

Abstract
Clostridium perfringens beta-toxin causes dermonecrosis and oedema in the dorsal skin of animals. In the present study, we investigated the mechanisms of oedema induced by the toxin.
The toxin induced plasma extravasation in the dorsal skin of Balb/c mice.
The extravasation was significantly inhibited by diphenhydramine, a histamine 1 receptor antagonist. However, the toxin did not cause the release of histamine from mouse mastocytoma cells.
Tachykinin NK1 receptor antagonists, [D-Pro2, D-Trp7,9]-SP, [D-Pro4, D-Trp7,9]-SP and spantide, inhibited the toxin-induced leakage in a dose-dependent manner. Furthermore, the non-peptide tachykinin NK1 receptor antagonist, SR140333, markedly inhibited the toxin-induced leakage.
The leakage induced by the toxin was markedly reduced in capsaicin-pretreated mouse skin but the leakage was not affected by systemic pretreatment with a calcitonin gene-related peptide receptor antagonist (CGRP8-37).
The toxin-induced leakage was significantly inhibited by the N-type Ca2+ channel blocker, ω-conotoxin MVIIA, and the bradykinin B2 receptor antagonist, HOE140 (D-Arg-[Hyp3, Thi5, D-Tic7, Oic8]-bradykinin), but was not affected by the selective L-type Ca2+ channel blocker, verapamil, the P-type Ca2+ channel blocker, ω-agatoxin IVA, tetrodotoxin (TTX), the TTX-resistant Na+ channel blocker, carbamazepine, or the sensory nerve conduction blocker, lignocaine.
These results suggest that plasma extravasation induced by beta-toxin in mouse skin is mediated via a mechanism involving tachykinin NK1 receptors.
Keywords: Clostridium perfringens, beta-toxin, plasma extravasation, tachykinin NK1 receptor, sensory nerve
Introduction
Clostridium perfringens type C strains cause haemorrhagic noxious ulceration or superficial mucousal necrosis of the small intestine in humans, pigs, cattle and chickens (McDonel, 1986; Sakurai, 1995; Songer, 1996; Sakurai et al., 1997). Acute and sudden deaths frequently occur in these animals (El-Idrissi et al., 1992). Prior to death, signs of neurological involvements such as tetany and opisthotonus (severe convulsion) may also occur (Songer, 1996; Sakurai et al., 1997). Administration of the beta-toxin toxoid which was detoxified with formalin but possessed immunogenicity to Papua New Guinea tribespeople resulted in a marked reduction in the incidence of necrotic enteritis (Lawrence et al., 1979). Furthermore, a beta-toxin toxoid administered to infant pigs during an outbreak of necrotizing enterocolitis reduced mortality by about 30% (Kennedy et al., 1977). Therefore beta-toxin is thought to be an important agent in the necrotic enteritis caused by type C strains. We have also reported that the toxin acts on the autonomic nervous system and produces arterial constriction (Sakurai et al., 1981, 1984). We have now extensively purified the beta-toxin produced by the type C strains and elucidated some of the physico-chemical properties of the toxin (Sakurai & Duncan, 1977, 1978; Sakurai & Fujii, 1987). We have also found that the toxin is inhibited by sulphydryl group reagents and oxidizing agents, that the toxin treated with p-chloromercuribenzoate and oxidizing agents is reactivated by reduced glutathione and reductants, respectively, and that the number of thiol groups is 1 mol−1 of beta-toxin from C. perfringens (Sakurai et al., 1980, 1992). More recently, the beta-toxin gene from C. perfringens was cloned and sequenced, with the suggestion that beta-toxin is a pore-forming toxin on the basis of weak similarities between the primary structure of beta-toxin and alpha- and gamma-haemolysin and the leukocidin from Staphylococcus aureus (Hunter et al., 1993). The deduced amino acid sequence showed that the toxin contains only one cysteine residue at position 265. However, we reported that replacement of Cys-265 had no effect on the activity of the toxin. In addition, the replacement of Tyr-266, Leu-268 and Trp-275 near the cysteine residue resulted in complete loss of the activity, suggesting that the site essential for the activity is close to the cysteine residue (Nagahama et al., 1999). The primary amino acid sequence surrounding Cys-265 in beta-toxin (positions 255–276) shows homology to that at positions 245–267 in the C-terminus of Staphylococcus aureus alpha-toxin (a conserved 11-amino acid sequence) (Walker & Bayley, 1995). It appears that Cys-265 in the beta-toxin corresponds to Asp-255 in the alpha-toxin. Walker & Bayley (1995) reported that treatment of D254C and D255C (variant toxins of the alpha-toxin) with sulphydryl reagent, 4′-acetamido-4-((iodoacetyl)amino)stilbene-2,2′-disulphonate, resulted in a significant reduction or complete loss of binding, oligomer formation and haemolytic activity, suggesting that the C-terminus of the alpha-toxin is implicated in binding to cells. It is possible that the region surrounding Cys-265 in beta-toxin is required for binding to the receptor of beta-toxin or formation of oligomerization. Steinthorsdottir et al. (2000) showed that beta-toxin formed oligomeric complexes on the membranes of human umbilical vein endothelial cells and induced the release of arachidonic acid and inositol from these cells. Shatursky et al. (2000) hypothesized that the lethal action of beta-toxin is based on the formation of cation-selective pores in susceptible cells. However, little is known about the precise mechanism of action of beta-toxin in vivo.
Previous studies have shown that beta-toxin produces a characteristic purplish dermonecrosis when intradermally injected in guinea-pig skin. To understand the action of beta-toxin in vivo, the effect of beta-toxin on mouse dorsal skin was investigated. The results presented show that beta-toxin activates a mechanism involving tachykinin NK1 receptors and induces plasma extravasation.
Methods
Animals and materials
Male Balb/c mice weighing approximately 30 g were obtained from Nippon SLC (Hamamatsu, Japan). The animals were housed in plastic cages under controlled environmental conditions (temperature 22±2°C, humidity 55±5%). Food and water were freely available. All experiments were approved by the Institute Animal Care and Use Committee, Tokushima Bunri University. Diphenhydramine hydrochloride, CGRP8-37, capsaicin (8-methyl N-vanillyl-6-nonenamide), carbamazepine, compound 48/80, histamine hydrochloride, tetrodotoxin, verapamil, ω-conotoxin MVIIA, capsazepine, Evans blue, Substance P (SP), septide ([pGlu6,Pro9]-SP(6-11) and bovine serum albumin (BSA) were purchased from Sigma (St. Louis, MO, U.S.A.). Spantide ([D-Asp1,DTrp7,9,Lue11]-SP), [D-Pro2,D-Trp7,9]-SP, [D-Pro4,D-Trp7,9]-SP, HOE140 (D-Arg-[Hyp3, Thi5, DTic7, Oic8]-bradykinin), ω-agatoxin IVA and bradykinin were purchased from Peptide Ins. Inc. (Osaka, Japan). Diethyl ether, Tween 80, N, N′-dimethyl formamide and o-phthalaldehyde were obtained from Nacalai tesque (Kyoto, Japan). SR140333 was kindly provided by Sanofi (Toulouse, France). [125I]-Bolton-Hunter reagent and Sephadex G-75 were obtained from Amersham Pharmacia Biotech (Tokyo, Japan). A mastocytoma cell line (P-815) was obtained from Riken Cell Bank (Tsukuba, Japan). RPMI-1640 medium was purchased from GIBCO BRL (New York, NY, U.S.A.). All other chemicals were of the highest grade available from commercial sources.
125I-labelled BSA was prepared by incubating 50 μg of the protein in 50 μl of 0.1 M Borate buffer (pH 8.5) with 250 μCi of Bolton–Hunter reagent for 1 h at 4°C as described previously (Nagahama & Sakurai, 1991). To remove free reagent from the mixture, the solution was filtrated through a Sephadex G-75 column (1×30 cm), equilibrated with 50 mM phosphate buffer (pH 7.5) containing 0.9% NaCl.
Beta-toxin
The expression and purification of recombinant beta-toxin was performed as described previously (Nagahama et al., 1999).
Measurements of plasma extravasation
Mice were anaesthetized with sodium pentobarbitone (Sagatal, 50 mg kg−1, i.p.). The dorsal skin of the mice was shaved and prepared for intradermal (i.d.) injection (up to four sites per mouse, each in a randomly allocated balanced site pattern). A mixture of 125I-BSA and Evans blue dye (0.1 ml of 2.5% solution) was injected in the tail vein. After 5 min, beta-toxin (5–100 ng) was injected i.d. (50 μl site−1). Various agents were given as pretreatments (i.d. or i.v. 5 min before i.d. injection of the toxin) when required. After 1 h, a blood sample (0.1 ml) was taken from the tail vein. The mouse was killed by cervical dissociation and 10 mm-diameter skin pieces were punched out. Plasma samples and the skin pieces were placed in a gamma-counter (Aloka Basic Scaler, Aloka Co., Ltd., Tokyo, Japan). Plasma extravasation at each site was expressed as microliters of plasma by dividing skin sample 125I counts by 125I counts in 1 μl of plasma (Williams, 1979). Then, the skin samples were placed in 1 ml of N, N′-dimethyl formamide. The extravasated dye was extracted at 55°C for 12 h. The Evans blue content of the samples was determined with a 96-well microplate reader (Spectramax 340 PC, Molecular Divices, Sunnyvale, CA, U.S.A.) at 620 nm (100 μl sample−1 well−1). Extravasation of Evans blue was expressed as μg Evans blue/skin site, by comparing the experimental values with a known standard.
Capsaicin desensitization protocol
A 10% (w v−1) solution of capsaicin was prepared in absolute ethanol (capsaicin stock solution). The solution was further diluted 1 : 1 : 8 (capsaicin stock solution : Tween 80 : 0.15 M NaCl) (Alber et al., 1989). After the dorsal skin of the mice was shaved, the capsaicin solution was painted three times on the first day of pretreatment and two times per day during the following 4 days. As a control, the diluent (ethanol and Tween 80) alone was painted on the dorsal skin of mice.
Histamine release from mastocytoma P-815 cells
Mastocytoma P-815 cells (2–4×105 cells ml−1 in 500 ml flasks) were maintained in suspension culture with RPMI-1640 medium containing 10% (v v−1) foetal bovine serum under 5% (v v−1) CO2 air at 37°C. P-815 cells (2–4×107 cells) were harvested by centrifuging 50 ml of the suspension and washing the cells three times with phosphate-buffered saline (PBS). Mastocytoma cells (4–6×106 cells) in 1 ml of PBS were incubated for 20 min after treatment with compound 48/80, beta-toxin or substance P (Teshigawara & Moriya, 1994). The release of histamine was measured by the o-phthalaldehyde spectrometric procedure as described previously (Sakurai & Fujii, 1987).
Histopathological evaluation
Eight male mice, weighing 25–30 g, were used. Beta-toxin (30 ng) or saline (each 50 μl site−1) was injected i.d. in the shaved dorsal skin of mice. These animals were killed 12 h after injection of the toxin. For light microscopy, the skin tissues were fixed in 10% neutralized buffered formalin solution (pH 7.4). After fixation, the tissues were dehydrated through graded ethanol, and paraffin sections were prepared by the routine method. These tissue sections were stained with haematoxylin and eosin for light microscopy.
Statistical analysis
All values were expressed as the mean±s.e.mean. Student's umpaired t-test and one-way analysis of variance (ANOVA) were used for statistical analysis. P-values less than 0.05 were considered to be statistically significant.
Results
Oedema evoked by intradermal injection of beta-toxin in mouse skin
We investigated if the toxin induces plasma extravasation in mouse dorsal skin. As shown in Figure 1A, the toxin (5–100 ng site−1) dose-dependently caused plasma extravasation and blueing lesions (6–14 mm in diameter) (data not shown). Less than 1 ng site−1 of the toxin caused no plasma extravasation (data not shown). The time-course for plasma extravasation caused by beta-toxin was investigated (Figure 1B). Plasma extravasation was formed rapidly within 15 min, and reached a maximum within 120 min. Next, we investigated the histopathological lesions caused by intradermal injection of the toxin (50 ng site−1) in mouse skin (Figure 2). Necrosis, congestion and oedema were observed in subcutaneous tissue and in perimysium internum 12 h after the injection. These results suggested that beta-toxin possessed two effects, indicating that the toxin induced early oedema formation and late necrosis in skin. The toxin-induced extravasation was reduced by co-injection with diphenhydramine, a histamine 1 receptor antagonist (Simons et al., 2001; Ishida et al., 2000) (0.1 μg site−1, P<0.01; 0.5 μg site−1, P<0.001) in a dose-dependent manner, but was not completely diminished (Figure 3). The plasma extravasation induced by histamine was significantly reduced by co-injection of diphenhydramine (Figure 3). It therefore is likely that the toxin-induced plasma leakage is entirely related to histamine release. To analyse the effect of the toxin on mast cells, mouse mastocytoma P-815 cells (5×108) were treated with the toxin (300 μg ml−1) or compound 48/80 (50 μg ml−1) for 30 min, and the histamine in the supernatant was measured. The percentage of histamine release in the cells was as follows: PBS (vehicle), 4.5±1.8%; beta-toxin, 5.1±2.2%; compound 48/80, 72.5±6.8% * (mean±s.e.mean, n=5; *P<0.01, compared with vehicle). The result indicated that beta-toxin cannot induce the release of histamine from the cells. Our previous report also showed that the toxin does not induce the release of histamine from rat mast cells (Sakurai & Fujii, 1987). It therefore appears that the toxin does not directly act on mast cells.
Figure 1.
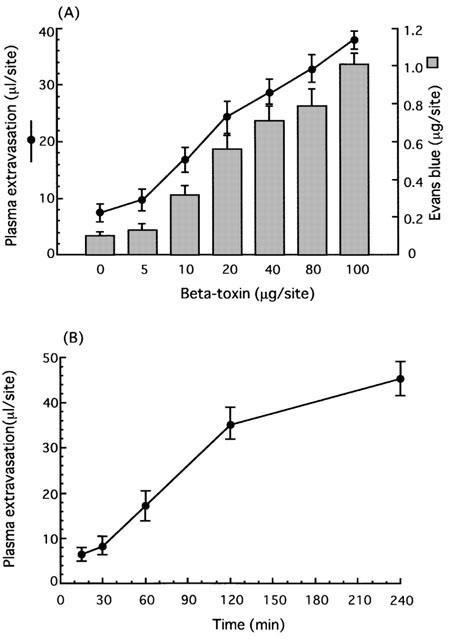
Local plasma extravasation induced by beta-toxin in mouse dorsal skin. (A) Dose-dependence of beta-toxin-induced plasma extravasation. A mixture of 125I-BSA and Evans blue dye (0.1 ml of 2.5% solution) was injected into the tail vein. After 5 min, the beta-toxin (5–100 ng) was injected i.d. (50 μl site−1). Plasma extravasation was measured 60 min after the injection of beta-toxin. (B) Time-course of beta-toxin-induced plasma extravasation. A mixture of 125I-BSA and Evans blue dye (0.1 ml of 2.5% solution) was injected in the tail vein. After 5 min, the beta-toxin (20 ng site−1) was injected i.d. (50 μl site−1). Plasma extravasation was measured various time after the injection of beta-toxin. Values are the mean±s.e.mean, n=6.
Figure 2.
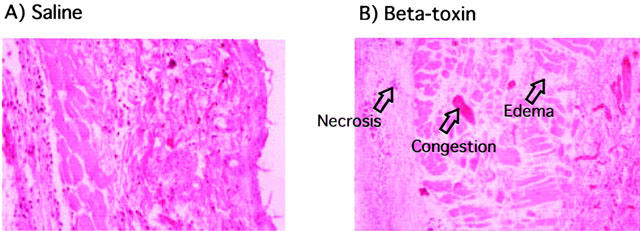
Effect of beta-toxin on mouse dermal tissue. Saline (A) or beta-toxin (50 ng site−1) (B) was injected i.d. into the dorsal skin of mice. After 12 h, dermal tissues from the dorsal skin were fixed in formalin and sections were stained with haematoxylin and eosin.
Figure 3.
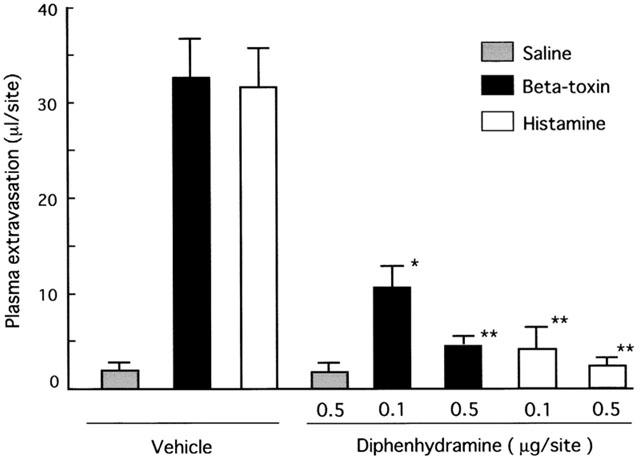
Effect of diphenhydramine on plasma extravasation induced by beta-toxin or histamine in dorsal skin of mice. A mixture of 125I-BSA and Evans blue dye (0.1 ml of 2.5% solution) was injected into the tail vein. After 5 min, beta-toxin (50 ng site−1) or histamine (5 μg site−1) and diphenhydramine (0.1 or 0.5 μg site−1) were simultaneously injected i.d. into the dorsal skin of mice. Plasma extravasation was measured 60 min after the injection of beta-toxin. Values are the mean±s.e.mean, n=6. *P<0.01, compared with control,**P<0.001, compared with saline.
The effect of tachykinin receptor antagonist and capsaicin on the toxin-induced plasma extravasation
To test if the toxin-induced plasma extravasation is related to tachykinins, the effect of tachykinin NK1 antagonist, [D-Pro2, D-Trp7,9]-SP, [D-Pro4, D-Trp7,9]-SP and spantide on the toxin-induced plasma leakage was investigated. Figure 4 shows that co-injection of these NK1 antagonists resulted in a reduction in the toxin-induced leakage in a dose-dependent manner (5.0–10 μg site−1). Intradermal injection of a selective NK1 receptor agonist, septide (1.0 nmol site−1), induced plasma extravasation in a dose-related manner. The extravasation induced by septide was significantly reduced by co-injection of NK1 antagonists (Figure 4). [D-Pro4, D-Trp7,9]-SP, an NK1 antagonist, exhibited the same potency in inhibiting the toxin- or septide-induced plasma leakage (data not shown). Next, the effect of the non-peptide long-lasting tachykinin neurokinin-1 antagonist, SR140333, on the toxin-induced plasma extravasation was investigated. Co-injection of SR140333 (0.1–1.0 nmol site−1, or 250–500 nmole kg−1 i.v. 5 min before) dose-dependently inhibited the extravasation, as shown in Figure 5. The plasma extravasation induced by septide (1 nmole site−1 33±5.2 μl site−1) was significantly (P<0.01) inhibited by co-injection of diphenhydramine (0.5 μg site−1; 4.8±1.5 μl site−1) or SR140333 (1.0 nmol site−1; 3.1±1.8 μl site−1), but the histamine-induced plasma extravasation (5 μg site−1; 32±4.5 μl site−1) was not blocked by SR140333 (1.0 nmole site−1; 33±5.2 μl site−1). Septide (5 μM) induced the release of about 70% of histamine from P-815 cells. On the other hand, systemic treatment with 400 nmol kg−1 of CGRP8-37 (calcitonin gene-related peptide receptor antagonist) had no effect on the toxin-induced extravasation (Table 1). It has been reported that the treatment of sensory nerve fibres with capsaicin leads to the release of neuropeptides (e.g. tachykinins such as SP and calcitonin gene-related peptide) and to the depletion of neuropeptides in sensory nerves. To investigate the role of endogenous SP release from sensory nerve fibres on beta-toxin-induced plasma extravasation, the effect of a topical administration of capsaicin on the toxin-induced extravasation was tested (Gamse et al., 1980; Alber et al., 1989; Costa et al., 1997). Topical administration of 5% capsaicin onto the dorsal back skin of mice markedly inhibited the toxin-induced leakage (40–100 ng site−1), as shown in Figure 6A. To exclude that the reactivity of cutaneous mast cells, histamine receptor and NK1 receptor might be impaired after capsaicin pretreatment, we compared the effect of compound 48/80 in a capsaicin-pretreated skin site versus a control skin site. As illustrated (Figure 6B), compound 48/80 retained its effect after capsaicin treatment. Similar results were obtained on administration of histamine or septide into capsaicin-treated skin and untreated skin. This evidence demonstrates that capsaicin pretreatment does not affect mast cell function in mouse skin. There is also evidence that capsaicin pretreatment does not reduce vascular reactivity, as demonstrated by the challenge with histamine and septide.
Figure 4.
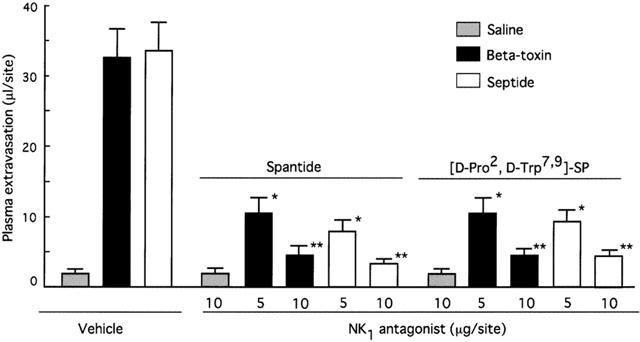
Effect of tachykinin NK1 receptor antagonists on plasma extravasation induced by beta-toxin in dorsal skin of mice. A mixture of 125I-BSA and Evans blue dye (0.1 ml of 2.5% solution) was injected into the tail vein. After 5 min, pretreatments with various amounts of spantide and [D-Pro2, D-Trp7, 9]SP were performed 1 min before beta-toxin (50 ng site−1) or septide (1 nmol site−1) challenge. Plasma extravasation was measured 60 min after the injection of beta-toxin. Values are the mean±s.e.mean, n=6. *P<0.05, compared with control, **P<0.01, compared with control.
Figure 5.
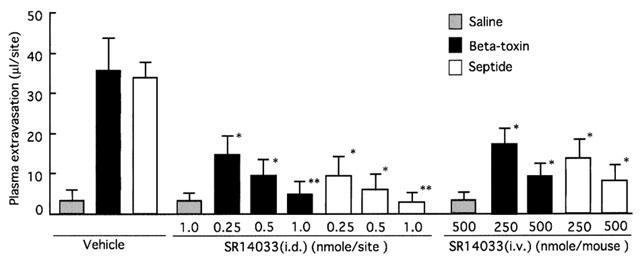
Effect of SR140333 treatment on plasma extravasation induced by beta-toxin in dorsal skin of mice. A mixture of 125I-BSA and Evans blue dye (0.1 ml of 2.5% solution) was injected into the tail vein. Various doses of SR140333 were given as pretreatments i.d. or i.v. 5 min before i.d. injection of toxin. Beta-toxin (50 ng site−1) and septide (1 nmole site−1) were injected i.d.. Plasma extravasation was measured 60 min after the injection of beta-toxin. Values are the mean±s.e.mean, n=6. *P<0.05, compared with vehicle, **P<0.01, compared with saline.
Table 1.
Effect of multiple drug treatments on plasma extravasation induced by beta-toxin in mouse dorsal skin

Figure 6.
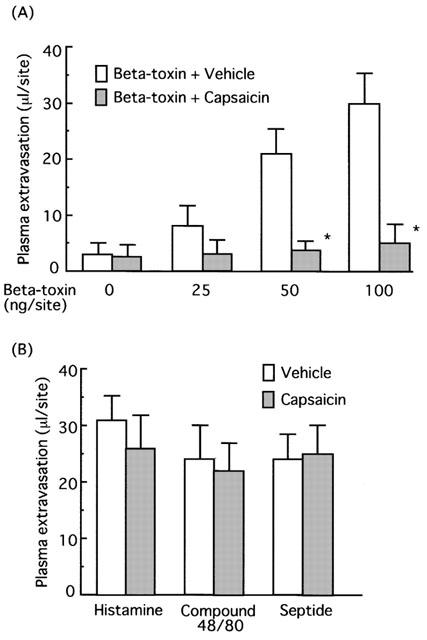
Effect of capsaicin on plasma extravasation induced by beta-toxin in dorsal skin of mice. After the dorsal skin was shaved, capsaicin solution (20 μg ml−1 in 10% ethanol solution containing 10% Tween 80) was painted twice a day for 4 days. As a control, the diluent alone was applied to the skin. A mixture of 125I-BSA and Evans blue dye (0.1 ml of 2.5% solution) was injected into the tail vein. Afer 5 min, beta-toxin (50 μg site−1) (A) and histamine (5 μg site−1), compound 48/80 (20 μg site−1) or septide (1 nmole site−1) (B) were injected intradermally into the skin. Plasma extravasation was measured 60 min after the injection of beta-toxin or agents. Values are the mean±s.e.mean, n=6. *P<0.01, compared with vehicle.
The effect of various agents that affect neurogenic inflammation on the toxin-induced extravasation
To investigate the toxin-induced plasma extravasation on skin afferent nerves, we tested various drugs that act on sensory nerves. The effects of L-, N- and P-type Ca2+ currents in the toxin-induced plasma extravasation were evaluated by treatment with various Ca2+ channel blockers (Table 1). Co-injection of the N-type Ca2+ channel blocker, ω-conotoxin MVIIA (Maggi et al., 1988) (3.2 μg kg−1, i.v. 5 min before), drastically reduced the toxin-induced plasma extravasation (P<0.01). On the other hand, neither systemic treatment with the selective L-type Ca2+ channel blocker, verapamil (Fox et al., 1987; Costa et al., 2000) (60 μg kg−1, i.v. 5 min before) nor co-injection of the P-type Ca2+ channel blocker, ω-agatoxin IVA (Baccei & Kocsis, 2000) (100 pmol site−1) produced a significantly different result from the control group. However, no significant change was noticed in the basal mean arterial blood pressure after treatment with verapamil (60 μg kg−1, i.v.) and ω-conotoxin MVIIA (3.2 μg kg−1, i.v.) (data not shown). Next, we tested different classes of blockers that act either via presynaptic receptors or via mechanisms located in sensory nerves, or postsynaptic receptors (calcitonin gene-related peptide receptor, or vanilloid receptor). The plasma extravasation induced by the toxin was significantly inhibited by HOE140 reported as a bradykinin B2 receptor antagonist by Palframan et al. (1996) and Cao et al. (2000). SR140333 (0.5 nmol site−1) significantly reduced oedema formation induced by bradykinin (1 nmol site−1) (data not shown). However, systemic treatment with capsazepine (120 μmol kg−1, i.v.) reported as a vanilloid receptor blocker by Perkins & Campbell (1992), TTX reported as a Na+ channel blocker by Akopian et al. (1996), carbamazepine known as a TTX-resistant Na+ channel blocker (Arbuckle & Docherty, 1995; Akopian et al., 1996) and lignocaine known as a sensory nerve conduction blocker (Escott et al., 1995), did not significantly inhibit the toxin-induced leakage, as shown in Table 1.
Discussion
C. perfringens beta-toxin injected in animal skin is known to cause a characteristic purplish dermonecrosis. In this study, histopathological analysis revealed that the toxin induced oedema formation and necrosis when injected in the mouse dorsal skin as shown in Figure 2. The data presented here are the first to be published showing that the toxin-induced plasma extravasation involves a tachykinin NK1 receptor-mediated mechanism.
After injection of beta-toxin into mouse, the mainly clinical manifestation is nervous signs including tetany and opisthotonus. We reported that the toxin acts on the autonomic nervous system and produces arterial constriction (Sakurai et al., 1981, 1984). On the basis of these results, we proposed that the toxin-induced oedema is dependent on action of the toxin on peripheral nerve systems in skin. When beta-toxin was injected i.d. in mouse skin, plasma extravasation was initially formed within 120 min and dermonecrosis was observed over 6 h, suggesting that the toxin-induced plasma extravasation results in reduction or block in support of nutrients and oxygen in the skin tissue and consequently, the toxin is destroyed to develop to dermonecrosis. However, the relationship between oedema formation and dermonecrosis is not clear.
Co-injection of the histamine H1 receptor antagonist, diphenhydramine (0.1 μg site−1), markedly inhibited the toxin-induced plasma extravasation, suggesting that the activity of the toxin is closely related to the release of histamine from skin mast cells. However, the toxin did not induce the release of histamine from rat mast cells (Sakurai & Fujii, 1987) and P-815 cells. It therefore is likely that the toxin indirectly acts on mast cells and induces the release of histamine from the cells.
Emonds-Alt et al. (1993) reported that SR140333 acts as a potent tachykinin NK1 receptor antagonist in vitro and in vivo in several species. Furthermore, Palframan et al. (1996) described the selectivity of SR140333 at the NK1 receptor, when injected intradermally in rat skin. In addition, it has been reported that capsaicin stimulates sensory nerve fibres to result in the release of neuropeptides such as tachykinins, showing that capsaicin pretreatment abolished neuropeptides in sensory nerve fibres (Gamse et al., 1980; Alber et al., 1989; Costa et al., 1997). To investigate the toxin-induced extravasation, we tested different classes of drugs that act on sensory nerves, presynaptic receptors or postsynaptic receptors. The results from the use of these blockers were as follows: (1) the toxin-induced plasma extravasation was inhibited by the SP antagonists, spantide, [D-Pro2, D-Trp7,9]-SP and [D-Pro4, D-Trp7,9]-SP; (2) the extravasation was markedly inhibited by the non peptide-selective tachykinin NK1 receptor antagonist, SR140333; (3) Holzer (1998) assumed that the release of multiple peptides from sensory nerves provides the potential for synergistic interactions. The toxin-induced extravasation was not blocked by co-injection of CGRP8–37. Therefore the vasodilator action of CGRP may potentiate the plasma extravasation induced by tachykinin NK1 receptor agonists in the skin, presumably by increasing blood flow at the site of leakage, as reported by Brain & Williams (1985, 1989). In this study, a lack of effect of the CGRP receptor antagonist, CGRP8-37, on the toxin-induced plasma extravasation was shown. Treatment with CGRP8-37 has been shown to inhibit cutaneous vasodilation induced by CGRP released from stimulated sensory nerves (Escott & Brain, 1993; Siney & Brain, 1996). Furthermore, this antagonist is the only one available and the peptide ligand is likely to be limited in its application. The result suggests that CGRP does not play a role in the response induced by beta-toxin; (4) Topical administration of capsaicin was used to deplete endogenous SP from sensory nerves and resulted in a complete loss of the toxin activity. On the other hand, the similar extent of plasma extravasation induced by histamine, compound 48/80 or the tachykinin NK1 agonist septide in both capsaicin- and vehicle-pretreated mice indicated that the tachykinin NK1 receptor, histamine H1 receptor, and cutaneous mast cell remain functional after capsaicin pretreatment. Alber et al. (1989) reported that the capsaicin pre-treatment induced the depletion of neurotransmitters in sensory nerve and Jancso et al. (1977) reported that the capsaicin pretreatment caused destruction of a large number of peripheral fibres in rats. Therefore, the observation suggests that sensory nerve-mediated mechanisms are involved in plasma extravasation induced by the toxin. Next, a vanilloid receptor antagonist, capsazepine, did not inhibit the toxin-induced plasma extravasation. It is assumed that capsazepine-insensitive vanilloid receptors exist on rat trigeminal ganglion neurons (Liu et al., 1998). Moreover, the results with capsazepine should be interpreted with caution since positive effects are not necessarily mediated by vanilloid receptors nor do negative data rule out the involvement of these receptors (Szallasi & Blumberg, 1999). Although capsazepine has been shown to have nonspecific actions at high concentrations (Santicioli et al., 1993), it is likely that capsazepine-sensitive vanilloid receptors do not precipitate the toxin-induced plasma extravasation on mouse skin; (5) plasma protein extravasation was reproduced by intradermal injection of the tachykinin NK1 receptor agonist, septide. These observations suggest that the action of the toxin is mainly dependent on the release of tachykinins such as SP via the tachykinin NK1 receptor. Furthermore, septide-induced plasma extravasation was inhibited by SR140333, but histamine-induced extravasation was not, suggesting that the toxin-elicited release of tachykinins such as SP occurs upstream of the histamine release. Thus these observations suggest that SP released from sensory nerves stimulates mast cells to release histamine.
Bradykinin is reported to produce oedema by increasing permeability in the microcirculation via the bradykinin B2 receptor. In addition, bradykinin can release SP from capsaicin-sensitive sensory neurons. The plasma extravasation induced by the toxin was significantly inhibited by the bradykinin B2 receptor antagonist, HOE140, suggesting that the toxin acts on a bradykinin B2 prejunctional receptor. However, we cannot exclude the possibilty that beta-toxin causes the release of endogeneous bradykinin. The observation indicates that the toxin stimulates sensory nerve fibres that contain tachykinins such as SP.
We investigated a range of agents that are known to influence the passage of ions into nerves. Voltage-sensitive Ca2+ channels such as L, P and Q types have been identified in a number of peripheral nerves in several species and are involved in the release of sensory neuropeptides such as tachykinin and CGRP (Fox et al., 1987; Maggi, 1991; Maggi et al., 1988; Costa et al., 2000). Animal toxins such as ω-agatoxins and ω-conotoxins have been reported to act on different types of Ca2+ channels (Maggi et al., 1988; Baccei & Kocsis, 2000). We investigated the effect of blockers, that are known to influence the passage of ions into nerves, on the toxin-induced plasma extravasation. The results showed that the toxin-induced plasma extravasation was markedly reduced by an N-type channel blocker (ω-conotoxin MVIIA), but did not by P (ω-agatoxin IVA)- and L (verapamil)-type channel blockers. Therefore the results appear to demonstrate an involvement of the N-type Ca2+ channel in the excitatory action of beta-toxin on sensory neurons.
Some substances such as Na+ channel blockers and local anesthetics have been known to inhibit the release of sensory neuropeptides by acting on other systems including sensory nerve conduction (Arbuckle & Docherty, 1995; Akopian et al., 1996; Costa et al., 2000). Local treatment with lignocaine resulted in no effect on the toxin-induced plasma extravasation. Furthermore, systemic treatment with tetrodotoxin (i.d.) and a tetrodotoxin-resistant Na+ channel blocker (carbamazepine, i.v.) failed to demonstrate an involvement of Na+ channels in the excitatory action of the toxin. These results suggest that Na+ channels act independent of the activation of sensory nerves by the toxin.
C. perfringens type C infection in sheep, lamb and goats exhibit a neurological involvement during the course of this disease (Songer, 1996). We have reported that beta-toxin induced arterial constriction and that the toxin-induced rise in blood pressure could be substantially reduced in rats treated with guanethidine or adrenal medullectomy, indicating that beta-toxin has a direct effect on the autonomic nervous system (Sakurai et al., 1984). In the present studies, we demonstrate that beta-toxin-induced plasma extravasation in mouse skin is mediated via stimulation of sensory nerve fibers. Based on these studies, it is possible that the drugs which modify sensory nerve systems may be worth pursing as a novel therapeutic approach in the clinic.
In conclusion, the present results indicate that beta-toxin stimulates sensory nerves via the bradykinin B2 prejunctional receptor or N-type Ca2+ channel in the neurons and then releases a tachykinin NK1 receptor agonist which is responsible for the subsequent neurogenic plasma extravasation. Further investigation of the mechanism of beta-toxin-induced plasma extravasation may result in a new model for disease involving neurogenic inflammation.
Acknowledgments
We thank Keiko Kobayashi for competent technical assistance. This research was supported in part by a Grant-in-Aid for Scientific Research (13670286) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Abbreviations
- CGRP
calcitonin-gene related peptide
- HOE140
DArg-[Hyp3, Thi5, D-Tic7, Oic8]-bradykinin; 125I-BSA, 125I-labelled bovine serum albumin
- PBS
phosphate-buffered saline
- SP
substance P
- TTX
tetrodotoxin
References
- AKOPIAN A.N., SIVILOTTI L., WOOD J.N. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- ALBER G., SCHEUBER P.H., RECK B., SAILER-KRAMER B., HARTMANN A., HAMMER D.K. Role of substance P in immediate-type skin reactions induced by staphylococcal enterotoxin B in unsensitized monkeys. J. Allergy. Clin. Immunol. 1989;84:880–885. doi: 10.1016/0091-6749(89)90383-7. [DOI] [PubMed] [Google Scholar]
- ARBUCKLE J.B., DOCHERTY R.J. Expression of tetrodotoxin-resistant sodium channels in capsaicin-sensitive dorsal root ganglion neurons of adult rats. Neurosci. Lett. 1995;185:70–73. doi: 10.1016/0304-3940(94)11227-a. [DOI] [PubMed] [Google Scholar]
- BACCEI M.L., KOCSIS J.D. Voltage-gated calcium currents in axotomized adult rat cutaneous afferent neurons. J. Neurophysiol. 2000;83:2227–2238. doi: 10.1152/jn.2000.83.4.2227. [DOI] [PubMed] [Google Scholar]
- BRAIN S.D., WILLIAMS T.J. Inflammatory oedema induced by synergism between calcitonin gene-related peptide (CGRP) and mediators of increased vascular permeability. Br. J. Pharmacol. 1985;86:855–860. doi: 10.1111/j.1476-5381.1985.tb11107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRAIN S.D., WILLIAMS T.J. Interactions between the tachykinins and calcitonin generelated peptide lead to the modulation of oedema formation and blood flow in rat skin. Br. J. Pharmacol. 1989;97:77–82. doi: 10.1111/j.1476-5381.1989.tb11926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAO T., PINTER E., AL-RASHED S., GERARD N., HOULT J.R., BRAIN S.D. Neurokinin-1 receptor agonists are involved in mediating neutrophil accumulation in the inflamed, but not normal, cutaneous microvasculature: an in vivo study using neurokinin-1 receptor knockout mice. J. Immunol. 2000;164:5424–5429. doi: 10.4049/jimmunol.164.10.5424. [DOI] [PubMed] [Google Scholar]
- COSTA S.K.P., DE NUCCI G., ANTUNES E., BRAIN S.D. Phoneutria nigriventer spider venom induces oedema in rat skin by activation of capsaicin sensitive sensory nerves. Eur. J. Pharmacol. 1997;339:223–226. doi: 10.1016/s0014-2999(97)01387-3. [DOI] [PubMed] [Google Scholar]
- COSTA S.K.P., DE NUCCI G., ANTUNES E., BRAIN S.D. Involvement of vanilloid receptors and purinoceptors in the Phoneutria nigriventer spider venom-induced plasma extravasation in rat skin. Eur. J. Pharmacol. 2000;391:305–315. doi: 10.1016/s0014-2999(00)00075-3. [DOI] [PubMed] [Google Scholar]
- EMONDS-ALT X., DOUTREMEPUICH J.D., HEAULME M., NELIAT G., SANTUCCI V., STEINBERG R., VILAIN P., BICHON D., DUCOUX J.P., PROIETTO V., BROECK D.V., SOUBRIE P., LE FUR G., BRELIERE J.C. In vitro and in vivo biological activities of SR140333, a novel potent non-peptide tachykinin NK1 receptor antagonist. Eur. J. Pharmacol. 1993;250:403–413. doi: 10.1016/0014-2999(93)90027-f. [DOI] [PubMed] [Google Scholar]
- EL-IDRISSI A.H., WARD G.E., JOHNSON D.W., BENKIRANE A., FASSI-FEHRI M.M. Bacteriological investigation of sudden sheep mortality in Morocco. Prev. Vet. Med. 1992;12:35–46. [Google Scholar]
- ESCOTT K.J., BRAIN S.D. Effect of a calcitonin gene-related peptide antagonist (CGRP8-37) on skin vasodilatation and oedema induced by stimulation of the rat saphenous nerve. Br. J. Pharmacol. 1993;110:772–776. doi: 10.1111/j.1476-5381.1993.tb13878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESCOTT K.J., BEATTIE D.T., CONNOR H.E., BRAIN S.D. The modulation of the increase in rat facial skin blood flow observed after trigeminal ganglion stimulation. Eur. J. Pharmacol. 1995;284:69–76. doi: 10.1016/0014-2999(95)00367-t. [DOI] [PubMed] [Google Scholar]
- FOX A.P., NOWYCKY M.C., TSIEN R.W. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurons. J. Physiol. 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAMSE R., HOLZER P., LEMBECK F. Decrease of substance P in primary afferent neurones and impairment of neurogenic plasma extravasation by capsaicin. Br. J. Pharmacol. 1980;68:207–213. doi: 10.1111/j.1476-5381.1980.tb10409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLZER P. Neurogenic vasodilatation and plasma leakage in the skin. Gen. Pharmacol. 1998;30:5–11. doi: 10.1016/s0306-3623(97)00078-5. [DOI] [PubMed] [Google Scholar]
- HUNTER S.E.C., BROWN J.E., OYSTON P.C.F., SAKURAI J., TITBALL R.W. Molecular genetic analysis of beta-toxin of Clostridium perfringens reveals sequence homology with alpha-toxin, gamma-toxin, and leukocidin of Staphylococcus aureus. Infect. Immun. 1993;61:3958–3965. doi: 10.1128/iai.61.9.3958-3965.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISHIDA H., FUJII E., IRIE K., YOSHIOKA T., MURAKI T., OGAWA R. Role of inflammatory mediators in lipid A analogue (ONO-4007)-induced vascular permeability change in mouse skin. Br. J. Pharmacol. 2000;130:1235–1240. doi: 10.1038/sj.bjp.0703425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANCSO G., KIRALY E., JANCSO-GABOR A. Pharmacologically induced selective degeneration of chemosensitive primary sensory neurones. Nature. 1977;270:741–743. doi: 10.1038/270741a0. [DOI] [PubMed] [Google Scholar]
- KENNEDY K.K., NORRIS S.J., BECKENHAUER W.H., WHITE R.G. Vaccination of cattle and sheep with combined Clostridium perfringens type C and type D toxoid. Am. J. Vet. Res. 1977;38:1515–1518. [PubMed] [Google Scholar]
- LAWRENCE G., SHANN F., FREESTONE D.S., WALKER P.D. Prevention of necrotizing enteritis in Papua New Guinea by active immunization. Lancet. 1979;1:227–230. doi: 10.1016/s0140-6736(79)90764-5. [DOI] [PubMed] [Google Scholar]
- LIU L., SZALLASI A., SIMON S.A. A non-pungent resiniferatoxin analogue, phorbol 12-phenylacetate 13 acetate 20-homovanillate, reveals vanilloid receptor subtypes on rat trigeminal ganglion neurons. Neuroscience. 1998;84:569–581. doi: 10.1016/s0306-4522(97)00523-x. [DOI] [PubMed] [Google Scholar]
- MAGGI C.A. The pharmacology of the efferent function of sensory nerves. J. Auton. Pharmacol. 1991;11:173–208. doi: 10.1111/j.1474-8673.1991.tb00317.x. [DOI] [PubMed] [Google Scholar]
- MAGGI C.A., PATACCHINI R., SANTICIOLI P., LIPPE I.T., GIULIANI S., GEPPETTI P., DEL BIANCO E., SELLERI S., MELI A. The effect of omega conotoxin GVIA, a peptide modulator of the N-type voltage sensitive calcium channels, on motor responses produced by activation of efferent and sensory nerves in mammalian smooth muscle. Naunyn-Schmiedebergs Arch. Pharmacol. 1988;338:107–113. doi: 10.1007/BF00174856. [DOI] [PubMed] [Google Scholar]
- MCDONEL J.L.Toxins of Clostridium perfringens types A, B, C, D and E Pharmacology of bacterial toxins 1986Oxford, Pergamon Press; 477–517.ed. F. Dorner & J. Drews [Google Scholar]
- NAGAHAMA M., KIHARA A., MIYAWAKI T., MUKAI M., SAKAGUCHI Y., OCHI S., SAKURAI J. Clostridium perfringens beta-toxin is sensitive to thiol-group modification but does not require a thiol group for lethal activity. Biochim. Biophys. Acta. 1999;1454:97–105. doi: 10.1016/s0925-4439(99)00026-5. [DOI] [PubMed] [Google Scholar]
- NAGAHAMA M., SAKURAI J. Distribution of labeled Clostridium perfringens epsilon toxin in mice. Toxicon. 1991;29:211–217. doi: 10.1016/0041-0101(91)90105-z. [DOI] [PubMed] [Google Scholar]
- PALFRAMAN R.T., COSTA S.K.P., WILSONCROFT P., ANTUNES E., DE NUCCI G., BRAIN S.D. The effect of a tachykinin NK1 receptor antagonist, SR140333, on oedema formation induced in rat skin by venom from the Phoneutria nigriventer spider. Br. J. Pharmacol. 1996;118:295–298. doi: 10.1111/j.1476-5381.1996.tb15402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERKINS M.N., CAMPBELL E.A. Capsazepine reversal of the antinociceptive action of capsaicin in vivo. Br. J. Pharmacol. 1992;107:329–333. doi: 10.1111/j.1476-5381.1992.tb12746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAKURAI J. Toxins of Clostridium perfringens. Rev. Medical. Microbiol. 1995;6:175–185. [Google Scholar]
- SAKURAI J., DUNCAN C.L. Purification of beta-toxin from Clostridium perfringens type C. Infect. Immun. 1977;18:741–745. doi: 10.1128/iai.18.3.741-745.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAKURAI J., DUNCAN C.L. Some properties of beta-toxin produced by Clostridium perfringens type C. Infect. Immun. 1978;21:678–680. doi: 10.1128/iai.21.2.678-680.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAKURAI J., FUJII Y. Purification and characterization of Clostridium perfringens beta toxin. Toxicon. 1987;25:1301–1310. doi: 10.1016/0041-0101(87)90008-0. [DOI] [PubMed] [Google Scholar]
- SAKURAI J., FUJII Y., DEZAKI K., ENDO K. Effect of Clostridium perfringens beta toxin on blood pressure of rats. Microbiol. Immunol. 1984;28:23–31. doi: 10.1111/j.1348-0421.1984.tb02944.x. [DOI] [PubMed] [Google Scholar]
- SAKURAI J., FUJII Y., MATSUURA M. Effect of oxidizing agents and sulfhydryl group reagents on beta toxin from Clostridium perfringens type C. Microbiol. Immunol. 1980;24:595–601. doi: 10.1111/j.1348-0421.1980.tb02862.x. [DOI] [PubMed] [Google Scholar]
- SAKURAI J., FUJII Y., MATSUURA M., ENDO K. Pharmacological effect of beta toxin of Clostridium perfringens type C on rats. Microbiol. Immunol. 1981;25:423–432. doi: 10.1111/j.1348-0421.1981.tb00045.x. [DOI] [PubMed] [Google Scholar]
- SAKURAI J., FUJII Y., NAGAHAMA M. Effect of p-chloromercuribenzoate on Clostridium perfringens beta toxin. Toxicon. 1992;30:323–330. doi: 10.1016/0041-0101(92)90872-3. [DOI] [PubMed] [Google Scholar]
- SAKURAI J., NAGAHAMA M., OCHI S. Major toxins of Clostridium perfringens. J. Toxicol.-Toxin Reviews. 1997;16:195–214. [Google Scholar]
- SANTICIOLI P., DEL BIANCO E., FIGINI M., BEVAN S., MAGGI C.A. Effect of capsazepine on the release of calcitonin gene-related peptide-like immunoreactivity (CGRP-LI) induced by low pH, capsaicin and potassium in rat soleus muscle. Br. J. Pharmacol. 1993;110:609–612. doi: 10.1111/j.1476-5381.1993.tb13854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHATURSKY O., BAYLES R., ROGERS M., JOST B.H., SONGER J.G., TWETEN R.K. Clostridium perfringens beta-toxin forms potential-dependent, cation-selective channels in lipid bilayers. Infect. Immun. 2000;68:5546–5551. doi: 10.1128/iai.68.10.5546-5551.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMONS F.E., SILVER N.A., GU X., SIMONS K.J. Skin concentrations of H1-receptor antagonists. J. Allergy. Clin. Immunol. 2001;107:526–530. doi: 10.1067/mai.2001.113080. [DOI] [PubMed] [Google Scholar]
- SINEY L., BRAIN S.D. Involvement of sensory neuropeptides in the development of plasma extravasation in rat dorsal skin following thermal injury. Br. J. Pharmacol. 1996;117:1065–1070. doi: 10.1111/j.1476-5381.1996.tb16698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONGER J.G. Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 1996;9:216–234. doi: 10.1128/cmr.9.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEINTHORSDOTTIR V., HALLDORSSON H., ANDRESSON O.S. Clostridium perfringens beta-toxin forms multimeric transmembrane pores in human endothelial cells. Microb. Pathog. 2000;28:45–50. doi: 10.1006/mpat.1999.0323. [DOI] [PubMed] [Google Scholar]
- SZALLASI A., BLUMBERG P.M. Vanilloid (Capsaicin) receptors and mechanisms. Pharmacol. Rev. 1999;51:159–212. [PubMed] [Google Scholar]
- TESHIGAWARA H., MORIYA Y. The role of leukotriene C4 in biphasic release of histamine from mouse mastocytoma cell line (P-815) Gen. Pharmacol. 1994;25:857–864. doi: 10.1016/0306-3623(94)90087-6. [DOI] [PubMed] [Google Scholar]
- WALKER B., BAYLEY H. Key residues for membrane binding, oligomerization, and pore forming activity of staphylococcal alpha-hemolysin identified by cysteine scanning mutagenesis and targeted chemical modification. J. Biol. Chem. 1995;270:23065–23071. doi: 10.1074/jbc.270.39.23065. [DOI] [PubMed] [Google Scholar]
- WILLIAMS T.J. Prostaglandin E2, prostaglandin I2 and the vascular changes of inflammation. Br. J. Pharmacol. 1979;65:517–524. doi: 10.1111/j.1476-5381.1979.tb07860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]