BEECH D.J., HILLE B. A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron. 1991;6:859–867. doi: 10.1016/0896-6273(91)90226-p. [DOI] [PubMed] [Google Scholar]
- BERNHEIM L., MATHIE A., HILLE B. Characterization of muscarinic receptor subtypes inhibiting Ca2+ current and M current in rat sympathetic neurons. Proc. Natl. Acad. Sci. U.S.A. 1992;89:9544–9548. doi: 10.1073/pnas.89.20.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIERVERT C., SCHROEDER B.C., KUBISCH C., BERKOVIC S.F., PROPPING P., JENTSCH T.J., STEINLEIN O.K. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- BROWN D.A., GAHWILER B.H., GRIFFITH W.H., HALLIWELL J.V. Membrane currents in hippocampal neurons. Prog. Brain Res. 1990;83:141–160. doi: 10.1016/s0079-6123(08)61247-9. [DOI] [PubMed] [Google Scholar]
- CHARLIER C., SINGH N.A., RYAN S.G., LEWIS T.B., REUS B.E., LEACH R.J., LEPPERT M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- COOPER E.C., HARRINGTON E., JAN Y.N., JAN L.Y. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J. Neurosci. 2001;21:9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DELMAS P., BROWN D.A., DAYRELL M., ABOGADIE F.C., CAULFIELD M.P., BUCKLEY N.J. On the role of endogenous G-protein beta gamma subunits in N-type Ca2+ current inhibition by neurotransmitters in rat sympathetic neurones. J. Physiol. 1998;506:319–329. doi: 10.1111/j.1469-7793.1998.319bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOYLE D.A., MORAIS CABRAL J., PFUETZNER R.A., KUO A., GULBIS J.M., COHEN S.L., CHAIT B.T., MACKINNON R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- HADLEY J.K., NODA M., SELYANKO A.A., WOOD I.C., ABOGADIE F.C., BROWN D.A. Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br. J. Pharmacol. 2000;129:413–415. doi: 10.1038/sj.bjp.0703086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALEY J.E., ABOGADIE F.C., DELMAS P., DAYRELL M., VALLIS Y., MILLIGAN G., CAULFIELD M.P., BROWN D.A., BUCKLEY N.J. The alpha subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons. J. Neurosci. 1998;18:4521–4531. doi: 10.1523/JNEUROSCI.18-12-04521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALLIWELL J.V., ADAMS P.R. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- HAMILTON S.E., LOOSE M.D., QI M., LEVEY A.I., HILLE B., MCKNIGHT G.S., IDZERDA R.L., NATHANSON N.M. Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13311–13316. doi: 10.1073/pnas.94.24.13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILLE B. Sunderland, Massachusetts, U.S.A.: Sinauer Associates, Inc; 2001. Ion channels of excitable membranes. [Google Scholar]
- HO S.N., HUNT H.D., HORTON R.M., PULLEN J.K., PEASE L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- JAKOBS K.H., LASCH P., MINUTH M., AKTORIES K., SCHULTZ G. Uncoupling of alpha-adrenoceptor-mediated inhibition of human platelet adenylate cyclase by N-ethylmaleimide. J. Biol. Chem. 1982;257:2829–2833. [PubMed] [Google Scholar]
- JENTSCH T.J. Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- JONES S., BROWN D.A., MILLIGAN G., WILLER E., BUCKLEY N.J., CAULFIELD M.P. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and Gαq/11. Neuron. 1995;14:399–405. doi: 10.1016/0896-6273(95)90295-3. [DOI] [PubMed] [Google Scholar]
- KANEVSKY M., ALDRICH R.W. Determinants of voltage-dependent gating and open-state stability in the S5 segment of Shaker potassium channels. J. Gen. Physiol. 1999;114:215–242. doi: 10.1085/jgp.114.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUBISCH C., SCHROEDER B.C., FRIEDRICH T., LUTJOHANN B., EL-AMRAOUI A., MARLIN S., PETIT C., JENTSCH T.J. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- LERCHE C., SCHERER C.R., SEEBOHM G., DERST C., WEI A.D., BUSCH A.E., STEINMEYER K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J. Biol. Chem. 2000;275:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- LERCHE H., JURKAT-ROTT K., LEHMANN-HORN F. Ion channels and epilepsy. Am. J. Med. Genet. 2001;106:146–159. doi: 10.1002/ajmg.1582. [DOI] [PubMed] [Google Scholar]
- MACKINNON R., YELLEN G. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science. 1990;250:276–279. doi: 10.1126/science.2218530. [DOI] [PubMed] [Google Scholar]
- MAIN M.J., CRYAN J.E., DUPERE J.R., COX B., CLARE J.J., BURBIDGE S.A. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol. Pharmacol. 2000;58:253–262. doi: 10.1124/mol.58.2.253. [DOI] [PubMed] [Google Scholar]
- MILLIGAN G. Techniques used in the identification and analysis of function of pertussis toxin-sensitive guanine nucleotide binding proteins. Biochem. J. 1988;255:1–13. doi: 10.1042/bj2550001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHROEDER B.C., HECHENBERGER M., WEINREICH F., KUBISCH C., JENTSCH T.J. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- SCHROEDER B.C., KUBISCH C., STEIN V., JENTSCH T.J. Moderate loss of function of cyclin-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature. 1998;396:687–690. doi: 10.1038/25367. [DOI] [PubMed] [Google Scholar]
- SCHWEITZER P., MADAMBA S., CHAMPAGNAT J., SIGGINS G.R. Somatostatin inhibition of hippocampal CA1 pyramidal neurons: mediation by arachidonic acid and its metabolites. J. Neurosci. 1993;13:2033–2049. doi: 10.1523/JNEUROSCI.13-05-02033.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHWEITZER P., MADAMBA S., SIGGINS G.R. Arachidonic acid metabolites as mediators of somatostatin-induced increase of neuronal M-current. Nature. 1990;346:464–467. doi: 10.1038/346464a0. [DOI] [PubMed] [Google Scholar]
- SELYANKO A.A., HADLEY J.K., BROWN D.A. Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J. Physiol. 2001;534:15–24. doi: 10.1111/j.1469-7793.2001.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SELYANKO A.A., HADLEY J.K., WOOD I.C., ABOGADIE F.C., JENTSCH T.J., BROWN D.A. Inhibition of KCNQ1-4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J. Physiol. 2000;522:349–355. doi: 10.1111/j.1469-7793.2000.t01-2-00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SELYANKO A.A., STANSFELD C.E., BROWN D.A. Closure of potassium M-channels by muscarinic acetylcholine-receptor stimulants requires a diffusible messenger. Proc. R. Soc. Lond. B. Biol. Sci. 1992;250:119–125. doi: 10.1098/rspb.1992.0139. [DOI] [PubMed] [Google Scholar]
- SHALABY F.Y., LEVESQUE P.C., YANG W.P., LITTLE W.A., CONDER M.L., JENKINS-WEST T., BLANAR M.A. Dominant-negative KvLQT1 mutations underlie the LQT1 form of long QT syndrome. Circulation. 1997;96:1733–1736. doi: 10.1161/01.cir.96.6.1733. [DOI] [PubMed] [Google Scholar]
- SHAPIRO M.S., ROCHE J.P., KAFTAN E.J., CRUZBLANCA H., MACKIE K., HILLE B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J. Neurosci. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAPIRO M.S., WOLLMUTH L.P., HILLE B. Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N-ethylmaleimide in rat sympathetic neurons. J. Neurosci. 1994;14:7109–7116. doi: 10.1523/JNEUROSCI.14-11-07109.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIEH C.C., KLEMIC K.G., KIRSCH G.E. Role of transmembrane segment S5 on gating of voltage-dependent K+ channels. J. Gen. Physiol. 1997;109:767–778. doi: 10.1085/jgp.109.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINGH N.A., CHARLIER C., STAUFFER D., DUPONT B.R., LEACH R.J., MELIS R., RONEN G.M., BJERRE I., QUATTLEBAUM T., MURPHY J.V., MCHARG M.L., GAGNON D., ROSALES T.O., PEIFFER A., ANDERSON V.E., LEPPERT M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- TATULIAN L., DELMAS P., ABOGADIE F.C., BROWN D.A. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J. Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TWITCHELL W., BROWN S., MACKIE K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- WANG H.S., MCKINNON D. Potassium currents in rat prevertebral and paravertebral sympathetic neurones: control of firing properties. J. Physiol. 1995;485:319–335. doi: 10.1113/jphysiol.1995.sp020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG H.S., PAN Z., SHI W., BROWN B.S., WYMORE R.S., COHEN I.S., DIXON J.E., MCKINNON D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- WANG Q., CURRAN M.E., SPLAWSKI I., BURN T.C., MILLHOLLAND J.M., VANRAAY T.J., SHEN J., TIMOTHY K.W., VINCENT G.M., DE JAGER T., SCHWARTZ P.J., TOUBIN J.A., MOSS A.J., ATKINSON D.L., LANDES G.M., CONNORS T.D., KEATING M.T. Positional cloning of a novel potassium channel gene: KvLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- WICKENDEN A.D., YU W., ZOU A., JEGLA T., WAGONER P.K. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol. Pharmacol. 2000;58:591–600. doi: 10.1124/mol.58.3.591. [DOI] [PubMed] [Google Scholar]
- YU S.P., KERCHNER G.A. Endogenous voltage-gated potassium channels in human embryonic kidney (HEK293) cells. J. Neurosci. Res. 1998;52:612–617. doi: 10.1002/(SICI)1097-4547(19980601)52:5<612::AID-JNR13>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]