

Abstract
We examined the effects of eprosartan, an AT1 receptor antagonist, on the progression of left ventricular (LV) dysfunction and remodelling in dogs with heart failure (HF) produced by intracoronary microembolizations (LV ejection fraction, EF 30 to 40%).
Dogs were randomized to 3 months of oral therapy with low-dose eprosartan (600 mg once daily, n=8), high-dose eprosartan (1200 mg once daily, n=8), or placebo (n=8).
In the placebo group, LV end-diastolic (EDV) and end-systolic (ESV) volumes increased after 3 months (68±7 vs 82±9 ml, P<0.004, 43±1 vs 58±7 ml, P<0.003, respectively), and EF decreased (37±1 vs 29±1%, P<0.001). In dogs treated with low-dose eprosartan, EF, EDV, and ESV remained unchanged over the course of therapy, whereas in dogs treated with high-dose eprosartan, EF increased (38±1 vs 42±1%, P<0.004) and ESV decreased (41±1 vs 37±1 ml, P<0.006), Eprosartan also decreased interstitial fibrosis and cardiomyocyte hypertrophy.
We conclude that eprosartan prevents progressive LV dysfunction and attenuates progressive LV remodelling in dogs with moderate HF and may be useful in treating patients with chronic HF.
Keywords: Ejection fraction, microembolization, norepinephrine
Introduction
The process of LV remodelling during heart failure (HF) can lead to progressive left ventricular (LV) dysfunction and is ultimately associated with a poor prognosis. Activation of neurohormones, including the renin-angiotensin system, plays a key role in the ventricular remodelling process. In the setting of HF, angiotensin-II exerts a number of harmful effects on the cardiovascular system via activation of AT1 receptors. The trophic effect of angiotensin-II produced locally by the myocardium is believed to contribute to overload-induced myocardial mass augmentation, and has been shown to stimulate myocyte hypertrophy and fibroblast hyperplasia in vitro (de lannoy et al., 1998; Dell'italia et al., 1997; Sadoshima & Izumo, 1993). In addition, angiotensin-II causes apoptosis and/or necrosis of myocytes under certain conditions (Kajstura et al., 1997; Tan et al., 1991).
The sympathetic nervous system and renin-angiotensin system are also thought to be closely interrelated (Gilbert et al., 1993; Litwin & Morgan, 1992). Angiotensin-II enhances norepinephrine outflow by activating prejunctional AT1 receptors located on the sympathetic nerve terminals (Clemson et al., 1994; Hatton & Clough, 1982; Rump et al., 1994). Exposure to increased levels of norepinephrine can lead to downregulation and uncoupling of β1-receptors, which may contribute to myocardial dysfunction (Brodde et al., 1998; Gengo et al., 1992; Steinfath et al., 1991). Norepinephrine has also been shown to be cytotoxic and capable of inducing apoptosis in cardiac myocytes (Communal et al., 1998). Regulation of AT1 receptors could potentially modulate the progression of HF through inhibition of norepinephrine release.
Long-term treatment with angiotensin converting enzyme (ACE) inhibitors has been shown to ameliorate HF in various animal models, as well as in humans (Goldstein et al., 1995; Konstam et al., 1992; McKelvie et al., 1999; Sabbah et al., 1994). The beneficial effects of ACE inhibitors may arise from blockade of angiotensin-II formation and/or through prevention of the degradation of bradykinins. Conversely, the effects of AT1 receptor antagonists on LV dysfunction and LV remodelling in HF have not been fully established. In the Evaluation of Losartan in the Elderly Study (ELITE II), losartan reduced all-cause mortality, cardiovascular mortality and hospitalization for HF with equal efficacy compared with patients treated with the ACE inhibitor captopril (Pitt et al., 2000). Similarly, in the Randomized Evaluation of Strategies for Left Ventricular Dysfunction Pilot Study (RESOLVD), candesartan attenuated LV remodelling and increased LVEF as effectively as enalapril. In addition, coadministration of candesartan and enalapril attenuated LV remodelling as evidenced by a lesser increase in LV end-systolic and end-diastolic volumes compared to candesartan alone or enalapril alone (McKelvie et al., 1999).
The present study was designed to determine the effects of early, long-term monotherapy with another AT1 receptor antagonist, eprosartan, on the progression of LV dysfunction and LV remodelling. Eprosartan has a unique chemical structure which makes it highly selective for the AT1 receptor subtype and is a competitive antagonist and can block prejunctional angiotensin II receptors (Brooks & Ruffolo, 1999; Brooks et al., 1999; Ohlstein et al., 1997). Blockade of prejunctional angiotensin II receptors may decrease norepinephrine release, thus possibly attenuating the aforementioned detrimental effects of both angiotensin II and norepinephrine on the myocardium. Eprosartan was previously shown to prevent a decrease in stroke volume and ejection fraction and increase in ventricular chamber volume in spontaneously hypertensive rats (Barone et al., 2001). The addition of the eprosartan to an ACE inhibitor in patients with heart failure was shown to decrease blood pressure without affecting LV ejection fraction (Murdoch et al., 2001). In the present study we did not test the effects of combined therapy with eprosartan and ACE inhibition, and therefore, a direct comparison with the above clinical trial is not possible. Instead, our objective was to determine whether eprosartan alone could attenuate the progression of heart failure.
Methods
Animal model
The canine model of chronic HF used in the present study was previously described in detail (Sabbah et al., 1991). Chronic LV dysfunction and failure were produced by multiple sequential intracoronary embolizations with polystyrene Latex microspheres (70–102 μm in diameter), which results in loss of viable myocardium. In the present study, 24 healthy mongrel dogs underwent intracoronary microembolizations to produce HF. Embolizations were performed 1 to 3 weeks apart and were discontinued when LV ejection fraction, determined angiographically, was between 30 and 40%. Microembolizations were performed during cardiac catheterization under general anaesthesia and sterile conditions. The anaesthesia regimen used consisted of a combination of an intravenous injection of oxymorphone (0.22 mg kg−1), diazepam (0.17 mg kg−1), and sodium pentobarbital (150–250 mg to effect). The study was approved by the Henry Ford Health System Institutional Animal Care and Use Committee and conformed to the ‘Position of the American Heart Association on Research Animal Use' and the Guiding Principles of the American Physiological Society.
Determination of doses of eprosartan
The high and low doses of eprosartan used in the study were selected based on studies performed on three dogs with heart failure instrumented for intra-arterial blood pressure measurement. In each dog, systolic pressure was measured at baseline and after a 3-min intravenous administration of exogenous angiotensin-II (50 ng kg min−1) sufficient to increase blood pressure by 30 mmHg or more. Following this initial pressor test, each dog received 1200 mg of oral eprosartan. The pressor response with exogenous angiotensin-II was then repeated at 1, 4, 8, 12 and 24 h after administration of eprosartan (Figure 1). With this dose of eprosartan, the pressor response decreased by 90% at 4 h, 67% at 12 h and 36% at 24 h. This degree of suppression of the pressor response was considered a surrogate to acceptable blockade of the AT1 receptor. Based on these data, two fixed doses of eprosartan were chosen for the study, namely a low dose of 600 mg once daily and a high dose of 1200 mg once daily.
Figure 1.
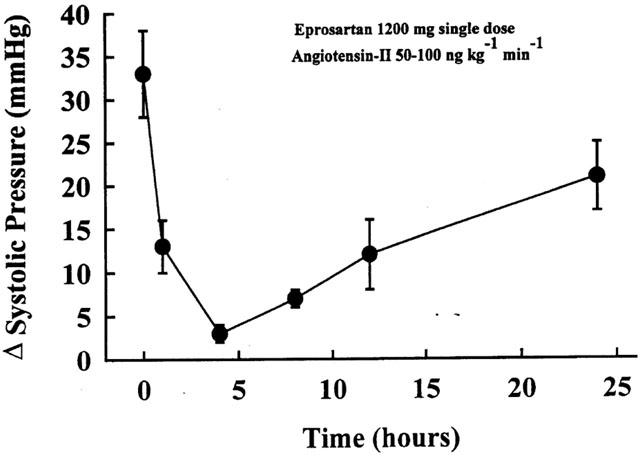
Temporal change in pressor response to intravenous administration of angiotensin-II after oral administration of a single dose of eprosartan (1200 mg). The pressor response was performed at 0, 1, 4, 8, 12 and 24 h after administration of eprosartan.
Study protocol
Two weeks after the last embolization procedure, dogs underwent a pre-randomization left and right heart catheterization. One day after cardiac catheterization, dogs were randomized to 3 months of oral therapy with high-dose eprosartan (1200 mg, once daily, n=8), low-dose eprosartan (600 mg, once daily, n=8), or to placebo (vehicle once daily, n=8). Haemodynamic, angiographic, echocardiographic, and neurohormonal measurements were made at baseline, at the time of randomization and prior to initiation of therapy and at 3 months after initiation of therapy.
Haemodynamic and angiographic measurements
Aortic and LV pressures were measured with catheter-tip micromanometers (Millar Instruments, Houston, TX, U.S.A.). Cardiac output was measured in duplicate with a Swan-Ganz catheter using the thermodilution method. Cardiac index was calculated as the ratio of cardiac output to body surface area. Systemic vascular resistance was calculated as the difference between mean aortic pressure and mean right atrial pressure times 80 divided by cardiac output.
Left ventriculograms were obtained during each catheterization after completion of the haemodynamic measurements with the dog placed on its right side. Ventriculograms were recorded on 35-mm cine at 30 frames per second during the injection of 20 ml of contrast material (RENO-M-60 Squibb, Princeton, N.J., U.S.A.). Correction for image magnification was made with a calibrated radiopaque grid placed at the level of the LV. LV end-diastolic and end-systolic volumes were calculated from ventricular silhouettes using the area-length method (Dodge et al., 1966). LV ejection fraction was calculated as the ratio of the difference between end-diastolic and end-systolic volumes divided by end-diastolic volume times 100. Extrasystolic and post-extrasystolic beats were excluded from the angiographic analysis.
Echocardiographic measurements
Echocardiographic studies were performed using a Hewlett-Packard model 77020A ultrasound system with a 3.5-MHz transducer. Measurements were made during cardiac catheterization with the dog placed in the right lateral decubitous position. Echocardiograms were recorded on a Panasonic 6300 VHS recorder. The thickness of the intraventricular septum and the thickness of the LV posterior wall were determined using M-mode echocardiography according to the recommendation of the American Society of Echocardiography (Sahn et al., 1978). The thickness of the intraventricular septum and the LV posterior wall were summed and averaged to obtain a single representative measure of the LV wall thickness. The end-diastolic LV major and minor semiaxes at the midwall were measured from two-dimensional echocardiograms using the apical four-chamber views. LV end-diastolic circumferential wall stress was calculated as previously described (Grossman, 1991).
Neurohormonal measurements
Venous blood samples were obtained from conscious dogs one day before cardiac catheterization for evaluation of plasma concentration of norepinephrine and angiotensin II. Blood for norepinephrine analysis was collected in tubes containing EGTA and reduced glutathione. Blood for angiotensin II analysis was collected in tubes containing ACE and protease inhibitors. To minimize possible variations, blood samples were always obtained between 0800 and 1000 h. Plasma norepinephrine concentration was measured using high performance liquid chromatography (HPLC) (Candito et al., 1990). Plasma immunoreactive angiotensin II was measured by radioimmunoassay after extraction by reversible absorption by phenyl silica and HPLC (Meng et al., 1993; Nussberger et al., 1985).
Histologic and morphometric assessments
Samples for histological assessment were prepared as described previously by Querejeta et al. (2000). Myocardial samples from the LV free wall were fixed in 10% buffered formalin, embedded in paraffin and sectioned into 4-μm thick sections. Sections were then stained with collagen-specific picosirius red (Sirius red F3BA in aqueous picric acid) according to Dolber and Spach (Dolber & Spach, 1987). Collagen volume fraction was determined by quantitative morphometry with an automated image analysis system (Mocha, Jandel Scientific). Sections were analysed under a microscope by selecting 10 random fields remote from old infarcts. Stained collagen areas were segmented by interactive gray-level thresholding of shading corrected images. Collagen volume fraction was calculated as the sum of all connective tissue areas divided by the sum of all connective tissue and muscle areas in all the fields analysed in each section. Paraffin embedded sections approximately 5-μm thick were prepared and stained with Gordon and Sweets' method for reticulin fibres (Gordon & Sweets, 1936) and used to delineate the myocyte border. Ten radially oriented, scar free, microscopic fields (×40) were selected at random from each section and used to measure myocyte cross-sectional area by computer-assisted planimetry (Liu et al., 1997). For comparison, tissue samples from seven normal dogs were obtained, prepared and studied in an identical manner.
Determination of brain or b-type natriuretic peptide (BNP) mRNA
Total RNA from frozen LV specimens was isolated using an RNA Stat-60 kit (Tel-Test ‘B' Inc, Friendwoods, TX, U.S.A.). The concentration and the quality of the isolated RNA were determined as previously described (Gupta et al., 1999). Isolated total RNA (2 μg) was reverse transcribed using oligo (dT) primers. The single-stranded cDNA was amplified by PCR using Platinum Taq DNA-polymerase (Invitrogen, Carlsbad, CA, U.S.A.). The thermal profile consisted of a denaturation step at 94°C for 30 sec, an annealing step of 55°C for 45 sec, and an extension step of 72°C for 45 sec, repeated for 30 cycles. The sequences for BNP sense and antisense primers were 5′-GAACCCCTTCTGGGTTTGTT-3′ and 5′-AGCCGATCTGGATGTTTGAG-3′, respectively. The same samples were subjected to PCR amplification for GAPDH cDNA as an internal standard to confirm that equal amounts of RNA from each sample were being analysed. The sequences of the primers for GAPDH were 5′-ACCACCATGGAGAAGGCTGG-3′ for the sense strand and 5′-CTCAGTGTAGCCCAGGAT-3′ for the antisense strand. PCR amplification produced 400-bp and 528-bp fragments originating from BNP mRNA and GAPDH mRNA, respectively. PCR products were identified by electrophoresis using a 1% agarose-ethidium bromide gel. The density of the bands were quantified by using a Bio-Rad model GS-670 imaging densitometer and expressed as densitometric units. BNP values were normalized to the housekeeping gene GAPDH whose expression is not altered in heart failure (Igarashi-Saito et al., 1999; Smith et al., 1998; Zarain-Herzberg et al., 1996). For comparison, LV tissue samples from seven normal dogs were obtained, prepared and studied in an identical manner.
Data analysis
Intragroup comparisons of haemodynamics, angiographic, echocardiographic, and neurohormonal variables within each of the three study groups were made between measurements obtained just before initiation of therapy and measurements made after completion of 3 months of therapy. For these comparisons, a Student's paired t-test was used, and a probability value less than 0.05 was considered significant. Study measures were tested at baseline before any embolization and at the time of randomization before initiation of therapy. Intergroup comparisons were made using a t-statistic for two means. To assess treatment effect, the change (Δ) in each measure from pre-treatment to post-treatment was calculated for each of the three study arms. Comparisons were made between the placebo group and each of the active treatment groups using a t-statistic for two means. For these tests, a probability less than 0.05 was considered significant. The statistical analysis for histomorphologic measures and for BNP mRNA was conducted separately. The data was examined using a one way ANOVA with alpha set at 0.05. If significance was achieved, pairwise comparisons were performed using the Student-Newman-Keuls test with P<0.05 considered significant. All data are reported as the mean±s.e.mean.
Results
At baseline, all dogs in the study had haemodynamic and angiographic findings that were within normal limits for mongrel dogs in our laboratory. Baseline data for all three study groups are shown in Table 1. There were no significant differences in any of the baseline parameters between dogs that were subsequently randomized to placebo to active treatment with either low-dose or high-dose eprosartan. Similarly, there were no significant differences between the three study groups in most of the parameters obtained just prior to treatment (Table 2). Three measures, however, were significantly different between placebo and high dose eprosartan, specifically, LV end-diastolic pressure, peak LV+dP/dt and end-diastolic wall stress. All three were lower in the eprosartan group.
Table 1.
Baseline haemodynamic, angiographic and echocardiographic measurements
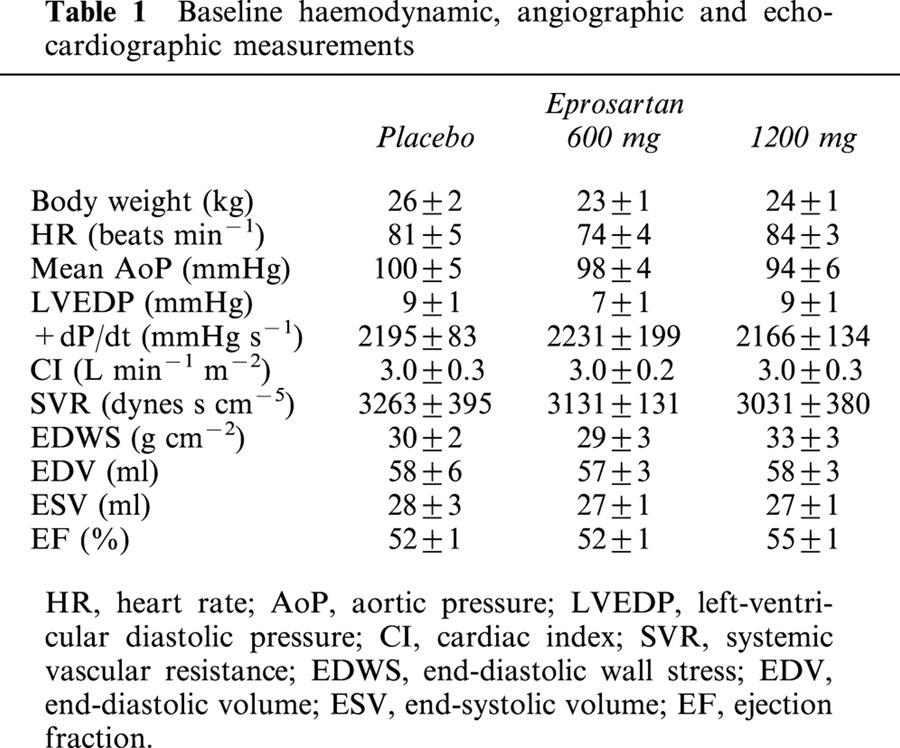
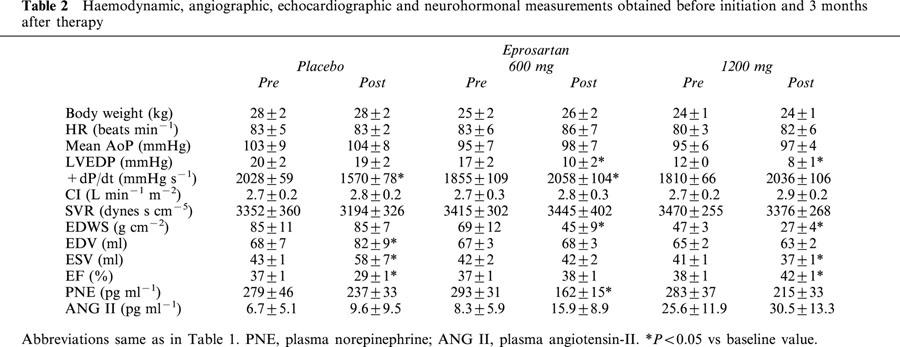
Table 2.
Haemodynamic, angiographic, echocardiographic and neurohormonal measurements obtained before initiation and 3 months after therapy
Effects of placebo on the progression of LV dysfunction and remodelling
In dogs receiving placebo, LV ejection fraction decreased significantly during the 3 months of follow-up (Table 2, Figure 2). This was accompanied by an increase in both LV end-diastolic and end-systolic volumes (Table 2). Dogs treated with placebo also showed a significant decline in peak +dP/dt (Table 2); while body weight, heart rate, mean aortic pressure, cardiac index, LV end-diastolic pressure and systemic vascular resistance remained unchanged after 3 months compared with measurements made at the time of randomization.
Figure 2.
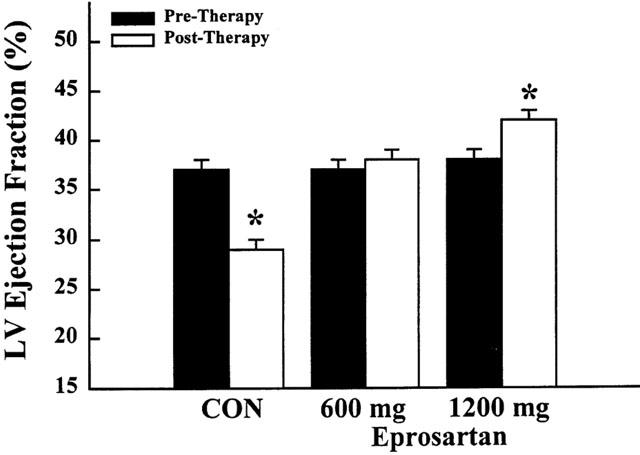
Bar graph depicting left ventricular (LV) ejection fraction before initiation of therapy and at the end of 3 months of therapy. Values are means±s.e.mean for untreated dogs (con, control), dogs treated with low-dose eprosartan (600 mg) administered once daily, and dogs treated with high-dose eprosartan (1200 mg) administered once daily. Probabilities refer to within-group comparisons between pre-therapy and post-therapy with *P<0.05.
Effects of monotherapy with low-dose eprosartan
In dogs treated with low-dose eprosartan (600 mg, once daily), LV ejection fraction remained unchanged during 3 months of treatment (Table 2, Figure 2). There were no progressive increases in either LV end-systolic or end-diastolic volume over the course of 3 months of therapy (Table 2). In this active treatment group, body weight, heart rate, mean aortic pressure, cardiac index and systemic vascular resistance also remained unchanged while LV end-diastolic pressure decreased significantly as did LV end-diastolic wall stress (Table 2).
Effects of monotherapy with high-dose eprosartan
In dogs treated with high-dose eprosartan (1200 mg once daily), LV ejection fraction increased significantly during the course of 3 months of therapy (Table 2, Figure 2). This increase was accompanied by a significant reduction of LV end-systolic volume and a reduction of LV end-diastolic volume; the latter, however, did not reach statistical significance (Table 2). The increase in peak +dP/dt was also not significant. LV end-diastolic pressure decreased significantly, as did end-diastolic wall stress (Table 2). Body weight, heart rate, mean aortic pressure, cardiac index and systemic vascular resistance remained essentially unchanged (Table 2).
Norepinephrine and angiotensin II
There were no significant differences between any of the groups in plasma norepinephrine at pre- and post-treatment except for animals treated with 600 mg eprosartan (Tables 2 and 3). In this group there was a significant reduction in plasma norepinephrine at post-treatment compared with pre-treatment. There were also no significant differences between any of the groups in plasma angiotensin II levels at pre- and post-treatment (Tables 2 and 3), indicating that eprosartan did not increase circulating levels of angiotensin-II.
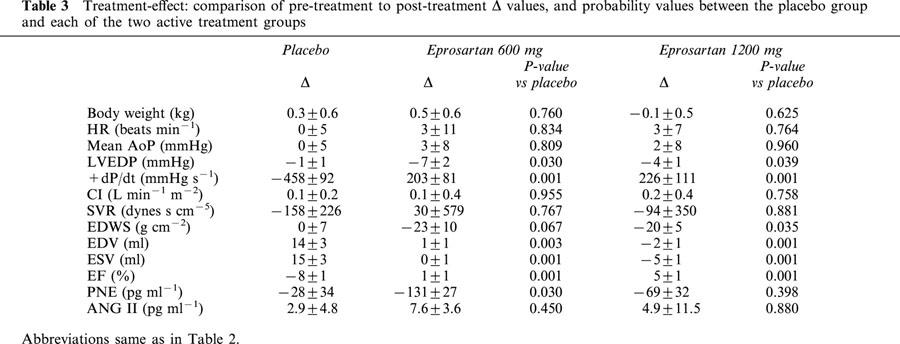
Table 3.
Treatment-effect: comparison of pre-treatment to post-treatment Δ values, and probability values between the placebo group and each of the two active treatment groups
Comparisons of treatment effect
In the post-treatment analysis, comparisons were made between the three treatment groups. Probability values based on a t-statistic for two means are shown in Table 3. Both low- and high-dose eprosartan significantly increased LV ejection fraction and LV peak +dP/dt compared with dogs receiving placebo. Both LV end-diastolic and end-systolic volumes were significantly lower in both eprosartan dose groups compared to placebo. No significant differences were noted between the three groups with respect to body weight, heart rate, mean aortic pressure, cardiac index and systemic vascular resistance. LV end-diastolic pressure and end-diastolic wall stress were lower in active treatment groups regardless of dose compared to placebo.
Compared to normal dogs, volume fraction of interstitial fibrosis was significantly increased in placebo dogs (3.5±0.3 vs 11.9±0.8%, P<0.05). Both low dose and high dose eprosartan significantly (P<0.05) decreased interstitial fibrosis compared with placebo (4.8±0.6% and 6.8±0.9% respectively) (Figure 3a–c). Compared to normal dogs, cardiomyocyte cross-sectional area was larger in placebo treated dogs (316±18 vs 693±31 μm2, P<0.05) (Figure 3d–f). Low-dose and high-dose eprosartan significantly (P<0.05) decreased cardiomyocyte size compared with placebo (386±22 μm2 and 368±36 μm2 respectively). Compared to normal dogs, mRNA for BNP was significantly increased in placebo dogs (Figure 4). Eprosartan significantly decreased mRNA for BNP in a dose-dependent manner compared to placebo (Figure 4). BNP mRNA was significantly lower in dogs treated with high dose eprosartan compared to dogs treated with low dose.
Figure 3.
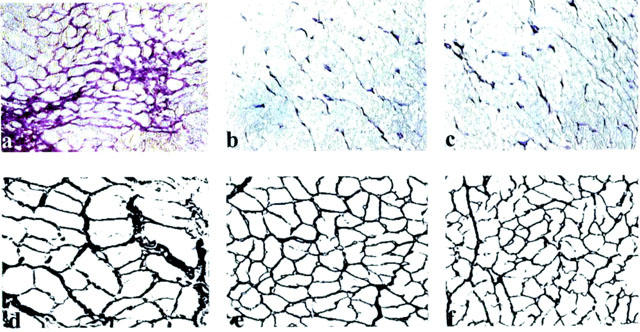
a–c: Representative staining for interstitial fibrosis in LV of control (a), low-dose eprosartan (b) and high-dose eprosartan (c) treated dogs. Interstitial fibrosis is indicated by dark purple staining. Original magnification 20×. d–f: Representative photograph depicting cardiomyocyte hypertrophy in LV of control (a), low-dose eprosartan (b) and high-dose eprosartan (c) treated dogs. Original magnification 40×.
Figure 4.
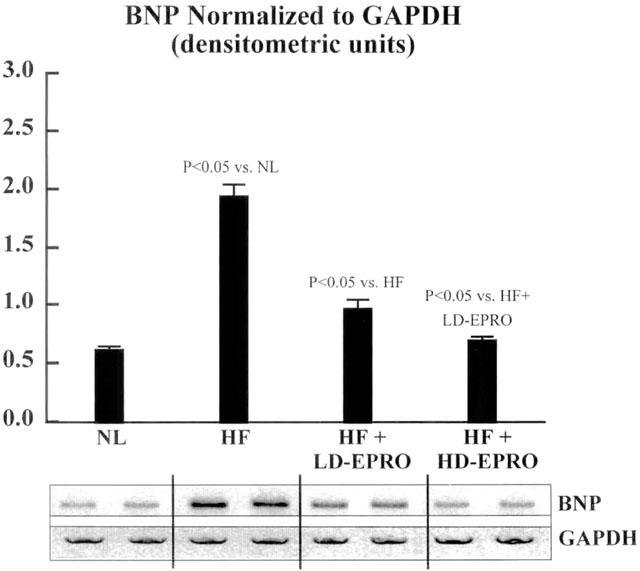
Top panel: Bar graph depicting changes in expression of BNP mRNA in normal (NL) dogs, untreated heart failure (HF) dogs, heart failure dogs treated with low dose eprosartan (LD-EPRO) and dogs treated with high dose eprosartan (HD-EPRO). Bottom panel: representative RT–PCR gel depicting changes in expression of BNP in all four groups and the lack of any change in the housekeeping gene GAPDH.
Discussion
The present work demonstrates that dogs with moderate HF randomized to monotherapy with low- or high-dose eprosartan do not develop progressive LV dysfunction and remodelling. LV ejection fraction and parameters of LV remodelling remained unchanged during 3 months therapy in dogs treated with low-dose eprosartan (600 mg once daily), while dogs treated with high-dose eprosartan (1200 mg once daily) showed a significant reduction of LV of end-systolic volume but not end-diastolic volume and a significant increase of LV ejection fraction. Both low- and high-dose eprosartan also significantly decreased interstitial fibrosis and cardiomyocyte cross-sectional area, a measure of myocyte hypertrophy. While cellular measures of LV remodelling based on cardiomyocyte hypertrophy and interstitial fibrosis did not directly correlate with the observed improvement in LV ejection fraction and reduction in LV end-systolic volume seen with high dose eprosartan, expression of BNP in LV tissue did correlate. We observed a dose-dependent reduction in the expression of BNP. BNP is a marker of LV dilation and, as such, is expected to rise in tissue with progressive LV enlargement and fall in association with reduction in LV size (Cataliotti et al., 2001; Nishikimi et al., 1996).
Several studies have examined the effects of AT1 receptor antagonists in animal models of heart failure. The AT1 antagonist, L-158,809 attenuated infarct expansion and early LV remodelling in dogs with acute myocardial infarction induced by coronary ligation (Ford et al., 1998). Positive results have also been obtained from studies in which these drugs acutely improved haemodynamic parameters in canine models of HF induced by coronary microembolizations (Wang et al., 1999), coronary ligation, or rapid ventricular pacing (Yamamoto et al., 1997). Though chronic studies in rats have demonstrated that AT1 receptor antagonists ameliorate LV dysfunction and ventricular remodelling (Liu et al., 1997; Richer et al., 1999; Sanbe & Takeo, 1995), the benefits of these drugs in chronic models of canine HF have not been as apparent.
Our laboratory had previously reported that early, long-term treatment with low-dose valsartan (400 mg b.i.d.) prevents the fall in LV ejection fraction in canine heart failure, but does not inhibit ventricular remodelling, as evidenced by a lack of reduction in LV chamber volumes as well as myocyte hypertrophy and interstitial fibrosis (Tanimura et al., 1999). High-dose valsartan (800 mg b.i.d.) did not attenuate the progressive decline in LV ejection fraction seen in untreated dogs and was associated with a significant increase in circulating plasma angiotensin-II levels (Tanimura et al., 1999). The enhanced selectivity of eprosartan for the AT1 receptor, and its ability to block pre-junctional AT1 receptors may account for the beneficial effects of eprosartan in our model of heart failure, which were not observed with valsartan. In the Valsartan Heart Failure Trial (Val-Heft), valsartan had no effect on mortality in patients with heart failure but significantly improved the combined end-point of event free survival. The latter included death, cardiac arrest with resuscitation, hospitalization for worsening heart failure, or therapy with intravenous inotropes (Cohn & Tognoni, 2001. In Val-Heft, however, valsartan when combined with an angiotensin converting enzyme (ACE) inhibitor and a beta-blocker caused increased adverse events (Cohn & Tognoni, 2001). Whether eprosartan will have the same adverse effects when combined with an ACE inhibitor or beta-blocker remains to be determined.
One of the proposed advantages of AT1 receptor blockade versus ACE inhibition is that it may not interfere with the postulated beneficial effects of AT2 receptor stimulation. Although the function of the AT2 receptor subtype has not been clearly established, it has been suggested that it exerts an anti-trophic action on cardiomyocytes and fibroblasts (Siragy, 1999). AT2 receptors are predominant in foetal rat brain, suggesting that the AT2 receptors may also be involved in growth, development, and differentiation (Cook et al., 1991; Zemel et al., 1990) or that the AT2 subtype may be a precursor of the AT1 subtype (Cook et al., 1991). Selective AT1 blockade would not interfere with any of these processes, nor would it affect angiotensin-II production, leaving it available to interact with AT2 receptors.
The ability of eprosartan to inhibit norepinephrine release from sympathetic nerve terminals may also account for its cardioprotective actions (Brooks & Ruffolo, 1999; Brooks et al., 1999; Ohlstein et al., 1997). β-adrenergic desensitization is one pathophysiologically important alteration that occurs during HF (Bristow et al., 1982; Packer, 1988). It has been shown that the sympathetic nervous system and renin-angiotensin system are closely associated (Gilbert et al., 1993; Litwin & Morgan, 1992). Angiotensin-II acts as a potent neuromodulator of norepinephrine release (Clemson et al., 1994; Hatton & Clough, 1982; Isaacson & Reid, 1990; Rump et al., 1994) and an inhibitor of norepinephrine reuptake into the sympathetic nerve terminal (Hughes & Roth, 1971; Kiran & Khairallah, 1969). Some studies have shown that the reduction in angiotensin-II formation by ACE inhibitors attenuates sympathetic drive to the heart and may lead to the restoration of β-adrenergic receptor density (Gilbert et al., 1993; Sanbe & Takeo, 1995). Other studies have suggested that other AT1 receptor antagonists are also able to block sympathetic nerve activity (Hughes & Roth, 1971; Murakami et al., 1997). However, these agents may only inhibit sympathetic outflow at doses high enough to increase plasma angiotensin-II level, while eprosartan acts at doses that do not affect circulating angiotensin-II levels.
In the present study we observed significant differences in LV end-diastolic pressure, peak +dP/dt and end-diastolic wall stress at pre-treatment between placebo and high dose eprosartan. Variability in physiologic measures is an inherent feature of studies using in vivo models. Table 3, in which comparison of treatment effect is examined, eliminates some of the potential bias that may arise when pre-treatment values are significantly different, such as in the present study. In addition to these variables, high dose eprosartan consistently improved the other haemodynamic variables measured and significantly attenuated remodelling. The fact that low-dose eprosartan also significantly improved function and remodelling lends further support to the final conclusion that eprosartan was beneficial to the failing heart in our model. We also observed that low dose eprosartan significantly reduced plasma norepinephrine concentration whereas high dose eprosartan did not. We have no explanation for this divergent finding except that plasma norepinephrine concentration is quite variable as suggested by the rather high values of standard error of the mean for this measurement.
In conclusion, early, long-term therapy with eprosartan prevents progressive LV dysfunction and attenuates LV remodelling. These beneficial effects of eprosartan compared to other AT1 receptor antagonists may be due largely to high selectivity for the AT1 receptor subtype, its action as a competitive antagonist and its ability to block prejunctional angiotensin-II receptors. Furthermore, since eprosartan is effective at doses that do not affect circulating levels of angiotensin-II, it is less likely to stimulate or interfere with activity of the AT2 receptor subtype. Eprosartan also did not affect mean aortic pressure, systemic vascular resistance or heart rate, indicating that its cardioprotective effects were not due to chronotropic or afterload modulation. It was also recently shown that combined therapy with an ACE inhibitor and eprosartan increased cardiac output in patients with severe heart failure compared to those treated with an ACE inhibitor alone (Gremmler et al., 2000). Together with the present study, these data suggest that eprosartan may be another potential pharmacological therapy to ameliorate heart failure.
Acknowledgments
Supported, in part, by a grant from SmithKlein Beecham and by a grant from the National Heart, Lung, and Blood Institute, HL49090-07.
Abbreviations
- ACE
angiotensin converting enzyme
- AoP
aortic pressure
- AT
angiotensin
- CI
cardiac index
- EDP
end-diastolic pressure
- EDV
end-diastolic volume
- EDWS
end-diastolic wall stress
- EF
ejection fraction
- ESV
end-systolic volume
- HF
heart failure
- HR
heart rate
- LV
left ventricular
- PNE
plasma norepinephrine
- SVR
systemic vascular resistance
References
- BARONE F.C., COATNEY R.W., CHANDRA S., SARKAR S.K., NELSON A.H., CONTINO L.C., BROOKS D.P., CAMPBELL W.G., JR, OHLSTEIN E.H., WILLETTE R.N. Eprosartan reduces cardiac hypertrophy, protects heart and kidney, and prevents early mortality in severely hypertensive stroke-prone rats. Cardiovasc. Res. 2001;50:525–537. doi: 10.1016/s0008-6363(01)00257-7. [DOI] [PubMed] [Google Scholar]
- BRISTOW M.R., GINSBURG R., MINOBE W., CUBICCIOTTI R.S., SAGEMAN W.S., LURIE K., BILLINGHAM M.E., HARRISON D.C., STINSON E.B. Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- BRODDE O.E., VOGELSANG M., BROEDE A., MICHEL-REHER M., BEISENBUSCH-SCHAFER E., HAKIM K., ZERKOWSKI H.R. Diminished responsiveness of Gs-coupled receptors in severely failing human hearts: no difference in dilated versus ischemic cardiomyopathy. J. Cardiovasc. Pharmacol. 1998;31:585–594. doi: 10.1097/00005344-199804000-00018. [DOI] [PubMed] [Google Scholar]
- BROOKS D.P., OHLSTEIN E.H., RUFFOLO R.R. Pharmacology of eprosartan, an angiotensin II receptor antagonist: exploring hypotheses from clinical data. Am. Heart J. 1999;138:S246–S251. doi: 10.1016/s0002-8703(99)70317-0. [DOI] [PubMed] [Google Scholar]
- BROOKS D.P., RUFFOLO R.R. Pharmacological mechanism of angiotensin II receptor antagonists: implications for the treatment of elevated systolic blood pressure. J. Hypertension. 1999;17 Suppl. 2:S27–S32. [PubMed] [Google Scholar]
- CANDITO M., KRSTULOVIC C., SBIRAZZUOLI V., CHAMBRON P. Proposal for the standardization of the calibration method for the assay of plasma catecholamines. J. Chromatog. 1990;526:194–202. doi: 10.1016/s0378-4347(00)82498-6. [DOI] [PubMed] [Google Scholar]
- CATALIOTTI A., MALATINO L.S., JOUGASKI M., ZOCCALI C., CASTELLINO P., GIACONE G., BELLANUOVA I., TRIPEPI R., SEMINARA G., PARLONGO S., STANCANELLI B., BONANNO G., FATUZZO P., RAPISARDA F., BELLUARDO P., SIGNORELLI S.S., HEUBLEIN D.M., LAINCHBURY J.G., LESKINEN H.K., BAILEY K.R., REDFIELD M.M., BURNETT J.C., JR Circulating natriuretic peptide concentrations in patients with end-stage renal disease: role of brain natriuretic peptide as a biomarker for ventricular remodeling. Mayo Clinic Proceedings. 2001;76:1111–1119. doi: 10.4065/76.11.1111. [DOI] [PubMed] [Google Scholar]
- CLEMSON B., GAUL L., GUBIN S.S., CAMPSEY D.M., MCCONVILLE J., NUSSBERGER J., ZELIS R. Prejunctional angiotensin II receptors: facilitation of norepinephrine release in the human forearm. J. Clin. Invest. 1994;93:684–691. doi: 10.1172/JCI117021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHN J.N., TOGNONI G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N. Engl. J. Med. 2001;345:1667–1675. doi: 10.1056/NEJMoa010713. [DOI] [PubMed] [Google Scholar]
- COMMUNAL C., SINGH K., PIMENTEL D.R., COLUCCI W.S. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 1998;98:1329–1334. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- COOK V.I., GROVE K.L., MCMENAMIN K.M., CARTER M.R., HARDING J.W., SPETH R.C. The AT2 angiotensin receptor subtype predominates in the 18 day gestation fetal rat brain. Brain Res. 1991;560:334–336. doi: 10.1016/0006-8993(91)91254-x. [DOI] [PubMed] [Google Scholar]
- DE LANNOY L.M., DANSER A.H., BOUHUIZEN A.M., SAXENA P.R., SCHALEKAMP M.A. Localization and production of angiotensin II in the isolated perfused rat heart. Hypertension. 1998;31:1111–1117. doi: 10.1161/01.hyp.31.5.1111. [DOI] [PubMed] [Google Scholar]
- DELL'ITALIA L.J., MENG Q.C., BALCELLS E., WEI C.-C., PALMER R., HAGEMAN G.R., DURAND J., HANKES G.H., OPARIL S. Compartmentalization of angiotensin II generation in the dog heart: evidence for independent mechanisms in intravascular and interstitial spaces. J. Clin. Invest. 1997;100:253–258. doi: 10.1172/JCI119529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DODGE H.T., SANDLER H., BAXLEY W.A., HAWLEY R.R. Usefulness and limitations of radiographic methods for determining left ventricular volume. Am. J. Cardiol. 1966;18:10–24. doi: 10.1016/0002-9149(66)90191-3. [DOI] [PubMed] [Google Scholar]
- DOLBER P.C., SPACH M.S. Picrosirius red staining of cardiac muscle following phosphomolybdic acid treatment. Stain Technol. 1987;62:23–26. doi: 10.3109/10520298709107961. [DOI] [PubMed] [Google Scholar]
- FORD W.R., MILLAN M.A., FEUILLAN P., AGUILERA G. Effect of the novel angiotensin II type I receptor antagonist L-158,809 on acute infarct expansion and acute anterior myocardial infarction in the dog. Can. J. Cardiol. 1998;14:73–80. [PubMed] [Google Scholar]
- GENGO P.J., SABBAH H.N., STEFFEN R.P., SHARPE J.K., KONO T., STEIN P.D., GOLDSTEIN S. Myocardial beta adrenoceptor and voltage sensitive calcium channel changes in a canine model of chronic heart failure. J. Mol. Cell. Cardiol. 1992;24:1361–1369. doi: 10.1016/0022-2828(92)93100-x. [DOI] [PubMed] [Google Scholar]
- GILBERT E.M., SANDOVAL A., LARRABEE P., RENLUND D.G., O'CONNELL J.B., BRISTOW M.R. Lisinopril lowers cardiac adrenergic drive and increases β-receptor density in the failing human heart. Circulation. 1993;88:472–480. doi: 10.1161/01.cir.88.2.472. [DOI] [PubMed] [Google Scholar]
- GREMMLER B., KUNERT M., SCHLEIRING H., ULBRICHT L.J. Improvement of cardiac output in patients with severe heart failure by use of ACE-inhibitors combined with the AT1-antagonist eprosartan. Eur. J. Heart Failure. 2000;2:183–187. doi: 10.1016/s1388-9842(00)00060-x. [DOI] [PubMed] [Google Scholar]
- GOLDSTEIN S., SHAROV V.G., COOK J.M., SABBAH H.N. Ventricular remodeling: insights from pharmacologic interventions with angiotensin-converting enzyme inhibitors. Mol. Cell. Biochem. 1995;147:51–55. doi: 10.1007/978-1-4615-2005-4_7. [DOI] [PubMed] [Google Scholar]
- GORDON H., SWEETS H.H. A simple method for the silver impregnation of reticulum. Am. J. Pathol. 1936;12:545–548. [PMC free article] [PubMed] [Google Scholar]
- GROSSMAN W.Pressure measurement Cardiac Catheterization Angiography, and Intervention. 1991Philadelphia: Lea & Febiger; 123ed. Grossman, W., Baim, D.S. p [Google Scholar]
- GUPTA R.C., MISHRA S., MISHIMA T., GOLDSTEIN S., SABBAH H.N. Reduced sarcoplasmic reticulum Ca2+-uptake and phospholamban expression in ventricular myocardium of dogs with heart failure. J. Mol. Cell. Cardiol. 1999;31:1381–1389. doi: 10.1006/jmcc.1999.0970. [DOI] [PubMed] [Google Scholar]
- HATTON R., CLOUGH D.P. Captopril interferes with neurogenic vasoconstriction in the pithed rat by angiotensin-dependent mechanisms. J. Cardiovasc. Pharmacol. 1982;4:116–123. doi: 10.1097/00005344-198201000-00019. [DOI] [PubMed] [Google Scholar]
- HUGHES J., ROTH R.H. Evidence that angiotensin enhances transmitter release during sympathetic nerve stimulation. Br. J. Pharmacol. 1971;41:239–255. doi: 10.1111/j.1476-5381.1971.tb08025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IGARASHI-SAITO K., TSUTSUI H., TAKAHASHI M., KINUGAWA S., EGASHIRA K., TAKESHITA A. Endocardial versus epicardial differences of sarcoplasmic reticulum Ca2+-ATPase gene expression in the canine failing myocardium. Basic Res. Cardiol. 1999;94:267–273. doi: 10.1007/s003950050152. [DOI] [PubMed] [Google Scholar]
- ISAACSON J.S., REID I.A. Importance of endogenous angiotensin II in the cardiovascular responses to sympathetic stimulation in conscious rabbits. Circ. Res. 1990;66:662–671. doi: 10.1161/01.res.66.3.662. [DOI] [PubMed] [Google Scholar]
- KAJSTURA J., CIGOLA E., MALHOTRA A., LI P., CHENG W., MEGGS L.G., ANVERSA P. Angiotensin II induces apoptosis of adult ventricular myocytes in vitro. J. Mol. Cell. Cardiol. 1997;29:859–870. doi: 10.1006/jmcc.1996.0333. [DOI] [PubMed] [Google Scholar]
- KIRAN B.K., KHAIRALLAH P.A. Angiotensin and norepinephrine efflux. Eur. J. Pharmacol. 1969;6:102–108. doi: 10.1016/0014-2999(69)90204-0. [DOI] [PubMed] [Google Scholar]
- KONSTAM M.A., ROUSSEAU M.F., KRONENBERG M.W., UDELSON J.E., MELIN J., STEWART D., DOLAN N., EDENS T.R., AHN S., KINAN D., HOWE D.M., KILCOYNE L., METHERALL J., BENEDICT C., YUSUF S., POULEUR H. Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dysfunction in patients with heart failure. Circulation. 1992;86:431–438. doi: 10.1161/01.cir.86.2.431. [DOI] [PubMed] [Google Scholar]
- LITWIN S.E., MORGAN J.P. Captopril enhances intracellular calcium handling and β-adrenergic responsiveness of myocardium from rats with postinfarction failure. Circ. Res. 1992;71:797–807. doi: 10.1161/01.res.71.4.797. [DOI] [PubMed] [Google Scholar]
- LIU Y.H., YANG X.P., SHAROV V.G., NASS O., SABBAH H.N., PETERSON E., CARRETERO O.A. Effects of angiotensin-converting enzyme inhibitors on angiotensin II type 1 receptor antagonists in rats with heart failure. J. Clin. Invest. 1997;99:1926–1935. doi: 10.1172/JCI119360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCKELVIE R.S., YUSUF S., PERICAK D., AVEZUM A., BURNS R.J., PROBSTFIELD J., TSUYUKI R.T., WHITE M., ROULEAU J., LATINI R., MAGGIONI A., YOUNG J., POGUE J. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. Circulation. 1999;100:1056–1064. doi: 10.1161/01.cir.100.10.1056. [DOI] [PubMed] [Google Scholar]
- MENG Q.C., DURAND J., CHEN Y.F., OPARIL S. Simplified method for quantitation of angiotensin peptides in tissue. J. Chromatog. 1993;614:19–25. doi: 10.1016/0378-4347(93)80219-t. [DOI] [PubMed] [Google Scholar]
- MURAKAMI H., LIU J.-L., ZUCKER I.H. Angiotensin II enhances baroflex control of sympathetic outflow in heart failure. Hypertension. 1997;29:564–569. doi: 10.1161/01.hyp.29.2.564. [DOI] [PubMed] [Google Scholar]
- MURDOCH D.R., MCDONAGH T.A., FARMER R., MORTON J.J., MCMURRAY J.J., DARGIE H.J. ADEPT: Addition of the AT1 receptor antagonist eprosartan to ACE inhibitor therapy in chronic heart failure trial: hemodynamic and neurohumoral effects. Am. Heart J. 2001;141:800–807. doi: 10.1067/mhj.2001.114802. [DOI] [PubMed] [Google Scholar]
- NISHIKIMI T., YOSHIHARA F., MORIMOTO A., ISHIKAWA K., ISHIMITSU T., SAITO Y., KANGAWA K., MATSUO H., OMAE T., MATSUOKA H. Relationship between left ventricular geometry and natriuretic peptide levels in essential hypertension. Hypertension. 1996;28:22–30. doi: 10.1161/01.hyp.28.1.22. [DOI] [PubMed] [Google Scholar]
- NUSSBERGER J., BRUNNER D.B., WAEBER B., BRUNNER H.R. True versus immunoreactive angiotensin II in human plasma. Hypertension. 1985;7 Suppl. I:I-1–I-7. doi: 10.1161/01.hyp.7.3_pt_2.i1. [DOI] [PubMed] [Google Scholar]
- OHLSTEIN E.H., BROOKS D.P., FEUERSTEIN G.Z., RUFFOLO R.R. Inhibition of sympathetic outflow by the angiotensin II receptor antagonist, eprosartan, but not by losartan, valsartan or irbesartan: relationship to differences in prejunctional angiotensin II receptor blockade. Pharmacology. 1997;55:244–251. doi: 10.1159/000139534. [DOI] [PubMed] [Google Scholar]
- PACKER M. Neurohumoral interactions and adaptations in congestive heart failure. Circulation. 1988;77:721–730. doi: 10.1161/01.cir.77.4.721. [DOI] [PubMed] [Google Scholar]
- PITT B., POOLE-WILSON P.A., SEGAL R., MARTINEZ F.A., DICKSTEIN K., CAMM A.J., KONSTAM M.A., RIEGGER G., KLINGER G.H., NEATON J., SHARMA D., THIYAGARAJAN B. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial – the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000;355:1582–1587. doi: 10.1016/s0140-6736(00)02213-3. [DOI] [PubMed] [Google Scholar]
- QUEREJETA R., VARO N., LOPEZ B., LARMAN M., ARTINANO E., ETAYO J.C., UBAGO J.L.M., GUTIERREZ-STAMPA M., EMPARANZA J.I., GIL M.J., MONREAL I., MINDAN J.P., DIEZ J. Serum carboxy-terminal propeptide of procollagen type I is a marker of myocardial fibrosis in hypertensive heart disease. Circulation. 2000;101:1729–1735. doi: 10.1161/01.cir.101.14.1729. [DOI] [PubMed] [Google Scholar]
- RICHER C., FORNES P., CAZAUBON C., DOMERGUE V., NISATO D., GIUDICELLI J.-F. Effects of long-term angiotensin II AT1 receptor blockade on survival, hemodynamics and cardiac remodeling in chronic heart failure in rats. Cardiovasc. Res. 1999;41:100–108. doi: 10.1016/s0008-6363(98)00227-2. [DOI] [PubMed] [Google Scholar]
- RUMP L.C., SCHWERTFEGER E., SCHAIBLE U., FRAEDRICH G., SCHOLLMEYER P. β2-adrenergic receptor and angiotensin II receptor modulation of sympathetic neurotranmission in human atria. Circ. Res. 1994;74:434–440. doi: 10.1161/01.res.74.3.434. [DOI] [PubMed] [Google Scholar]
- SABBAH H.N., SHIMOYAMA H., KONO T., GUPTA R.C., SHAROV V.G., SCICLI G., LEVINE T.B., GOLDSTEIN S. Effects of long-term monotherapy with enalapril, metoprolol, and digoxin on the progression of left ventricular dysfunction and dilation in dogs with reduced ejection fraction. Circulation. 1994;89:2852–2859. doi: 10.1161/01.cir.89.6.2852. [DOI] [PubMed] [Google Scholar]
- SABBAH H.N., STEIN P.D., KONO T., GHEORGHIADE M., LEVINE T.B., JAFRI S., HAWKINS E.T., GOLDSTEIN S. A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. Am. J. Physiol. 1991;260:H1379–H1384. doi: 10.1152/ajpheart.1991.260.4.H1379. [DOI] [PubMed] [Google Scholar]
- SADOSHIMA J., IZUMO S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts: critical role of the AT1 receptor subtype. Circ. Res. 1993;73:413–423. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- SAHN D.J., DEMARIA A., KISSLO J., WEYMAN A. Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation. 1978;58:1072–1083. doi: 10.1161/01.cir.58.6.1072. [DOI] [PubMed] [Google Scholar]
- SANBE A., TAKEO S. Long-term treatment with angiotensin I-converting enzyme inhibitors attenuates the loss of cardiac β-adrenoceptor responses in rats with chronic heart failure. Circulation. 1995;92:2666–2675. doi: 10.1161/01.cir.92.9.2666. [DOI] [PubMed] [Google Scholar]
- SIRAGY H. Angiotensin II receptor blockers: review of the binding characteristics. Am. J. Cardiol. 1999;84:3S–8S. doi: 10.1016/s0002-9149(99)00727-4. [DOI] [PubMed] [Google Scholar]
- SMITH C.J., HE J., RICKETTS S.G., DING J.Z., MOGGIO R.A., HINTZE T.H. Downregulation of right ventricular phosphodiesterase PDE-3A mRNA and protein before the development of canine heart failure. Cell. Biochem. Biophys. 1998;29:67–88. doi: 10.1007/BF02737829. [DOI] [PubMed] [Google Scholar]
- STEINFATH M., GEERTZ B., SCHMITZ W., SCHOLZ H., HAVERICH A., BREIL I., HANRATH P., REUPCKE C., SIGMUND M., LO H.B. Distinct down-regulation of cardiac beta 1- and beta 2-adrenoceptors in different human heart diseases. Naunyn Schmiedebergs Arch. Pharmacol. 1991;343:217–220. doi: 10.1007/BF00168613. [DOI] [PubMed] [Google Scholar]
- TAN L.-B., JALIL J.E., PICK R., JANICKI J.S., WEBER K.T. Cardiac myocyte necrosis induced by angiotensin II. Circ. Res. 1991;69:1185–1195. doi: 10.1161/01.res.69.5.1185. [DOI] [PubMed] [Google Scholar]
- TANIMURA M., SHAROV V.G., SHIMOYAMA H., MISHIMA T., LEVINE T.B., GOLDSTEIN S., SABBAH H.N. Effects of AT1-receptor blockade on progression of left ventricular dysfunction in dogs with heart failure. Am. J. Physiol. 1999;276:H1385–H1392. doi: 10.1152/ajpheart.1999.276.4.H1385. [DOI] [PubMed] [Google Scholar]
- WANG J., YI G.H., ZHU S.M., GU A.G., POPILSKIS S., ZHANG H., BURKHOFF D. The role of angiotensin II AT1 receptor in the maintenance of hemodynamics in a canine model of coronary microembolization-induced heart failure. J. Cardiovasc. Pharmacol. 1999;33:335–340. doi: 10.1097/00005344-199902000-00023. [DOI] [PubMed] [Google Scholar]
- YAMAMOTO S., HAYAHI N., KOMENTANI M., NAKAO K. Pharmacological profile of valsartan, a non-peptide angiotensin II type I receptor antagonist. 5th communication: hemodynamic effects of valsartan in dog heart failure models. Arzneimittel-Forschung. 1997;47:630–634. [PubMed] [Google Scholar]
- ZARAIN-HERZBERG A., AFZAL N., ELIMBAN V., DHALLA N.S. Decreased expression of cardiac sarcoplasmic reticulum Ca2+-pump ATPase in congestive heart failure due to myocardial infarction. Mol. Cell Biochem. 1996;163–164:285–290. doi: 10.1007/BF00408669. [DOI] [PubMed] [Google Scholar]
- ZEMEL S., MILLAN M.A., FEUILLAN P., AGUILERA G. Characterization and distribution of angiotensin-II receptors in the primate fetus. J. Clin. Endocrinol. Metab. 1990;71:1003–1007. doi: 10.1210/jcem-71-4-1003. [DOI] [PubMed] [Google Scholar]