

Abstract
The presynaptic interactions between facilitatory β-adrenoreceptors and inhibitory 5-hydroxytryptamine (5-HT) receptors modulating glutamate release from cerebrocortical nerve terminals were examined.
4-Aminopyridine (4-AP, 1 mM)-evoked glutamate release was facilitated by the membrane permeant cyclic-3′,5′-adenosine monophosphate (cAMP) analogue, 8-bromo-cAMP (8-Br-cAMP), used to directly activate cAMP-dependent protein kinase (PKA).
The β-adrenoreceptor agonist, isoprenaline (ISO), effected a concentration-dependent potentiation of 4-AP-evoked glutamate release which was abolished by the β-adrenoreceptor antagonist, propranolol, and the PKA inhibitor, Rp-cyclic-3′,5′-adenosine-monophosphothioate (Rp-cAMPS).
5-HT receptor activation by 100 μM 5-HT produced an inhibition of 4-AP-evoked glutamate release in nerve terminals. The inhibitory effect of 5-HT could be mimicked by the selective 5-HT1A receptor agonist, 8-hydroxy-dipropylaminotetralin (8-OH-DPAT) and antagonized by 1-(2-methoxyphenyl)-4-(4-phthalimidobutyl)piperazine (NAN-190).
When 5-HT (or 8-OH-DPAT) was used in conjunction with ISO or 8-Br-cAMP, the β-adrenoreceptor- and PKA-mediated potentiation of glutamate release was abrogated.
The inhibitory crosstalk of 5-HT1A receptors to β-adrenoceptor-mediated facilitation of glutamate release was abolished in the presence of NAN-190.
Examination of voltage-dependent Ca2+ influx revealed that, while ISO and 5-HT alone caused a respective potentiation and diminution of the 4-AP-evoked increase in [Ca2+]c, the co-presence of 5-HT abolished the ISO mediated potentiation of Ca2+ influx.
Together, these results suggest that β-adrenoreceptors and 5-HT1A receptors coexist on the cerebrocortical nerve terminals and that the cross-talk between the two receptor signalling pathways occurs at a locus downstream from cAMP production, possibly at the level of voltage-dependent Ca2+ influx.
Keywords: Adenylyl cyclase, calcium, glutamate release, 5-hydroxytryptamine, isoprenaline, cAMP-dependent protein kinase (PKA), presynaptic G-protein-coupled receptors, phosphorylation, synaptosomes, voltage-dependent calcium channel
Introduction
Metabotropic signalling cascades initiated by extracellular ligands or first messengers typically involve the posttranslational modification of proteins by phosphorylation/dephosphosphorylation and represent fundamental means for the regulation of cellular function. In the mammalian central nervous system, a number of neurotransmitters act via heterotrimeric G-protein-coupled receptors (GPCRs) to affect excitatory and inhibitory synaptic transmission by both phosphorylation-dependent and independent mechanisms. Presynaptically, together with ionotropic receptors (Macdermott et al., 1999), coincidence and interaction of GPCR-mediated signalling cascades potentially form the basis for modulation of neurotransmitter release underlying synaptic plasticity. With respect to glutamate release, facilitatory and inhibitory auto- and hetero-receptor GPCRs have been shown to produce opposing effects on release, as well as cross-regulating each other's function within the same terminal (Herrero et al., 1992; Coffey et al., 1994; Vazquez et al., 1995; Budd & Nicholls, 1995). Perhaps the most compelling evidence for the role of GPCR signalling cascades in synaptic plasticity has been obtained for GPCRs that activate adenylyl cyclase (AC) to produce the prototypic second-messenger, cyclic-3′,5′-adenosine monophosphate (cAMP). Apart from the demonstration that mouse knockouts of AC lead to impairment of synaptic potentiation (Storm et al., 1998), the target for cAMP, cAMP-dependent protein kinase (PKA), and its pre- and postsynaptic protein phosphorylation substrates, have been implicated in a number of models addressing synaptic plasticity. Thus, activation of β-adrenoreceptors, that are positively coupled to AC through a Gs protein, has been shown to enhance synaptic transmission in slice preparations (Gereau & Conn, 1994; Huang et al., 1996). In direct support of a presynaptic mechanism of action of β-adrenoreceptor agonists, aβ-adrenoreceptor/cAMP/cAMP-dependent protein kinase (PKA)-dependent pathway has been ascribed a facilitatory role in studies looking at glutamate release from cerebrocortical nerve terminals (Herrero & Sanchez-Prieto, 1996). Along the lines of a reciprocating GPCR signalling paradigm, 5-hydroxytryptamine (5-HT), has emerged as an important inhibitory neurotransmitter involved in central neuromodulation. Indeed, 5-HT1A receptors, that have been shown to be negatively coupled to AC through a pertussis toxin-sensitive Gi protein, depress synaptic transmission in the locus coeruleus and basolateral amygdala (Bobker & Williams, 1989; Cheng et al., 1998). Consistent with a presynaptic mechanism of actions, data from in vitro release experiments as well as in vivo microdialysis studies have shown that 5-HT receptor activation can mediate the downregulation of the release of a number of neurotransmitters (Raiteri et al., 1986; Srkalovic et al., 1994; Maura & Raiteri, 1996; Maura et al., 1998). Consequently, presynaptic coincidence of β-adrenoreceptor and 5-HT receptor signalling has been inferred in the PKA-dependent modulation of synaptic enhancement observed in slice preparations of the basolateral amygdala (Wang et al., 1999). In the current study we therefore sought to determine whether we could obtain direct evidence for this type of cross-talk within nerve terminals.
The isolated cerebrocortical nerve terminal preparation (synaptosomes) represents a well established model for the specific study of presynaptic regulatory pathways and their possible cross-talk, in the absence of any complications of interpretation produced by concomitant postsynaptic effects (Nicholls, 1993; Sanchez-Prieto et al., 1996). Using this model, we have addressed the question whether facilitatory β-adrenoreceptors and inhibitory 5-HT1A receptors coexist in the same glutamatergic nerve terminals and whether the two signal transduction pathways initiated by these, respectively Gs- and Gi-linked, receptors can cross-talk in their modulation of neurotransmitter glutamate release. We examined cAMP/PKA-dependent regulation of 4-AP-evoked glutamate release from cerebrocortical synaptosomes using exogenous cAMP or the β-adrenoreceptor agonist, isoprenaline (ISO), to stimulate AC and produce endogenous cAMP. Furthermore, we investigated the effect of 5-HT receptor activation on the PKA-mediated modulation of glutamate release. Finally, we addressed the mechanism of the β-adrenoreceptor and 5-HT receptor interaction by looking at the effects of the agonists on voltage-dependent Ca2+ entry into nerve terminals. We propose that the interaction between the intracellular signalling pathways activated by β-adrenoreceptors and 5-HT1A receptors occurs at a locus downstream from cAMP production, at the level of nerve terminal excitability and/or voltage-dependent Ca2+ channels (VDCCs). Our findings contribute fundamental clues to the understanding of the presynaptic changes occurring during synaptic plasticity mediated with coincident facilitatory and inhibitory inputs, respectively coupled by Gs and Gi, to provide opposing modulation of neurotransmitter glutamate release.
Methods
Preparation of synaptosomes
Synaptosomes were prepared from the cerebral cortices of 2-month-old male Sprague–Dawley rats as described previously (Sihra, 1997). The final synaptosomal fraction was resuspended in approximately 2–3 ml HEPES-buffered incubation medium (HBM (mM): NaCl 140, KCl 5, NaHCO3 5, MgCl2·6H2O 1, Na2HPO4 1.2, glucose 10, HEPES 20, pH 7.4) and protein concentration determined using the Bradford assay. Synaptosomes were centrifuged in the final wash to obtain synaptosomal pellets with 0.3 mg protein. Synaptosomal pellets were stored on ice and used within 2–3 h.
Glutamate release assay
Glutamate release was assayed by on-line fluorimetry (Nicholls & Sihra, 1986). Pelleted synaptosomes were resuspended at a protein concentration of 0.3 mg ml−1 in HBM containing 16 μM bovine serum albumin (BSA) and incubated in a stirred and thermostatted cuvette at 37°C in a Perkin-Elmer LS-3B spectrofluorimeter. NADP+ (1 mM), glutamate dehydrogenase (50 units ml−1) and CaCl2 (1 mM) were added after 3 min. After a further 7 min of incubation, 1 mM 4-AP was added to stimulate glutamate release. The oxidative decarboxylation of released glutamate leading to the reduction of NADP+ was monitored by measuring NADPH fluorescence at excitation and emission wavelengths of 340 and 460 nm, respectively. Data were accumulated at 2-s intervals. A standard of exogenous glutamate (5 nmol) was added at the end of each experiment and the fluorescence change produced by the standard addition used to calculate the released glutamate as nmol glutamate released per mg synaptosomal protein (nmol mg−1). Release traces are shifted vertically to align the point of depolarization as zero release. Unless otherwise indicated, release values quoted in the text are levels attained at ‘steady-state' after 5 min of depolarization (nmol mg−1 5 min−1). Cumulative data were analysed using Lotus 1-2-3 and MicroCal Origin. Statistical analysis was performed by two-tailed Student's t-tests.
Cytosolic Ca2+ measurements using Fura-2
Synaptosomes (0.3 mg ml−1) were preincubated in HBM containing 5 μM Fura-2-acetoxymethyl ester, 0.1 mM CaCl2, and 16 μM BSA for 25 min at 37°C in a stirred test-tube. After Fura-2 loading, synaptosomes were centrifuged in a microfuge for 15-s, the pellet resuspended in HBM at 37°C and the synaptosomal suspension stirred in a thermostatted cuvette in a Perkin-Elmer LS-3B spectrofluorimeter. CaCl2 (1 mM) was added after 3 min and further additions were made after an additional 5 min, as described in the legends to the figures. Fluorescence data were accumulated at excitation wavelengths of 340 and 380 nm (emission wavelength 505 nm) at 3.5-s intervals. Cytosolic free Ca2+ concentration ([Ca2+]c, nM) was calculated following calibration procedures (Sihra et al., 1993), using 0.1% sodium dodecyl sulphate to obtain the maximal fluorescence with Fura-2 saturation with Ca2+, followed by 10 mM EGTA (Tris buffered) to obtain minimum fluorescence in the absence of any Fura-2/Ca2+ complex. Cytosolic free Ca2+ concentration ([Ca2+]c, nM) was evaluated using 340/380 fluorescence ratios as described (Grynkiewicz et al., 1985). Cumulative data were analysed using Lotus 1-2-3 and MicroCal Origin. Statistical analysis was performed by two-tailed Student's t-tests.
Materials
8-OH-DPAT, 8-Br-cAMP, IBMX, Rp-cAMPS and propranolol were from Research Biochemicals International (Natick, MA, U.S.A.). Fura-2 acetoxymethyl ester (fura-2-AM) was from Molecular Probes (Eugene, OR, U.S.A.). Isoprenaline, 5-HT and all other regents were from Sigma (Poole, U.K.) or Merck (Poole, U.K.).
Results
Glutamate release from purified cerebrocortical synaptosomes was monitored on-line, using an assay employing exogenous glutamate dehydrogenase and NADP+ to couple the oxidative decarboxylation of the released glutamate to the generation of NADPH detected fluorometrically (Nicholls & Sihra, 1986). We first established the modulatory influence of cAMP-dependent protein kinase (PKA) on 4-AP-evoked glutamate release by activating the protein kinase using a cAMP analogue, 8-bromo-cAMP (8-Br-cAMP), to mimic the endogenous stimulation of AC. Under control conditions, 4-AP (1 mM) evoked a glutamate release of 7.76±0.54 nmol mg−1 5 min−1 from synaptosomes incubated in the presence of 1 mM CaCl2 (Figure 1(i)). Application of 8-Br-cAMP (150 μM) alone produced only slight potentiation of 4-AP-evoked glutamate release to 9.98±1.22 nmol mg−1 5 min−1 (Figure 1(ii)). The limited extent of this facilitation was possibly due to phosphodiesterase activity as well as the reported inhibitory action of ‘leaked' endogenous adenosine acting at adenosine A1 receptors (Pockett et al., 1993). Accordingly, when 8-Br-cAMP was coapplied with 3-isobutyl-1-methylxanthine (IBMX, 50 μM), which acts both as a phosphodiesterase inhibitor and an adenosine A1 receptor antagonist, 4-AP-evoked glutamate release was strongly potentiated by the cyclic nucleotide analogue to 18.74±1.66 nmol mg−1 5 min−1 (Figure 1(iii)).
Figure 1.
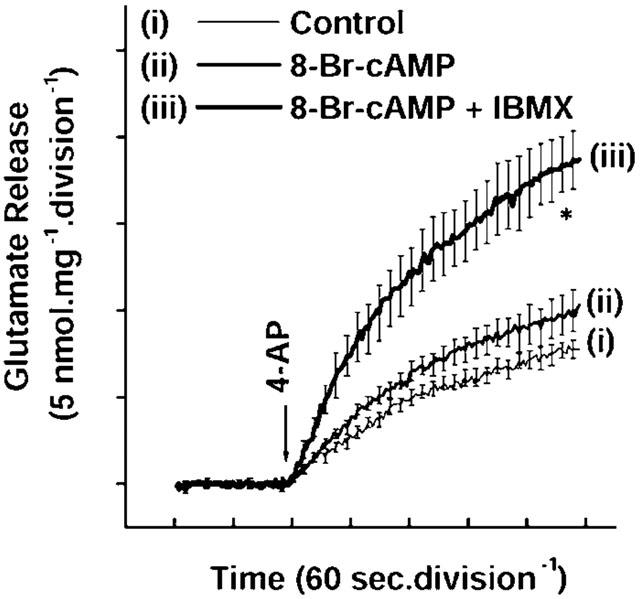
Facilitation of 4-AP-evoked glutamate release by the cAMP analogue 8-Br-cAMP. Glutamate release was evoked by the addition of 1 mM 4-AP in (i) the absence (control) and (ii) the presence of 100 μM 8-Br-cAMP, or (iii) 100 μM 8-Br-cAMP+100 μM IBMX, added 5 min before depolarization. Results are means±s.e.means of three to five independent experiments. Means and s.e.means were calculated at each time-point (2-s), but error bars are only shown every 10-s for clarity. *P<0.05; different from control.
We next examined whether stimulation of endogenous cAMP production through β-adrenoreceptor stimulation of AC could cause a facilitation of transmitter release similar to that seen with the cAMP analogue. Under control conditions, 4-AP (1 mM) evoked a glutamate release of 8.32±0.79 nmol mg−1 5 min−1 from synaptosomes incubated in the presence of 1 mM CaCl2 (Figure 2(i)). Preincubation with the β-adrenoreceptor agonist, isoprenaline (100 μM; ISO), significantly potentiated the 4-AP-evoked release to 23.32±0.74 nmol mg−1 5 min−1 (Figure 2a(ii)). The ISO-mediated potentiation of glutamate release was concentration-dependent, with 15, 50, and 100 μM ISO increasing glutamate release to 118.3±2.2%, 168.5±2.6%, and 233.6±2.4% of control values respectively (Figure 2b).
Figure 2.
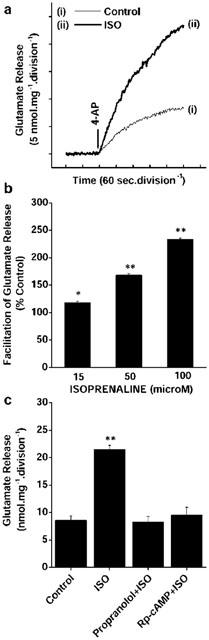
ISO-mediated potentiation of 4-AP-evoked release of glutamate: Concentration-dependence and sensitivity to β-adrenoreceptor and PKA antagonism. (a) Glutamate release was evoked by the addition of 1 mM 4-AP in the absence (control) and in the presence of 50 μM ISO, added 5 min before depolarization. (b) Concentration-dependent potentiation of 4-AP-evoked glutamate release by ISO. Results are means±s.e.means of four independent experiments. (c) 4-AP-evoked Ca2+-dependent glutamate release in control conditions (control) or in the presence of 50 μM ISO, 5 μM propanolol +50 μM ISO or 100 μM Rp-cAMP+50 μM ISO, added 5 min before 4-AP addition. Columns are the means±s.e.means of four independent experiments. *P<0.05; **P<0.01; different from control.
The ISO-mediated potentiation of glutamate release displayed classic β-adrenoreceptor pharmacology in that the facilitation of 4-AP (1 mM)-evoked release by 50 μM ISO was effectively blocked by the β-adrenoreceptor antagonist propranolol (5 μM) ((nmol mg−1 5 min−1): control 4-AP, 8.59±0.79; ISO (50 μM)+4-AP, 21.5±0.74; propranolol+ISO (50 μM)+4-AP, 8.3±0.99) (Figure 2c). To confirm that the β-adrenoreceptor-mediated potentiation of 4-AP-evoked glutamate release occurred as a consequence of the activation of PKA by cyclic nucleotide, we used the non-hydrolysable cAMP analogue, Rp-cyclic-3′,5′-adenosine-monophosphothioate (Rp-cAMPS), which acts to specifically inhibit PKA through competitive binding to the cAMP-binding sites on the regulatory subunits of the kinase. The potentiation of control 4-AP (1 mM)-evoked release by 50 μM ISO was effectively prevented by the application of 100 μM Rp-cAMPS (9.62±1.43 nmol mg−1 5 min−1) (Figure 2c). These results suggest that the presynaptic β-adrenoreceptor coupling to the facilitation of glutamate release from cerebrocortical nerve terminals is mediated by a cAMP/PKA signalling pathway.
In contrast to the facilitatory effect of β-adrenoreceptor activation on glutamate release, presynaptic 5-HT receptor stimulation produced an inhibition of neurotransmitter release. Control 4-AP (1 mM)-evoked release of 8.28±0.94 nmol mg−1 5 min−1 (Figure 3(i)) was attenuated by 5-HT (100 μM) to 5.84±0.4 nmol mg−1 5 min−1 (Figure 3(ii)). Looking for an interplay between facilitatory and inhibitory aminergic signalling pathways modulating glutamate release hitherto described, we next investigated the influence of 5-HT on the potentiation of 4-AP-evoked glutamate release obtained with ISO. Figure 4a compares glutamate release under control conditions (i), in the presence of 5-HT (ii) or ISO (iii) alone, and in the presence of the two receptor agonists together (iv). Pretreatment with 5-HT severely attenuated the potentiation of 4-AP-evoked glutamate release by ISO, with the magnitude of facilitated release decreasing from 23.4±0.94 nmol mg−1 5 min−1 with ISO alone, back to levels (8.98±1.26 nmol mg−1 5 min−1) resembling controls when ISO was applied in the presence of 5-HT (Figure 4b).
Figure 3.
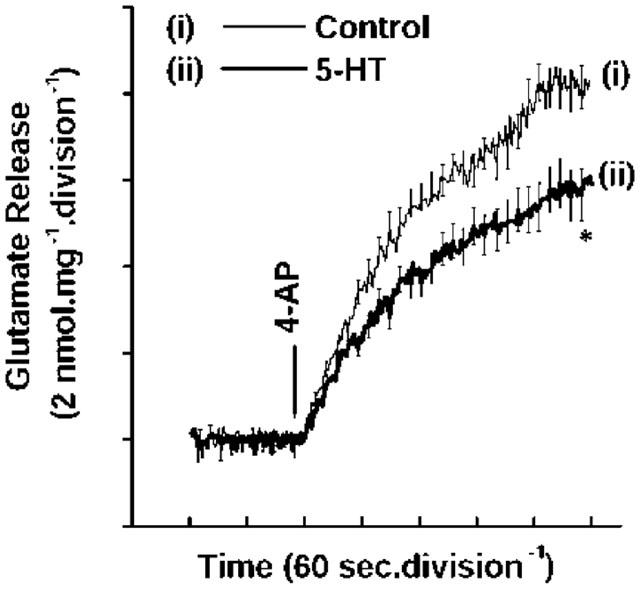
5-HT-mediated inhibition of 4-AP-evoked glutamate release. Glutamate release was evoked by the addition of 1 mM 4-AP in (i) the absence (control) and (ii) in the presence of 100 μM 5-HT. Results are means±s.e.means of three independent experiments. Means and s.e.means were calculated at each time-point (2-s), but error bars are only shown every 10-s for clarity. *P<0.05; different from control.
Figure 4.
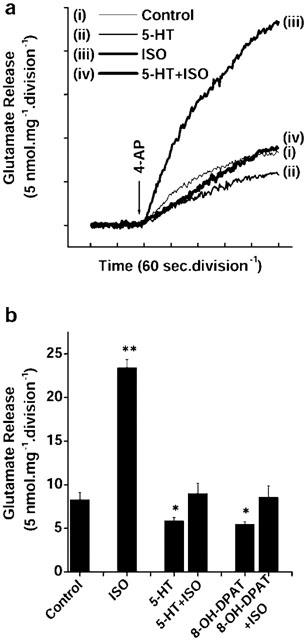
Abrogation of ISO-mediated potentiation by 5-HT and the selective 5-HT1A agonist, 8-OH-DPAT. (a) Glutamate release was evoked by the addition of 1 mM 4-AP in (i) the absence (control) and in the presence of (ii) 50 μM ISO, (iii) 100 μM 5-HT or (iv) 100 μM 5-HT+50 μM ISO, added 5 min before depolarization. (b) Quantitative comparison of the extent of glutamate release evoked by 1 mM 4-AP in the absence and presence of 50 μM ISO (added 5 min before depolarization) and absence and presence of 100 μM 5-HT or 10 μM 8-OH-DPAT (added 2 min before ISO). Results are means±s.e.means of four independent experiments. *P<0.05; **P<0.01; different from control.
To determine which 5-HT receptor subtype was involved in the ISO-mediated potentiation of 4-AP-evoked glutamate release, the effect of the selective 5-HT1A receptor agonist 8-hydroxy-dipropylaminotetralin (8-OH-DPAT; 10 μM) was also examined in parallel experiments. Control 4-AP-evoked release of 7.40±1.00 nmol mg−1 5 min−1 was attenuated by 8-OH-DPAT to 4.33±0.88 nmol mg−1 5 min−1 in similar manner to 5-HT (Figure 4b). Also as with 5-HT, in the presence of 8-OH-DPAT, ISO again failed to potentiate the 4-AP-evoked release of glutamate (8.57±1.4 nmol mg−1 5 min−1) (Figure 4b), suggesting that the effect of 5-HT may be mediated via 5-HT1A receptor activation.
To confirm the extent to which the observed modulation of glutamate release by 5-HT-receptor activation was occurring via specific stimulation of 5-HT1A receptors, we looked at the effect of 5-HT in the presence of 5-HT1A receptor antagonist 1-(2-methoxyphenyl)-4-(4-phthalimidobutyl)piperazine (NAN-190) (Penington & Kelly, 1990). While NAN-190 (2 μM) itself had no effect alone, it eliminated both the 5-HT (100 μM)-mediated inhibition of glutamate release, and the 5-HT-mediated abrogation of ISO (50 μM)-facilitation (Figure 5). The substantial extent of the latter effect implies that 5-HT1A receptors constitute a major proportion of the presynaptic 5-HT receptors being stimulated in our preparation.
Figure 5.
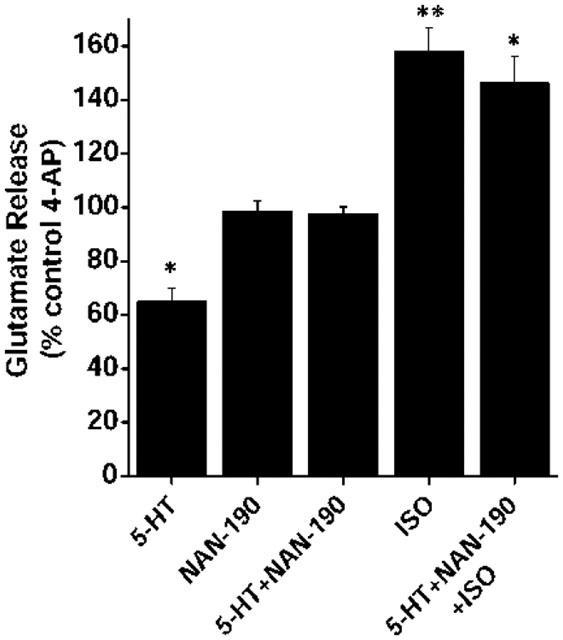
Elimination of 5-HT receptor-mediated abrogation of β-adrenoreceptor-mediated facilitation of glutamate by the 5-HT1A antagonist, NAN-190. Modulation of 1 mM 4-AP-evoked glutamate release by 100 μM 5-HT, in the absence or presence of 2 μM NAN-190 (added 5 min before 5-HT). Lack of effect of 100 μM 5-HT on the 50 μM ISO-induced facilitation in the presence of 2 μM NAN-190. Results are presented as per cent of the control 4-AP-evoked release and are means±s.e.means of three to five independent experiments. *P<0.05; **P<0.01; different from control.
The results hitherto suggest that there is antagonistic interaction between β-adrenoreceptors and 5-HT1A receptors with respect to the modulation of glutamate release from cerebrocortical synaptosomes. Interestingly, in common with this effect of presynaptic 5-HT receptors, we found a similar abrogatory effect on ISO-mediated potentiation of 4AP-evoked glutamate release with the activation of inhibitory GABAB receptors localized in nerve terminals (Perkinton & Sihra, 1998; Wang & Sihra, unpublished observations). Next, to identify the locus in the second messenger cascade at which the cross-talk between β-adrenoreceptors and 5-HT receptors takes place, we looked at the effect of 5-HT on the exogenous cAMP-mediated potentiation of glutamate release. If cross-talk occurs at locus downstream of cAMP production, 5-HT should eliminate the effect of cAMP analogue on 4-AP-evoked glutamate release. However, if it occurs more proximally, at the level of Gi modulation of AC activity for instance, then the activation of PKA effected by the cAMP analogue should persist in causing the facilitation by 8-Br-cAMP observed in Figure 1. As before (Figure 1), these experiments employing cAMP analogues to stimulate PKA were performed in the presence of IBMX to both prevent the degradation of cAMP analogue, and obviate the inhibitory action of adenosine A1 receptors. Figure 6 shows that the potentiation of control 4-AP-evoked release of 6.77±1.77 nmol mg−1 5 min−1 (Figure 6(i)) to 18.3±3.04 nmol mg−1 5 min−1 produced by 8-Br-cAMP+IBMX (Figure 6(ii)) was effectively blocked in synaptosomes pretreated with 5-HT to produce release indistinguishable from control levels (5.95±0.75 nmol mg−1 5 min−1, Figure 6(iii)). These results indicated that the interaction between the β-adrenoreceptor and 5-HT receptor likely occurs downstream of cAMP production and possibly at the level of PKA-dependent modulation of events, such as nerve terminal excitation, Ca2+ influx, or the processes leading up to and possibly including exocytosis itself.
Figure 6.
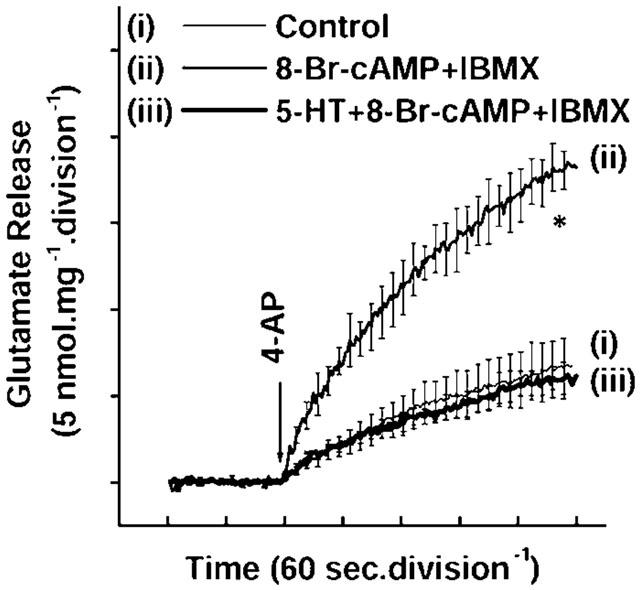
Antagonism of 8-Br-cAMP-mediated potentiation of glutamate release by 5-HT. Glutamate release was evoked by the addition of 1 mM 4-AP in (i) the absence (control) and in the presence of (ii) 100 μM 8-Br-cAMP or (iii) 100 μM 5-HT+100 μM 8-Br-cAMP, added 5 min before depolarization. In the experiments with 5-HT, the agonist was added 2 min before 8-Br-cAMP. Results are means±s.e.means of four to five independent experiments. Means and s.e.means were calculated at each time-point (2-s), but error bars are only shown every 10-s for clarity. *P<0.05; different from control.
Looking directly at the involvement of the modulation of Ca2+ influx in the cross-talk between β-adrenoreceptors and 5-HT receptors, we used Fura-2 to assess the effects of ISO and 5-HT on the 4-AP-evoked increase of [Ca2+]c. 4-AP (1 mM) caused a rise in [Ca2+]c to a plateau level of 128.3±10.7 nM (Figure 7(i)). This 4-AP-evoked rise in [Ca2+]c was increased by 61.9 nM with ISO (50 μM) preincubation (Figure 7(ii)) and slightly decreased by 13.7 nM in the presence of 5-HT (100 μM) (Figure 7(iii)). Despite the modest inhibitory effect of 5-HT alone on Ca2+ influx, the agonist markedly reduced the substantial potentiation produced by ISO (Figure 7(iv)). While ISO preincubation also caused a small but notable increase in the basal levels of [Ca2+]c prior to 4-AP-mediated depolarization, this effect was statistically not significant (Figure 7(ii)).
Figure 7.
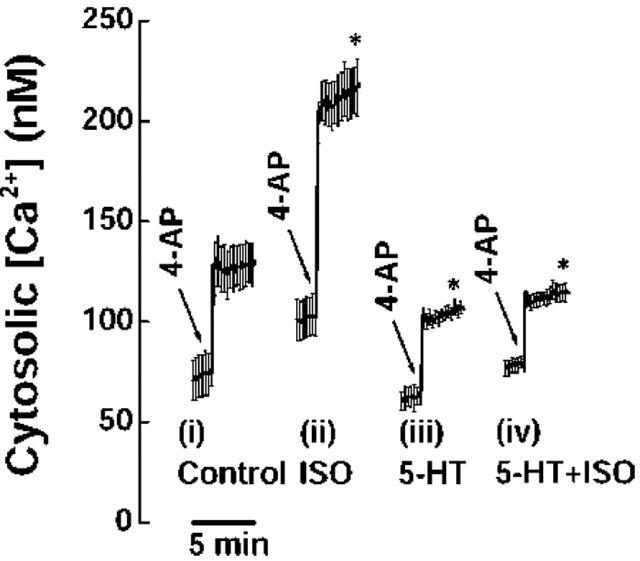
Facilitation of 4-AP-evoked Ca2+ influx by β-adrenoreceptor activation is suppressed by 5-HT. Cytosolic free Ca2+ concentration (nM) was monitored using Fura-2. Synaptosomes were stimulated with 1 mM 4-AP in (i) the absence (control) and in the presence of (ii) 50 μM ISO, (iii) 100 μM 5-HT or (iv) 100 μM 5-HT+50 μM ISO, added 5 min before stimulation. Results are means±s.e.means of five independent experiments. Means and s.e.means were calculated at each 3.75-s time point, but error bars are shown for every fourth reading for clarity. *P<0.05; different from control.
Discussion
Our results demonstrate that 5-HT strongly suppresses the facilitatory effects of exogenous cAMP and the β-adrenoreceptor agonist, isoprenaline (ISO), on glutamate release from cerebrocortical nerve terminals. This suggests that there is antagonistic interaction or cross-talk, between presynaptic β-adrenoreceptor and 5-HT receptor signalling cascades. While previous electrophysiological studies in the basolateral amygdala had evinced presynaptic interaction indirectly by looking at changes in postsynaptic responses (Wang et al., 1999), using the synaptosomal model, we provide direct evidence for a presynaptic locus and mechanism for the modulation of glutamatergic transmission by cross-regulation of these two GPCR signalling systems.
β-Adrenoreceptor-mediated facilitation of glutamate release
Electrophysiological studies have shown that elevation of cAMP and PKA stimulation can enhance excitatory transmission in the Schaeffer collateral-CA1 (Chavez-Noriega & Stevens, 1992; Gereau & Conn, 1994; Huang & Kandel, 1994) and mossy fibre-CA3 (Weisskopf et al., 1994) pathways of the hippocampus, as well as in the basolateral amygdala (Huang et al., 1996; 1998). Here, the β-adrenoreceptor agonist ISO was found to potentiate 4-AP-evoked glutamate release from cerebrocortical synaptosomes. Sensitivity of this effect to the PKA inhibitor, Rp-cAMPS, and the ability of the cAMP analogue, 8-Br-cAMP, to mimick the effect of β-adrenoreceptor activation, implicates an AC/cAMP/PKA signalling cascade in the enhancement of glutamate release (Herrero & Sanchez-Prieto, 1996).
Posttranslational modification of proteins by phosphorylation may regulate neurotransmitter release at multiple loci in the stimulus-exocytosis cascade including: ion-channels modulating nerve terminal excitability (Frace & Hartzell, 1993; Roeper & Pongs, 1996; Dascal & Lotan, 1991; Li et al., 1993), voltage-dependent Ca2+ channels (VDCCs) and, downstream of Ca2+ entry, components of the synaptic vesicle trafficking and exocytotic apparatus (Thompson et al., 1993; Trudeau et al., 1996; Sihra & Nichols, 1993). Given the significant effect of ISO on the 4-AP-evoked increase in [Ca2+]c observed here, it is tempting to postulate a PKA-mediated effect on either channels involved in determining synaptosomal excitability or VDCC subunits themselves, to upregulate Ca2+ influx/glutamate release.
In the absence of direct evidence of phosphorylation, a number of synaptosomal studies have suggested a delayed rectifier K+-channel target for the modulation of nerve terminal excitability by protein phosphorylation (Sanchez-Prieto et al., 1996). An effect of PKA at this locus has been suggested based on the enhancement of facilitation of 50 μM 4-AP-mediated depolarization by forskolin (Herrero & Sanchez-Prieto, 1996). Notably however, in contrast to our clear demonstration of 100 μM ISO-mediated facilitation of 1 mM 4-AP-evoked Ca2+ entry and glutamate release, no modulation was obtained with 1 mM 4-AP in this previous study. Although the reasons for this discrepancy are not immediately apparent, one obvious notable difference, apart from our use of a percoll-gradient purified synaptosome preparation (cf. Herrero & Sanchez-Prieto, 1996), was the significantly higher resting [Ca2+]c reported (∼160 nM, Herrero & Sanchez-Prieto, 1996 versus ∼70 nM, current study). One consequence of raised [Ca2+]c would be the activation of nerve terminal Ca2+-dependent protein kinases and phosphatases (Nichols et al., 1990; Sihra et al., 1995; Burley & Sihra, 2000). The inevitable alteration of the phosphorylation-state of key modulatory substrate proteins may subsequently affect the responsiveness of 4-AP-evoked release to modulation by PKA differentially, depending on the levels of voltage-dependent Ca2+-entry (Herrero & Sanchez-Prieto, 1996).
Our observation, using membrane potential-sensitive fluorophors (Perkinton & Sihra, 1998), that 1 mM 4-AP-evoked depolarization of synaptosomes is minimally affected even after substantive stimulation of the cAMP/PKA pathway using the AC activator forskolin (Wang and Sihra, unpublished observations), argues against a major effect of PKA on synaptosomal excitability. Several previous studies have used the differential mechanisms of depolarization with 4AP and high-[K+]external (high-KCl) to resolve effects on channels controlling synaptosomal excitability (4AP▪equals;high-KCl) as opposed to VDCCs (4AP≡high-KCl) (Barrie et al., 1991; Coffey et al., 1993; Sanchez-Prieto et al., 1996 for review). However, the strong, clamped, tetrodotoxin-insensitive depolarization produced by KCl causes ion-channel inactivation and evidently adversely affects modulatory influences known to impinge on VDCCs as we have observed (Perkinton & Sihra, 1998; Davies & Sihra, unpublished observations). A lack of effect of PKA activation on KCl-mediated glutamate release, as has been reported (Herrero & Sanchez-Prieto, 1996), may not therefore be unequivocal evidence against a modulatory effect on VDCCs and for an upstream regulation of synaptosomal excitability. Importantly, in electrophysiological studies directly addressing the involvement of ion-channel activities during synaptic transmission, using patched neurons under voltage-clamp, delayed rectifier K+-currents were reported to be recalcitrant to modulation by PKA, while a clear modulation of Ca2+ currents was demonstrable (Huang et al., 1998). While this did not exclude the action of PKA at other channel targets determining synaptosomal excitability, in this type of study, PKA could be directly implicated in the modulation of P-type VDCCs.
Given that glutamate exocytosis is triggered by a localized increase in [Ca2+]c (Sihra et al., 1992) via specific P/Q- and N-subtypes of VDCCs (Pocock & Nicholls, 1992; Turner et al., 1993), direct regulation of Ca2+ entry into the nerve terminals would be potent means to regulate synaptic transmission. Indeed, there is compelling evidence for the posttranslational modulation of VDCCs by direct phosphorylation of both α1- and β-subunits of VDCCs, and by a number of protein kinases, including PKA (Hell et al., 1995; Chien & Hosey, 1998; Catterall, 2000). With both P/Q-type and N-type VDCCs, receptor-mediated increases in the PKA-dependent phosphorylation appear to facilitate channel activity (Gray & Johnston, 1987; Catterall, 2000). Our postulate, that a β-adrenoreceptor/AC/cAMP/PKA signaling cascade may be enhancing glutamate release by action at the VDCC level, is therefore consistent with these observations and others, explicitly showing ISO-mediated enhancement of P- and Q-type Ca2+ currents recorded under voltage-clamp conditions (Huang et al., 1996; 1998).
5-HT Receptors mediate inhibition of glutamate release
In contrast to β-adrenoreceptor stimulation, 5-HT receptor activation in nerve terminals suppressed glutamate release. There is a consensus that presynaptic inhibition by 5-HT is mediated by the 5-HT1 receptor type and, depending on the system, specifically by 5-HT1A (Bobker & Williams, 1989; Cheng et al., 1998; Lin et al., 2001), 5-HT1B (Bobker & Williams, 1989; Tanaka & North, 1993; Singer et al., 1996) and 5-HT1D (Travagli & Williams, 1996; Maura & Raiteri, 1996; Maura et al., 1998) subtypes. In the current study, we found that the selective 5-HT1A receptor agonist, 8-OH-DPAT, mimicked the effect of 5-HT. On the other hand, we obtained no significant effect with the 5-HT1B/5-HT1D agonist sumatriptan (data not shown). Indeed, the reversal of inhibitory effect of 5HT alone, as well as its abrogation of the ISO-induced facilitation of release, by NAN-190, reinforces the involvement of presynaptic 5-HT1A receptors in the observed modulation of glutamate release. Interestingly, in contrast to these results with cerebrocortical synaptosomes, with cerebellar synaptosomes, a significant inhibition of glutamate release by sumatriptan coupled with a lack of effect of 5-HT1A receptor activation, has implicated 5-HT1D receptors presynaptically, with 5-HT1A receptors being proposed to have post-synaptic function (Maura & Raiteri, 1996).
The reduction of 4-AP-evoked voltage-dependent Ca2+ influx produced by 5-HT, points to the suppression of VDCCs as a potential mechanism underlying the inhibition of glutamate exocytosis by 5-HT1A receptor activation. Electrophysiological studies, in several neuronal systems, have demonstrated 5-HT-mediated inhibition of VDCCs, in general (Penington & Kelly, 1990; Rhee et al., 1996; Cheng et al., 1998), and of P/Q- and N-type VDCCs, specifically (Foehring, 1996; Bayliss et al., 1997; Lin et al., 2001). This suppression of Ca2+ influx by 5-HT1A receptor activation is thought to occur through a direct, membrane-delimited, Gi/o-coupled inhibition of VDCCs (Foehring, 1996; Cheng et al., 1998; Lin et al., 2001), and is akin to presynaptic inhibition produced by GPCRs like GABAB (Wu & Saggau, 1995; Perkinton & Sihra, 1998) and adenosine A1 receptors (Barrie et al., 1991; Wu & Saggau, 1994; Lu & Gean, 1999).
Cross-talk of β-adrenoreceptor and 5-HT signalling in nerve terminals
In addition to the inhibitory effect of 5-HT receptor stimulation on 4-AP-evoked release of glutamate, 5-HT effectively blocked the facilitatory effect of PKA activation on glutamate release, whether the kinase was stimulated by endogenous cAMP produced by ISO-mediated β-adrenoreceptor activation of AC, or by exogenous cyclic nucleotide, 8-Br-cAMP. Given that co-application of ISO and 5-HT did not produce additive effects–the substantial ISO-mediated potentiation of glutamate release and Ca2+ influx are completely abolished by 5-HT, despite the much more modest inhibitory effect of the latter alone–the interaction of β-adrenoreceptor and 5-HT receptor signalling pathways can be interpreted as the presynaptic coexistence and cross-talk of the two systems in a major population of cerebrocortical synaptosomes. This β-adrenoreceptor and 5-HT receptor cross-talk is consistent with a presynaptic origin for the previously reported inhibitory actions of 5-HT1A receptor activation on ISO-mediated enhancement of excitatory postsynaptic potentials recorded in amygdalar neurons (Wang et al., 1999).
The antagonistic cross-talk between β-adrenoreceptors and 5-HT receptors described could in principle occur at one of several loci in GPCR signal transduction, viz. G-protein/AC coupling, cAMP metabolism, PKA activation, or alternatively, at the substrate level of presynaptic ion channels and/or release processes. At the level of AC, opposing homotypic and heterotypic interactions between stimulatory Gs-and inhibitory Gi-linked receptors have been reported (Pedarzani & Storm, 1996; Lopes et al., 1999; Gerber & Gahwiler, 1994). Reciprocity with Ca2+/calmodulin-sensitive isoforms of AC (Mons & Cooper, 1995; Xia & Storm, 1997) may also occur by inhibitory receptors effecting membrane-delimited Gi/o-mediated inhibition of VDCCs (Wu & Saggau, 1997). In the current experiments however, interaction at the level of AC appears unlikely because the 5-HT suppression of 4-AP-evoked glutamate release occurred even with AC activation circumvented using exogenous 8-Br-cAMP to activate PKA directly. β-adrenoreceptors and 5-HT receptors therefore seem to cross-talk downstream of PKA activation and at the level of one or more of the protein kinase substrates.
Our observation that both ISO and 5-HT affect Ca2+-entry, invokes the possibility that the two GPCR signalling cascades stimulated by these agonists may interact at the level of VDCCs to regulate glutamate release. Overtly, in concert with the 5-HT suppression of ISO-mediated facilitation of glutamate release, 5-HT strongly abrogated the facilitation of Ca2+-influx by ISO, despite having a relatively modest inhibitory effect on Ca2+-influx/release by itself. One possible implication of this is that, Gi/o-linked GPCRs causing inhibition of Ca2+ influx, converge with Gs-linked/AC GPCRs at the level of presynaptic VDCCs. In support of this notion, baclofen, a GABAB receptor agonist known to inhibit presynaptic VDCCs (Wu & Saggau, 1995; Pfrieger et al., 1994; Perkinton & Sihra, 1998), also suppressed ISO-mediated potentiation of glutamate release similarly to 5-HT (Wu & Saggau, 1997; Wang & Sihra, unpublished observations). However, the mechanism of the proposed abrogation of PKA-mediated facilitation of Ca2+ entry, by inhibitory GPCRs negatively-coupled to VDCCs, remains to be elucidated.
Together with the well documented reciprocity of Gs- and Gi/o-coupled GPCRs at the level of AC, the current study suggests a presynaptic interaction of GPCR/Gs-protein/AC/PKA-mediated facilitation and GPCR/Gi-protein-mediated inhibition, possibly at the level of VDCCs, to affect Ca2+ entry and glutamate release. Physiologically, the coincidence and interplay between such opposing signalling cascades, by modulating nerve terminal activity, may play a crucial role in presynaptic plasticity. Pharmacologically, given that excitotoxic release of glutamate likely underlies the aetiology of a number of pathophysiological conditions (Meldrum & Garthwaite, 1990), the ability of 5-HT to moderate glutamate release suggests the potential clinical application of 5-HT1A receptors agonists as neuroprotective agents.
Acknowledgments
This work was supported by a Wellcome Trust Travelling Fellowship to S.-J. Wang and grant support from the Wellcome Trust to T.S. Sihra. We thank Dr Jasmina Jovanovic, Dr Andrew Ramage and Ms Jennifer Davies for helpful discussion.
Abbreviations
- AC
Adenylyl cyclase
- 4-AP
4-aminopyridine
- 8-Br-cAMP
8-bromo-cyclic-3′,5′-adenosine monophosphate
- cAMP
cyclic-3′,5′-adenosine monophosphate
- ISO
isoprenaline
- 5-HT
5-hydroxytryptamine
- GPCR
G-protein-coupled receptors (GPCRs)
- 8-OH-DPAT
8-hydroxy-dipropylaminotetralin
- IBMX
3-isobutyl-1-methylxanthine
- NAN-190
1-(2-methoxyphenyl)-4-(4-phthalimidobutyl)piperazine hydrobromide
- PKA
cAMP-dependent protein kinase
- PKC
protein kinase C
- Rp-cAMPS
Rp-cyclic-3′,5′-adenosine-monophosphothioate
- VDCC
voltage-dependent Ca2+ channels
References
- BARRIE A.P., NICHOLLS D.G., SANCHEZ-PRIETO J., SIHRA T.S. An ion channel locus for the protein kinase C potentiation of transmitter glutamate release from guinea pig cerebrocortical synaptosomes. J. Neurochem. 1991;57:1398–1404. doi: 10.1111/j.1471-4159.1991.tb08306.x. [DOI] [PubMed] [Google Scholar]
- BAYLISS D.A., LI Y.W., TALLEY E.M. Effects of serotonin on caudal raphe neurons: inhibition of N- and P/Q-type calcium channels and the afterhyperpolarization. J. Neurophysiol. 1997;77:1362–1374. doi: 10.1152/jn.1997.77.3.1362. [DOI] [PubMed] [Google Scholar]
- BOBKER D.H., WILLIAMS J.T. Serotonin agonists inhibit synaptic potentials in the rat locus ceruleus in vitro via 5-hydroxytryptamine 1A and 5-hydroxytryptamine 1B receptors. J. Pharmacol. Exp. Ther. 1989;250:37–43. [PubMed] [Google Scholar]
- BUDD D.C., NICHOLLS D.G. Protein kinase C-mediated suppression of the presynaptic adenosine A1 receptor by a facilitatory metabotropic glutamate receptor. J. Neurochem. 1995;65:615–621. doi: 10.1046/j.1471-4159.1995.65020615.x. [DOI] [PubMed] [Google Scholar]
- BURLEY J.R., SIHRA T.S. A modulatory role for protein phosphatase 2B (Calcineurin) in the regulation of Ca2+ entry. Eur. J. Neurosci. 2000;12:2881–2891. doi: 10.1046/j.1460-9568.2000.00178.x. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- CHAVEZ-NORIEGA L.E., STEVENS C.F. Modulation of synaptic efficacy in field CA1 of the rat hippocampus by forskolin. Brain Res. 1992;574:85–92. doi: 10.1016/0006-8993(92)90803-h. [DOI] [PubMed] [Google Scholar]
- CHENG L.L., WANG S.J., GEAN P.W. Serotonin depresses excitatory synaptic transmission and depolarization-evoked Ca2+ influx in rat basolateral amygdala via 5-HT1A receptors. Eur. J. Neurosci. 1998;10:2163–2172. doi: 10.1046/j.1460-9568.1998.00229.x. [DOI] [PubMed] [Google Scholar]
- CHIEN A.J., HOSEY M.M. Post-translational modifications of beta subunits of voltage-dependent calcium channels. J. Bioenerg. Biomembr. 1998;30:377–386. doi: 10.1023/a:1021941706726. [DOI] [PubMed] [Google Scholar]
- COFFEY E.T., HERRERO I., SIHRA T.S., SANCHEZ PRIETO J., NICHOLLS D.G. Glutamate exocytosis and MARCKS phosphorylation are enhanced by a metabotropic glutamate receptor coupled to a protein kinase C synergistically activated by diacylglycerol and arachidonic acid [Published erratum appears in J. Neurochem. 1995 Jan; 64, 471] J. Neurochem. 1994;63:1303–1310. doi: 10.1046/j.1471-4159.1994.63041303.x. [DOI] [PubMed] [Google Scholar]
- COFFEY E.T., SIHRA T.S., NICHOLLS D.G. Protein kinase C and the regulation of glutamate exocytosis from cerebrocortical synaptosomes. J. Biol. Chem. 1993;268:21060–21065. [PubMed] [Google Scholar]
- DASCAL N., LOTAN I. Activation of protein kinase C alters voltage dependence of a Na+ channel. Neuron. 1991;6:165–175. doi: 10.1016/0896-6273(91)90131-i. [DOI] [PubMed] [Google Scholar]
- FOEHRING R.C. Serotonin modulates N- and P-type calcium currents in neocortical pyramidal neurons via a membrane-delimited pathway. J. Neurophysiol. 1996;75:648–659. doi: 10.1152/jn.1996.75.2.648. [DOI] [PubMed] [Google Scholar]
- FRACE A.M., HARTZELL H.C. Opposite effects of phosphatase inhibitors on L-type calcium and delayed rectifier currents in frog cardiac myocytes. J. Physiol. Lond. 1993;472:305–326. doi: 10.1113/jphysiol.1993.sp019948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERBER U., GAHWILER B.H. GABAB and adenosine receptors mediate enhancement of the K+ current, IAHP, by reducing adenylyl cyclase activity in rat CA3 hippocampal neurons. J. Neurophysiol. 1994;72:2360–2367. doi: 10.1152/jn.1994.72.5.2360. [DOI] [PubMed] [Google Scholar]
- GEREAU R.W., CONN P.J. Presynaptic enhancement of excitatory synaptic transmission by beta-adrenergic receptor activation. J. Neurophysiol. 1994;72:1438–1442. doi: 10.1152/jn.1994.72.3.1438. [DOI] [PubMed] [Google Scholar]
- GRAY R., JOHNSTON D. Noradrenaline and beta-adrenoceptor agonists increase activity of voltage-dependent calcium channels in hippocampal neurons. Nature. 1987;327:620–622. doi: 10.1038/327620a0. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- HELL J.W., YOKOYAMA C.T., BREEZE L.J., CHAVKIN C., CATTERALL W.A. Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. EMBO J. 1995;14:3036–3044. doi: 10.1002/j.1460-2075.1995.tb07306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERRERO I., MIRAS-PORTUGAL M.T., SANCHEZ-PRIETO J. Positive feedback of glutamate exocytosis by metabotropic presynaptic receptor stimulation [see comments] Nature. 1992;360:163–166. doi: 10.1038/360163a0. [DOI] [PubMed] [Google Scholar]
- HERRERO I., SANCHEZ-PRIETO J. cAMP-dependent facilitation of glutamate release by beta-adrenergic receptors in cerebrocortical nerve terminals. J. Biol. Chem. 1996;271:30554–30560. doi: 10.1074/jbc.271.48.30554. [DOI] [PubMed] [Google Scholar]
- HUANG C.C., HSU K.S., GEAN P.W. Isoproterenol potentiates synaptic transmission primarily by enhancing presynaptic calcium influx via P- and/or Q-type calcium channels in the rat amygdala. J. Neurosci. 1996;16:1026–1033. doi: 10.1523/JNEUROSCI.16-03-01026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG C.C., WANG S.J., GEAN P.W. Selective enhancement of P-type calcium currents by isoproterenol in the rat amygdala. J. Neurosci. 1998;18:2276–2282. doi: 10.1523/JNEUROSCI.18-06-02276.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG Y.Y., KANDEL E.R. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn. Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- LI M., WEST J.W., NUMANN R., MURPHY B.J., SCHEUER T., CATTERALL W.A. Convergent regulation of sodium channels by protein kinase C and cAMP-dependent protein kinase. Science. 1993;261:1439–1442. doi: 10.1126/science.8396273. [DOI] [PubMed] [Google Scholar]
- LIN C.H., HUANG Y.C., TSAI J.J., GEAN P.W. Modulation of voltage-dependent calcium currents by serotonin in acutely isolated rat amygdala neurons. Synapse. 2001;41:351–359. doi: 10.1002/syn.1092. [DOI] [PubMed] [Google Scholar]
- LOPES L.V., CUNHA R.A., RIBEIRO J.A. Cross talk between A(1) and A(2A) adenosine receptors in the hippocampus and cortex of young adult and old rats. J. Neurophysiol. 1999;82:3196–3203. doi: 10.1152/jn.1999.82.6.3196. [DOI] [PubMed] [Google Scholar]
- LU K.T., GEAN P.W. Masking of forskolin-induced long-term potentiation by adenosine accumulation in area CA1 of the rat hippocampus. Neuroscience. 1999;88:69–78. doi: 10.1016/s0306-4522(98)00200-0. [DOI] [PubMed] [Google Scholar]
- MACDERMOTT A.B., ROLE L.W., SIEGELBAUM S.A. Presynaptic ionotropic receptors and the control of transmitter release. Annu. Rev. Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- MAURA G., MARCOLI M., TORTAROLO M., ANDRIOLI G.C., RAITERI M. Glutamate release in human cerebral cortex and its modulation by 5-hydroxytryptamine acting at h 5-HT1D receptors. Br. J. Pharmacol. 1998;123:45–50. doi: 10.1038/sj.bjp.0701581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAURA G., RAITERI M. Serotonin 5-HT1D and 5-HT1A receptors respectively mediate inhibition of glutamate release and inhibition of cyclic GMP production in rat cerebellum in vitro. J. Neurochem. 1996;66:203–209. doi: 10.1046/j.1471-4159.1996.66010203.x. [DOI] [PubMed] [Google Scholar]
- MELDRUM B., GARTHWAITE J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends. Pharmacol. Sci. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- MONS N., COOPER D.M. Adenylate cyclases: critical foci in neuronal signaling. Trends. Neurosci. 1995;18:536–542. doi: 10.1016/0166-2236(95)98375-9. [DOI] [PubMed] [Google Scholar]
- NICHOLLS D.G. The glutamatergic nerve terminal. Eur. J. Biochem. 1993;212:613–631. doi: 10.1111/j.1432-1033.1993.tb17700.x. [DOI] [PubMed] [Google Scholar]
- NICHOLLS D.G., SIHRA T.S. Synaptosomes possess an exocytotic pool of glutamate. Nature. 1986;321:772–773. doi: 10.1038/321772a0. [DOI] [PubMed] [Google Scholar]
- NICHOLS R.A., SIHRA T.S., CZERNIK A.J., NAIRN A.C., GREENGARD P. Calcium/calmodulin-dependent protein kinase II increases glutamate and noradrenaline release from synaptosomes. Nature. 1990;343:647–651. doi: 10.1038/343647a0. [DOI] [PubMed] [Google Scholar]
- PEDARZANI P., STORM J.F. Interaction between alpha- and beta-adrenergic receptor agonists modulating the slow Ca2+-activated K+ current IAHP in hippocampal neurons. Eur. J. Neurosci. 1996;8:2098–2110. doi: 10.1111/j.1460-9568.1996.tb00731.x. [DOI] [PubMed] [Google Scholar]
- PENINGTON N.J., KELLY J.S. Serotonin receptor activation reduces calcium current in an acutely dissociated adult central neuron. Neuron. 1990;4:751–758. doi: 10.1016/0896-6273(90)90201-p. [DOI] [PubMed] [Google Scholar]
- PERKINTON M.S., SIHRA T.S. Presynaptic GABAB receptor modulation of glutamate exocytosis from rat cerebrocortical nerve terminals: receptor decoupling by protein kinase C. J. Neurochem. 1998;70:1513–1522. doi: 10.1046/j.1471-4159.1998.70041513.x. [DOI] [PubMed] [Google Scholar]
- PFRIEGER F.W., GOTTMANN K., LUX H.D. Kinetics of GABAB receptor-mediated inhibition of calcium currents and excitatory synaptic transmission in hippocampal neurons in vitro. Neuron. 1994;12:97–107. doi: 10.1016/0896-6273(94)90155-4. [DOI] [PubMed] [Google Scholar]
- POCKETT S., SLACK J.R., PEACOCK S. Cyclic AMP and long-term potentiation in the CA1 region of rat hippocampus. Neuroscience. 1993;52:229–236. doi: 10.1016/0306-4522(93)90151-5. [DOI] [PubMed] [Google Scholar]
- POCOCK J.M., NICHOLLS D.G. A toxin (Aga-GI) from the venom of the spider agelenopsis aperta inhibits the mammalian presynaptic Ca2+ channel coupled to glutamate exocytosis. Eur. J. Pharmacol. 1992;226:343–350. doi: 10.1016/0922-4106(92)90052-w. [DOI] [PubMed] [Google Scholar]
- RAITERI M., MAURA G., BONANNO G., PITTALUGA A. Differential pharmacology and function of two 5-HT1 receptors modulating transmitter release in rat cerebellum. J. Pharmacol. Exp. Ther. 1986;237:644–648. [PubMed] [Google Scholar]
- RHEE J.S., ISHIBASHI H., AKAIKE N. Serotonin modulates high-voltage-activated Ca2+ channels in rat ventromedial hypothalamic neurons. Neuropharmacology. 1996;35:1093–1100. doi: 10.1016/s0028-3908(96)00052-4. [DOI] [PubMed] [Google Scholar]
- ROEPER J., PONGS O. Presynaptic potassium channels. Curr. Opin. Neurobiol. 1996;6:338–341. doi: 10.1016/s0959-4388(96)80117-6. [DOI] [PubMed] [Google Scholar]
- SANCHEZ-PRIETO J., BUDD D.C., HERRERO I., VAZQUEZ E., NICHOLLS D.G. Presynaptic receptors and the control of glutamate exocytosis. Trends. Neurosci. 1996;19:235–239. doi: 10.1016/0166-2236(96)10031-x. [DOI] [PubMed] [Google Scholar]
- SIHRA T.S.Protein phosphorylation and dephosphorylation in isolated nerve terminals (synaptosomes) Posttranslational modifications: techniques and protocols 1997Totowa: Humana Press; 67–119.Hemmings, H.C., ed. pp [Google Scholar]
- SIHRA T.S., BOGONEZ E., NICHOLLS D.G. Localized Ca2+ entry preferentially effects protein dephosphorylation, phosphorylation, and glutamate release. J. Biol. Chem. 1992;267:1983–1989. [PubMed] [Google Scholar]
- SIHRA T.S., NAIRN A.C., KLOPPENBURG P., LIN Z., POUZAT C. A role for calcineurin (Protein phosphatase-2B) in the regulation of glutamate release. Biochem. Biophys. Res. Commun. 1995;212:609–616. doi: 10.1006/bbrc.1995.2013. [DOI] [PubMed] [Google Scholar]
- SIHRA T.S., NICHOLS R.A. Mechanisms in the regulation of neurotransmitter release from brain nerve terminals: current hypotheses. Neurochem. Res. 1993;18:47–58. doi: 10.1007/BF00966922. [DOI] [PubMed] [Google Scholar]
- SIHRA T.S., PIOMELLI D., NICHOLS R.A. Barium evokes glutamate release from rat brain synaptosomes by membrane depolarization: involvement of K+, Na+, and Ca2+ channels. J. Neurochem. 1993;61:1220–1230. doi: 10.1111/j.1471-4159.1993.tb13612.x. [DOI] [PubMed] [Google Scholar]
- SINGER J.H., BELLINGHAM M.C., BERGER A.J. Presynaptic inhibition of glutamatergic synaptic transmission to rat motoneurons by serotonin. J. Neurophysiol. 1996;76:799–807. doi: 10.1152/jn.1996.76.2.799. [DOI] [PubMed] [Google Scholar]
- SRKALOVIC G., SELIM M., REA M.A., GLASS J.D. Serotonergic inhibition of extracellular glutamate in the suprachiasmatic nuclear region assessed using in vivo brain microdialysis. Brain Res. 1994;656:302–308. doi: 10.1016/0006-8993(94)91474-5. [DOI] [PubMed] [Google Scholar]
- STORM D.R., HANSEL C., HACKER B., PARENT A., LINDEN D.J. Impaired cerebellar long-term potentiation in type I adenylyl cyclase mutant mice. Neuron. 1998;20:1199–1210. doi: 10.1016/s0896-6273(00)80500-0. [DOI] [PubMed] [Google Scholar]
- TANAKA E., NORTH R.A. Actions of 5-hydroxytryptamine on neurons of the rat cingulate cortex. J. Neurophysiol. 1993;69:1749–1757. doi: 10.1152/jn.1993.69.5.1749. [DOI] [PubMed] [Google Scholar]
- THOMPSON S.M., CAPOGNA M., SCANZIANI M. Presynaptic inhibition in the hippocampus. Trends Neurosci. 1993;16:222–227. doi: 10.1016/0166-2236(93)90160-n. [DOI] [PubMed] [Google Scholar]
- TRAVAGLI R.A., WILLIAMS J.T.Endogenous monoamines inhibit glutamate transmission in the spinal trigeminal nucleus of the guinea-pig J. Physiol. 1996491177–185.Pt 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRUDEAU L.E., EMERY D.G., HAYDON P.G. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- TURNER T.J., ADAMS M.E., DUNLAP K. Multiple Ca2+ channel types coexist to regulate synaptosomal neurotransmitter release. Proc. Natl. Acad. Sci. U.S.A. 1993;90:9518–9522. doi: 10.1073/pnas.90.20.9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAZQUEZ E., BUDD D.C., HERRERO I., NICHOLLS D.G., SANCHEZ-PRIETO J. Co-existence and interaction between facilitatory and inhibitory metabotropic glutamate receptors and the inhibitory adenosine A1 receptor in cerebrocortical nerve terminals. Neuropharmacology. 1995;34:919–927. doi: 10.1016/0028-3908(95)00067-g. [DOI] [PubMed] [Google Scholar]
- WANG S.J., CHENG L.L., GEAN P.W. Cross-modulation of synaptic plasticity by beta-adrenergic and 5-HT1A receptors in the rat basolateral amygdala. J. Neurosci. 1999;19:570–577. doi: 10.1523/JNEUROSCI.19-02-00570.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEISSKOPF M.G., CASTILLO P.E., ZALUTSKY R.A., NICOLL R.A. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- WU L.G., SAGGAU P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994;12:1139–1148. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- WU L.G., SAGGAU P.GABAB receptor-mediated presynaptic inhibition in guinea-pig hippocampus is caused by reduction of presynaptic Ca2+ influx J. Physiol. (Lond.) 1995485649–657.Pt 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU L.G., SAGGAU P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- XIA Z., STORM D.R. Calmodulin-regulated adenylyl cyclases and neuromodulation. Curr. Opin. Neurobiol. 1997;7:391–396. doi: 10.1016/s0959-4388(97)80068-2. [DOI] [PubMed] [Google Scholar]