

Abstract
The α1-adrenergic responses of rat aorta and tail artery have been analysed measuring the contractility and the inositol phosphate (IP) formation induced by noradrenaline. Three antagonists, prazosin, 5-methylurapidil (α1A selective) and BMY 7378 (α1D selective) have been used in different experimental procedures.
Noradrenaline possesses a greater potency inducing contraction and IP accumulation in aorta (pEC50-contraction=7.32±0.04; pEC50-IPs=6.03±0.08) than in the tail artery (pEC50-contraction=5.71±0.07; pEC50-IPs=5.51±0.10). Although the maximum contraction was similar in both tissues (Emax-tail=619.1±55.6 mg; Emax-aorta-698.2±40.8 mg), there were marked differences in the ability of these tissues to generate intracellular second messengers the tail artery being more efficient (Emax-tail=1060±147%; Emax-aorta=108.1±16.9%).
Concentration response curves of noradrenaline in presence of antagonist together with concentration inhibition curves for antagonists added before (CICb) or after (CICa) noradrenaline-induced maximal response in Ca2+-containing or Ca2+-free medium have been performed. A comparative analysis of the different procedures as well as the mathematical approaches used in each case to calculate the antagonist potencies, were completed.
The CICa was the simplest method to characterize the predominant α1-adrenoceptor subtype involved in the functional response of a tissue.
In aorta, where constitutively active α1D-adrenoeptors are present, the use of different experimental procedures evidenced a complex equilibrium between α1D- and α1A-adrenoceptor subtypes.
The appropriate management of LiCl in IP accumulation studies allowed us to reproduce the different experimental procedures performed in contractile experiments giving more technical possibilities to this methodology.
Keywords: α1-Adrenoceptor subtypes, vascular smooth muscle, noradrenaline, BMY 7378, 5-methylurapidil, contraction, inositol phosphates, antagonist potencies
Introduction
In functional studies, in which an antagonist affects the response produced by an agonist on a tissue, the relation between antagonist, agonist and response observed is complex, even when analysing competitive antagonists. In fact, the binding may follow the law of mass action needed to apply the Cheng & Prusoff (1973) equation, but the functional response curve can be altered by non-linear amplification of the signal. This is specially true if different subtypes of receptors, with different subcellular localizations and a different degree of coupling to internal signals, are involved in the response, as occurs with α1-adrenoceptors (Schwinn et al., 1995; Theroux et al., 1996; McCune et al., 2000; Piascik & Perez, 2001) or, as has recently been shown, if a constitutively active receptor, such as α1D-adrenoceptor, intervenes in it (Noguera et al., 1996; García-Sáinz & Torres-Padilla, 1999; Gisbert et al., 2000; 2002; McCune et al., 2000; Ziani et al., 2002).
Until now, the Schild analysis is considered the most reliable method to calculate the equilibrium dissociation constant for a competitive antagonist in functional studies (Arunlakshana & Schild, 1959). This analysis needs the construction of full concentration-response curves for an agonist in the absence and in the presence of fixed antagonist concentrations and this makes the Schild analysis awkward but useful in contractile studies in isolated organ baths. However, some functional responses, notably those where a desensitization appears or those where second messenger generation was measured, are not susceptible to this kind of experimental design. The most common methodology in these situations is an inhibition curve design that involves antagonist titration in the presence of a fixed concentration of agonist. In this case, the equation of Cheng & Prusoff (1973) and its modifications for functional studies (Craig, 1993; Leff & Douglas, 1993) are the methods chosen to calculate the estimated Ki, although they are not considered a theoretically valid alternative to the Schild analysis (Craig, 1993; Leff & Douglas, 1993; Lazareno & Birdsall, 1993).
The aim of this study was to make a comparative analysis between the two different methodological approaches cited above and commonly used to calculate the antagonist potency in functional studies, and a new experimental procedure which we validate as the simplest alternative method when ethical reasons or difficulties to obtain tissue samples are considered. In order to do so, we have chosen the family of α1-adrenoceptors (α1-ARs) integrated by three different subtypes (α1A, α1B and α1D), with different cellular localization and different degrees of coupling to intracellular signals (Piascik & Perez, 2001). We studied the contractility of vascular smooth muscle (aorta and tail artery) as an indicator of its functional activity, and the inositol phosphate (IP) accumulation as the biochemical signal linked to its activation. We have chosen aorta and tail artery as tissues representative of the functionality of different subtypes of α1-ARs since it is well established that the α1D- and the α1A-ARs are mainly responsible for the contractile response to adrenergic stimulus in aorta and tail artery respectively (Kenny et al., 1995; Lachnit et al., 1997; Saussy et al., 1996; Hussain & Marshall, 1997; Gisbert et al., 2000). The evidence that α1D-ARs in native tissues such as aorta, exhibit constitutive activity (Noguera et al., 1996; Gisbert et al., 2000; 2002; Ziani et al., 2002), was also an important reason to choose the aorta in this study.
Methods
Functional studies in isolated organ bath
Rings of the aorta and tail artery (approx 3–5 mm in length) of female Wistar rats (200–220 g) were denuded of endothelium by gentle rubbing and suspended in a 10 ml organ bath containing physiological solution, maintained at 37°C and gassed with 95% O2 and 5% CO2. An initial load of 1 g was applied and maintained throughout a 75–90 min equilibration period. Tension was recorded isometrically by Grass FT03 force displacement transducers, and data were recorded on disc (McLab). The composition of the physiological Ca2+-containing solution was (mM): NaCl 118, KCl 4.75, CaCl2 1.8, MgCl2 1.2, KH2PO4 1.2, NaHCO3 25 and glucose 11. The Ca2+-free solution had the same composition, except that CaCl2 was omitted and EDTA (0.1 mM) was added.
The absence of relaxant response (10%) after acetylcholine (100 μM) addition to preparations precontracted with noradrenaline (10 or 1 μM in tail artery or aorta, respectively) indicated the absence of a functional endothelium in all the rings.
Three different experimental procedures were used:
Concentration-response curves to noradrenaline. These were performed by the addition of cumulative concentrations of the agonist (0.1 nM–100 μM) to tissues in the absence or presence of antagonist (incubated for 15 min). Contractions were expressed in mg of developed tension or as a percentage of the maximal contraction to the agonist in normal physiological solution (Emax). The concentration (−log [M]) of agonist required to produce 50% of the maximal response (pEC50) was obtained from a non-linear regression plot (Graph Pad Software; San Diego, CA, U.S.A.). Estimates of the antagonist affinity were determined from the negative logarithm of the antagonist dissociation constant (KB), determined from the equation KB=[A]/(DR-1) where the dose ratio (DR) was produced by a single concentration of antagonist [A].
Concentration-inhibition curves to selective α1-adrenoceptor antagonists added ‘before' agonist stimulation (CICb). CICb to Prazosin (0.001 nM–10 μM, BMY 7378 (0.001 nM–30 μM) and 5-methylurapidil (0.01 nM–10 μM) were obtained by the addition of one concentration of a compound 15 min before and during noradrenaline-induced contraction in Ca2+ containing, or Ca2+ free medium. In physiological Ca2+-containing solution, two successive additions of maximal concentrations of NA (10 μM in rat tail artery or 1 μM in rat aorta) give similar sustained contractile responses. After washing, a third addition of the agonist was made in the presence of a concentration of antagonist. The experimental procedure designed to study the actions of compounds on the contractile response to noradrenaline in Ca2+-free medium was as follows: noradrenaline (NA1 10 μM in rat tail artery or 1 μM in rat aorta) was added in physiological Ca2+-containing solution at 37°C and then the tissue was treated with Ca2+-free, EDTA-containing solution for 20 min. After this time, NA (NA2) was applied and the amplitude of the resulting contraction was monitored as a reference. A new addition of agonist did not reproduce this response because intracellular Ca2+ stores were depleted, then the tissue was incubated for 20 min in Krebs solution to refill the intracellular Ca2+ stores. After washing and 20 min of incubation in Ca2+-free solution, NA (NA3) was added again in the absence of Ca2+ and a response, similar to the first one obtained in Ca2+-free solution (NA2), was observed. The effect of different concentrations of each compound added 15 min before was tested on this last agonist-induced contraction in Ca2+-free medium. The magnitude of the noradrenaline-induced contraction in the presence of the different concentrations of the antagonists was expressed as a percentage of noradrenaline-induced response obtained in the absence of any agent.
- Concentration-inhibition curves to selective α1-adrenoceptor antagonists added ‘after' agonist stimulation (CICa). CICa to selective α1-adrenoceptor antagonists were performed by addition of cumulative concentrations of prazosin (0.001 nM–1 μM), BMY 7378 (0.001 nM–30 μM) and 5-methylurapidil (0.01 nM–10 μM) to tail artery and aortic rings in which sustained contractions had been induced by a maximal concentration of noradrenaline (10 or 1 μM respectively). Relaxations were expressed as a percentage of the maximum increment in tension obtained by agonist addition. The concentration of antagonist (−log [M]) needed to produce 50% relaxation (procedure CICa) or inhibition (procedure CICb) was obtained (pIC50) from a non-linear regression plot (Graph Pad Software; San Diego, CA, U.S.A.). Ki value was calculated from the IC50 estimates according to the Cheng & Prusof (1973) equation:
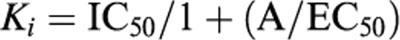
where A is the concentration of noradrenaline present in the assay and EC50 is the concentration of noradrenaline required for half-maximal stimulation in each tissue.
Accumulation of [3H]-inositol phosphates
The determination of the accumulation of inositol phosphates was adapted from Berridge et al. (1982). Briefly, rat tail arteries or rat thoracic aortas (four or five female Wistar rats 200–220 g were sacrificed) were cut into rings (2 mm for tail artery, 1 mm for aorta), pooled and incubated at 37°C for 30 min in physiological solution (composition in mM): NaCl 118, KCl 4.7, CaCl2 1.8, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, and glucose 11, aerated with 95% O2/5% CO2. The physiological solution was changed twice. Subsequently, both tissues were labelled in the presence of 10 μCi ml−1 of myo [3H]-inositol (specific activity 81.0 Ci mmol−1) in physiological solution for 2 h at 37°C with vigorous shaking, aerating the mixture every 15 min. After incubation, the samples were washed twice with physiological solution and two pieces of tail artery or four rings of aorta (0.1–0.2 mg of protein) were placed in individual tubes which were incubated at 37°C under an atmosphere of 95% O2 and 5% CO2 in a final volume of 500 μl of physiological solution. The tissues were incubated for 30 min with increasing concentrations of noradrenaline (0.01 μM–1 mM) in the presence of LiCl (10 mM) in order to inhibit the metabolism of inositol monophosphates.
The antagonist action of prazosin (0.1 nM–10 μM), BMY 7378 (1 nM–100 μM) and 5-methylurapidil (0.1 nM–100 μM) on noradrenaline-induced inositol phosphate accumulation was tested in two experimental conditions in order to reproduce the experiments carried out in organ bath studies: (i) In the CICb protocol, the tissues were incubated for 15 min with different concentrations of the antagonist and subsequently stimulated for 30 min with a maximal concentration of noradrenaline (10 μM), LiCl was added 30 s before agonist addition. (ii) In the CICa protocol, the tissues were stimulated with 10 μM for 30 min and allowed to incubate for an additional period of 15 min with different concentrations of the antagonists. LiCl (10 μM) was then added to the tubes during an additional incubation period of 30 min.
In all cases, unstimulated (basal) as well as noradrenaline-stimulated (maximal response) [3H]-IP formation was determined simultaneously. In the CICa procedure, a sample was included to determine the [3H]-IP accumulation induced by noradrenaline in the absence of LiCl. Each sample was performed in triplicate.
The reaction was stopped by the addition of 2 ml cold CH3OH/CHCl3/HCl 40 : 20 : 1 (v/v/v) mixture and the samples were sonicated for 45 min at 2–5°C in an ultrasonic water bath. After the addition of 0.63 ml of CHCl3 and 1.26 ml of distilled water, the samples were centrifuged at 1500×g for 10 min to facilitate phase separation. The aqueous layer was removed from the tubes for the [3H]-IP assay. Each sample was neutralized to pH 7–7.5 and run through a column containing Dowex AG 1X8 formate (100–200 mesh, ∼0.5 ml bed volume, Bio-Rad) ion exchange resin, previously equilibrated with 30 ml 10 mM Tris-formate, pH 7.4. The columns were washed with 6 ml distilled water and 6 ml 60 mM sodium formate/5 mM sodium tetraborate to eliminate free myo[3H]-inositol and glycerophosphoinositol, respectively. Total [3H]-IPs were eluted with 3 ml of 0.1 M formic acid in 1 M ammonium formate according to the method of Berridge et al. (1982), and counted for radioactivity. The lipid layer remaining after removal of the aqueous phase was used for measurement of [3H]-phosphatidylinositols. Aliquots of the lipid phase (200 μl) were removed and placed in scintillation vials, allowed to evaporate overnight, and counted for radioactivity in order to calculate total [3H]-inositol incorporated in each sample.
Accumulation of [3H]-IPs was routinely calculated as a percentage (d.p.m.%) of total [3H]-inositol labelled lipids in each individual sample to correct interexperimental variations in label incorporation and sample sizes or was expressed as a percentage over the unstimulated [3H]-IP accumulation (basal). The [3H]-IP accumulation in the presence of the different concentrations of the antagonists was expressed as percentages of the maximum increase obtained in presence of the agonist after subtracting the unstimulated [3H]-IP accumulation (basal).
Concentration-response data for both noradrenaline-induced [3H]-IP accumulation and antagonist inhibition were fitted by non-linear regression plot (Graph Pad Software; San Diego, CA, U.S.A.) and the pEC50 and pIC50 was obtained. The Ki value was calculated from the IC50 estimates according to the Cheng & Prusoff (1973) equation as described above.
Chemicals
The following drugs were obtained from SIGMA (St. Louis MO, U.S.A.): acetylcholine chloride, (−)-noradrenaline bitartrate, prazosin, litium chloride or Research Biochemicals International (Natick, MA, U.S.A.): BMY 7378 (8-[2-[4-(2-Methoxyphenyl)-1-piperazynil-8-azaspiro[4,5]decane-7,9-dione dihydrochloride), 5-methylurapidil. Myo-[3H]-inositol with PT6 was from Amersham (Buckinghamshire, U.K.). Other reagents were of analytical grade. All compounds were dissolved in distilled water.
Statistical analysis
The results are presented as the mean±s.e.mean or 95% confidence intervals for n determinations obtained from different animals. Where ANOVA showed significant differences (P<0.05), the results were further analysed using the Student–Newman–Keuls test and differences were considered significant when P<0.05.
Results
Effects of noradrenaline on contractile force and [3H]-inositol phosphate accumulation in rat tail artery and rat aorta
Noradrenaline (0.1 nM–1 mM) elicited concentration-dependent contractions and [3H]-IP accumulation in both aorta and tail artery (Figure 1). The pEC50 and Emax obtained are summarized in Table 1. The maximal contraction to noradrenaline is similar in both tissues and similar also to the maximal contractile response induced by a depolarizing solution (KCl 80 mM) (Tail: 610.7±33.3 mg, n=4; Aorta: 600.8±48.8 mg, n=6). However, the potency of noradrenaline was significantly higher in aorta than in tail artery (Table 1).
Figure 1.
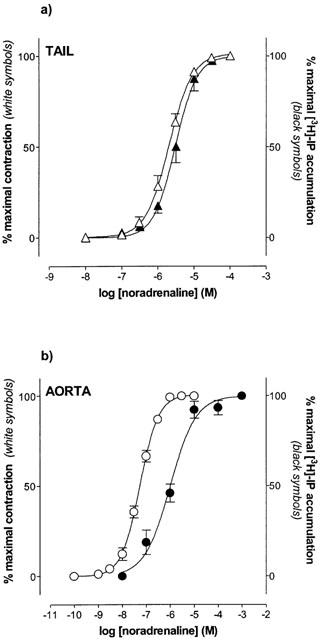
Comparative analysis of concentration-response curves of contraction and [3H]-inositol phosphate accumulation to noradrenaline in tail artery (a) and aorta (b). Data are means (expressed as a percentage of the maximal response to the agonist)±s.e.mean of 5–6 experiments.
Table 1.
pEC50 and Emax values for noradrenaline-induced contraction and [3H]-inositol phosphate accumulation in rat aorta and tail artery
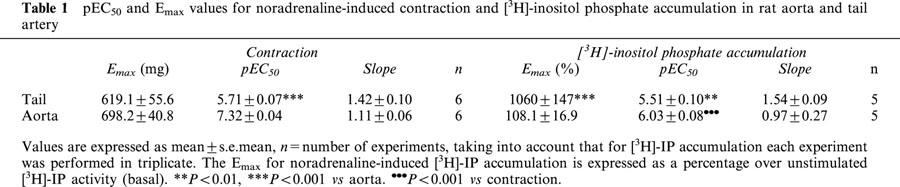
When we analysed the [3H]-IP accumulation as the intracellular signal linked to α1-AR activation, we found that the potency of noradrenaline was also significantly higher in aorta than in tail artery (Table 1) but, this increase in potency was accompanied by a lower efficiency of IP formation because the maximum [3H]-IP accumulation induced by noradrenaline was up to 10 times lower in aorta than in tail artery (Table 1).
These results evidence a close relationship between the contractile force and the [3H]-IP formation induced by noradrenaline in tail artery (Figure 1a). This relationship was not found in rat aorta where the pEC50 value for noradrenaline induced contraction was significantly higher than the pEC50 value for this agonist in stimulating [3H]-IPs formation (Figure 1b, Table 1). In fact, the maximum contractile response was obtained with noradrenaline 1 μM whereas the maximum [3H]-IP accumulation was obtained with noradrenaline 10 μM (Figure 1b).
Effects of α1-adrenoceptor antagonists on noradrenaline-induced contractile response
The pKB values obtained for prazosin (non selective antagonist between the three subtypes), BMY 7378 (α1D-selective) and 5-methylurapidil (α1A-selective) on rat tail artery and rat aorta are summarized in Tables 2 and 3, respectively, and are in the same range of the pA2/pKB values previously obtained by other authors in these tissues (see Table 5).
Table 2.
Functional potencies (pIC50 or pKB) of antagonists against noradrenaline-induced contraction in rat tail artery determined in different experimental conditions
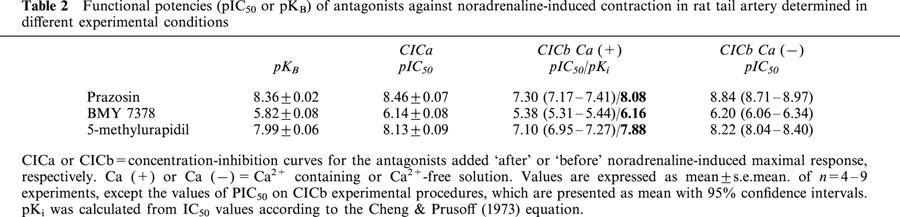
Table 3.
Functional potencies (pIC50 or pKB) of antagonists against noradrenaline-induced contraction in rat aorta determined in different experimental conditions
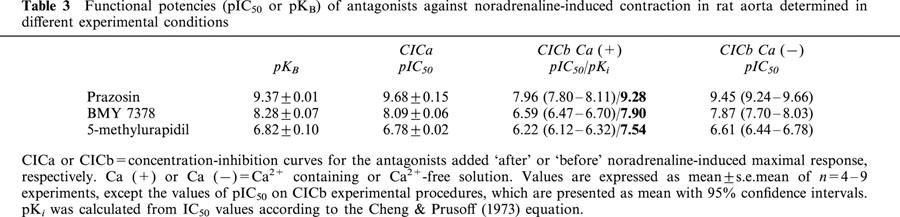
Table 5.
Comparison of published agonist pD2 or antagonist pA2 or pKB* values on rat blood vessels (tail or aorta) with their published pKi values calculated by use of measurements with cloned receptor subtypes (α1A-, α1B- and α1D-adrenoceptors)

The CICb protocol was performed in two experimental conditions: in Ca2+-containing or Ca2+-free solution and the results were different depending on the presence or not of this ion in the incubating medium (Tables 2 and 3, Figure 2). The pIC50 value of each antagonist determined in Ca2+-free medium was similar to the pKB value previously obtained (Tables 2 and 3). However, the pIC50 values determined in Ca2+-containing solution were lower than pKB, but similar values were obtained when the pIC50 was transformed to pKi according to the Cheng & Prusoff (1973) equation. An exception is the case of 5-methylurapidil in rat aorta, since this calculated pKi value from the CICb Ca (+) protocol was approximately one order of magnitude higher than the pKB (Table 3).
Figure 2.
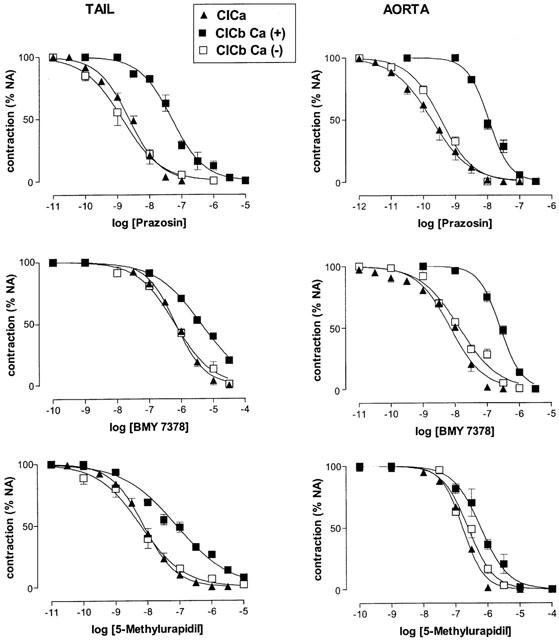
Concentration inhibition curves for prazosin, BMY 7378 and 5-methylurapidil, added after (CICa) or before (CICb) the maximal noradrenaline-induced contraction in tail artery and aorta, in Ca2+-containing (CICb Ca (+)) or Ca2+-free solution (CICB Ca (−)). Data are mean±s.e.mean of 5–9 experiments. The pIC50 values are shown in Tables 2 and 3.
CICa to the antagonists were obtained and shown in Figure 2. The estimate of the antagonist affinity expressed as pIC50 value in this case, was not significantly different from the pKB obtained previously (Tables 2 and 3).
In general, the results obtained evidence that the CICa and CICb obtained in Ca2+-free medium were similar but the CICb performed in Ca2+-containing solution was significantly shifted to the right (Figure 2).
Effects of α1-adrenoceptor antagonists on noradrenaline-induced [3H]-inositol phosphate accumulation
As described in Methods, different concentrations of each antagonist were added for 15 min before (CICb) or after (CICa) an incubation period (30 min) with the agonist as we performed in contraction studies.
A control response to the agonist demonstrates that there were no differences in the noradrenaline-induced [3H]-IP accumulation in both experimental conditions (tail artery: CICa=859.7±105.4% over basal, n=12; CICb= 860.4±75.3% over basal, n=15. Aorta: CICa= 136.0±16.5% over basal, n=9; CICb=126.1±8.5% over basal, n=16). When LiCl was not present during the incubation time in presence of noradrenaline, the accumulation of [3H]-IP due to this agonist was not detectable (n=9–12). The appropriate management of LiCl allowed us to reproduce in [3H]-IP accumulation studies the experimental conditions performed in contractile studies.
Figure 3 and Table 4 show the inhibitory effect of the tested compounds on noradrenaline-induced [3H]-IP formation and, as has been found in contractile studies, there was also a shift to the right in the CICb with respect to the CICa and subsequently, the pIC50 values obtained for the different antagonists were higher in the CICa protocol than in the CICb protocol, except for 5-methylurapidil in the aorta. As happens in contractile studies, application of the Cheng & Prusoff (1973) equation using the pIC50 values determined from the CICb gives pKi values that were similar to the pIC50 determined from CICa (except for 5-methylurapidil in the aorta).
Figure 3.
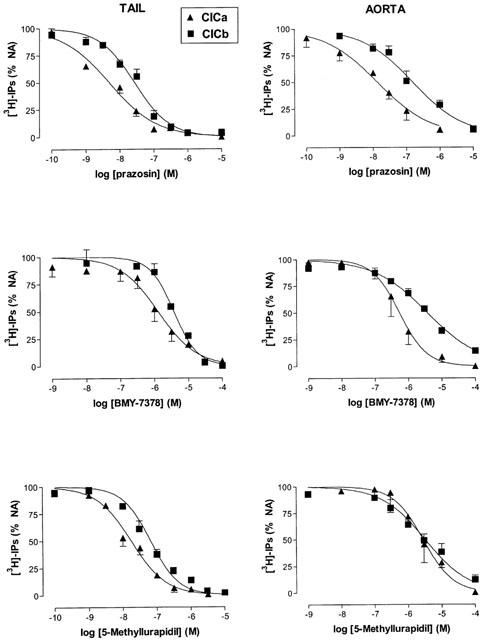
Concentration inhibition curves for prazosin, BMY 7378 and 5-methylurapidil added after (CICa) or before (CICb) the maximal noradrenaline-induced [3H]-inositol phosphate accumulation in tail artery and aorta. Data are mean±s.e.mean of 3–8 separate experiments performed in triplicate. The pIC50 values are shown in Table 4.
Table 4.
Functional potencies (pIC50) of antagonists against noradrenaline-induced [3H]-inositol phosphate accumulation in rat tail artery and aorta determined in different experimental conditions

In rat aorta, the estimated affinities for the three antagonists obtained in the different experimental procedures are lower when IPs accumulation was measured but as previously pointed out, the inhibition curves of the antagonists were performed against a concentration of noradrenaline that elicits the maximal response and this concentration is 10 times higher in IPs determinations (10 μM) than in contractile studies (1 μM).
Discussion
The results obtained show that the contractile response induced by noradrenaline was similar in tail artery and aorta, whereas there were marked differences in the ability of the tissues to generate intracellular second messengers, the α1-ARs present in tail artery being more efficient in increasing IP accumulation. This difference could be explained by the different subtypes implicated in the functional response of each vessel, α1A-AR in tail artery and α1D-AR in aorta (Kenny et al., 1995; Piascik et al., 1995; Buckner et al., 1996; Saussy et al., 1996; Lachnit et al., 1997; Hussain & Marshall, 1997; Hrometz et al., 1999; Gisbert et al., 2000; Ziani et al., 2002). Previous reports with cloned α1A-, α1B and α1D -ARs showed that all of them couple to phospholipase C but there were marked differences in the ability of each subtype to generate intracellular second messengers: the α1A was the most efficiently coupled to calcium release and IP production whereas the α1D was poorly coupled to intracellular signalling cascades (Schwinn et al., 1995; Theroux et al., 1996; Taguchi et al., 1998). Moreover, the experimental evidence that the α1D-ARs present in aorta induced a weak response in second messenger generation (IP accumulation and increase in intracellular Ca2+) compared to that produced by the α1A-ARs present in tail artery (Gisbert et al., 2000; submitted), but the contractile response was similar in the two tissues, suggests the existence of a sensitization process associated with α1D-ARs.
The results also showed that noradrenaline possesses a greater potency in producing contractile response and IP accumulation in aorta although as has been described the maximal response is similar (contraction) or even smaller (IP accumulation) than that obtained in tail artery. Another time this difference could be explained considering the subtype mainly involved in the response, since previous reports showed a higher affinity of noradrenaline for cloned α1D-ARs (see Table 5; Schwinn et al., 1995; Buckner et al., 1996). The constitutive activity of α1D-ARs (Noguera et al., 1996; García-Sáinz & Torres-Padilla, 1999; Gisbert et al., 2000; 2002); McCune et al., 2000; Ziani et al., 2002) would also explain the higher affinity of agonists for this subtype (Piascik & Perez, 2001).
However, it is also interesting to point out that in aorta, the potency of noradrenaline in stimulating IP formation was lower than in producing a contractile response. If we consider the theoretical possibility that, together with the α1D-AR, another α1-AR subtype would intervene in a small proportion of the response of aorta to noradrenaline, for example, the α1A subtype, we can suppose that, according to the above, the different efficiency of coupling between the α1A- and α1D-ARs to IP cascade can ‘apparently increase' the participation of the α1A-ARs with respect to the α1D-AR subtype in the IP signal but not in the contractile response, then, non parallelism must be expected between the concentration-response curves of contraction and IP accumulation with a lower potency of noradrenaline on the latter compared with the former. Following the same reasoning, in tail artery, where the α1A-AR is the subtype mainly responsible for the functional responses, and is also the subtype most efficiently coupled to the IP formation, a little participation of another subtype poorly coupled to the intracellular signal cascades can be uncertain, then the noradrenaline-induced concentration-response curves of contraction or IP accumulation must be identical. The results summarized in Figure 1 validate these theoretical considerations.
According to these results, if different subtypes with different efficiencies of coupling to signals coexist in a tissue, confusing results about participation of each subtype in a given response can be obtained and, in these circumstances, a complete analysis of different functional responses must be done in order to get decisive results.
The experiments performed using selective antagonists show that all of them were able to inhibit the contractile response and the IP production induced by noradrenaline in aorta and tail artery, but essential differences in the antagonist potencies were found depending on the procedure used and on the vessel studied. Another time, a more complex picture appears in aorta than in tail artery. In both tissues, the pKB values obtained (Tables 2 and 3) were similar to that described in the literature (see Table 5). If we compare these values with the pKi obtained in competition binding experiments on cloned α1-ARs (see Table 5), a main role of α1A or α1D-ARs in the contractile response of tail artery (lower pKB values for BMY 7378 and higher for 5-methylurapidil, see Table 2) or rat aorta (higher pKB values for BMY 7378 and lower for 5-methylurapidil, see Table 3) respectively, can be evidenced as has previously been published (Kenny et al., 1995; Lachnit et al., 1997; Saussy et al., 1996; Hussain & Marshall, 1997; Gisbert et al., 2000).
We also performed two other experimental approaches: concentration-inhibition curves obtained using different concentrations of antagonists added before (CICb protocol, see Methods) or after (CICa protocol, see Methods) the maximal noradrenaline-induced contractile response or IP accumulation.
When the contractile response in tail artery and aorta was analysed, the pIC50 values obtained for the different antagonist in the CICa protocol were similar to the pKB obtained in the same tissue. Moreover, the pIC50 obtained from these CICa experiments were similar to the pA2 obtained by other authors (see Table 5). These results are the experimental evidence of a good relationship between the antagonist potencies determined using the CICa procedure and the Schild plot analysis. Previous data obtained in our laboratory where we applied the CICa protocol to study the potencies of α1-AR antagonists (Noguera & D'ocon, 1993; Noguera et al., 1996; Gisbert et al., 2000; 2002; Ziani et al., 2002) but also other compounds such as serotonergic antagonists (ketanserine; Catret et al., 1998), Ca2+ channel blockers (nifedipine, nimodipine, diltiazem, verapamil; Ivorra et al., 1992; Noguera & D'ocon, 1993; Noguera et al., 1997), papaverine and other related benzylsioquinolines and aporphines (Ivorra et al., 1992; 1998; Chuliá et al., 1994; Valiente et al., 1998), confirm this point.
When we performed the CRCb protocol lower pIC50 values were obtained (contractile studies and IPs accumulation), but if we transform these pIC50 to the pKi according to the Cheng & Prusoff (1973) equation the resulting data were similar to the pKB or to the pIC50 values obtained using the CICa protocol. One exception to this was the behaviour of 5-methylurapidil in aorta, where a higher pKi value (approximately an order of magnitude) was obtained in the presence of calcium.
In order to know if the lower pIC50 obtained in the CICb as opposed to CICa experiments was due to just the methodological approach (antagonist addition ‘before' or ‘after' agonist addition), or responded to a more complex scenario in the interaction agonist–antagonist–receptor, we reproduced the CICb procedure in different experimental conditions, excluding Ca2+ from the physiological medium. In this case, the pIC50 values were similar to the pKB or to the pIC50 values obtained using the CICa protocol and a transformation of pIC50 to pKi values is not required.
Three main observations can be made from these results: (1) Similar results can be obtained by different experimental procedures if the mathematical method chosen to calculate the antagonist potency is appropriate. (2) The application of the Cheng & Prusoff (1973) equation when an ‘inhibition curve design' was performed is not a general rule because the pIC50 obtained depends not only on the protocol but also on the experimental conditions used as CICb protocol in presence or absence of calcium show. Why did the Cheng & Prusoff transformation give only rational results when CICb procedure was performed in presence of Ca2+ and is not necessary in the others? This is an open question that needs to be considered at least when drug-receptor interaction analysis of α1-ARs are performed. (3) The CICa experimental procedure offers the simplest alternative method to pharmacologically characterize the main receptor subtype/s implicated in a functional response of a given tissue offering clear advantages with respect to the classical Schild plot analysis, specially when ethical reasons or difficulties in obtaining tissue samples must be considered.
Special attention needs to be given to the behaviour of 5-methylurapidil in rat aorta. This antagonist exhibits a greater potency in the CICb procedure performed in the presence of calcium than in the other experimental methods. This anomalous behaviour, observed in contractile studies as well as in IP accumulation analysis, may reflect changes in the adrenoceptor subtype responsible for the response to noradrenaline in aorta, depending on the experimental procedure used. Taking account of the different localization of α1-AR subtypes (the α1A in the cell membrane and the constitutive α1D in a perinuclear orientation: McCune et al., 2000; Piascik & Perez, 2001; Chalothorn et al., 2002), the greater potency showed by 5-methylurapidil indicates a higher participation of the α1A subtype in the initial response elicited by a maximal concentration of noradrenaline in Ca2+-containing solution, participation that becomes less evident when time permits reaching an equilibrium between subtypes.
In conclusion, a complex interaction between agonists, antagonists and the α1-AR subtypes is evidenced using different experimental procedures, but in order to obtain more rational and simple knowledge of the predominant subtype involved in the functional response of a tissue, the CICa procedure seems to be the best, especially when ethical reasons are invoked. The use of this procedure in contractile studies has previously been established by us and other authors but is new in IP accumulation studies. As present results show, the appropriate management of LiCl allows us to reproduce the experimental conditions performed in the contractile studies increasing the possibilities of this methodology.
Acknowledgments
This work was supported by research grants from the Spanish Comisión Interministerial de Ciencia y Tecnologìa (SAF 2001-2656).
Abbreviations
- AR
adrenoceptor
- BMY 7378
8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl-8-azaspiro[4,5]decane-7,9-dione dihydrochloride
- Ca (+) or Ca (−)=
Ca2+ containing or Ca2+-free solution
- CICb or CICa=
concentration-inhibition curves for the antagonists added ‘before' or ‘after' noradrenaline-induced maximal response respectively
- IP
inositol phosphate
- NA
noradrenaline
References
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERRIDGE M.T., DAWSON R.M.C., DOWNES E.S., HESLOP J.P., IRVINE R.F. Changes in the levels of inositol phosphates after agonist-dependent hydrolysis of membrane phosphoinositides. Biochem. J. 1982;212:473–482. doi: 10.1042/bj2120473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUCKNER S.A., OHEIM K.W., MORSE P.A., KNEPPER S.M., HANDCOK A.A. Alpha 1-adrenoceptor-indued contractility in rat aorta is mediated by the alpha 1D subtype. Eur. J. Pharmacol. 1996;297:241–248. doi: 10.1016/0014-2999(95)00755-5. [DOI] [PubMed] [Google Scholar]
- CATRET M., IVORRA M.D., D'OCON M.P., ANSELMI E. The 5-HT and α-adrenoceptor antagonist effect of four benzylisoquinoline alkaloids on rat aorta. J. Pharm. Pharmacol. 1998;50:317–322. doi: 10.1111/j.2042-7158.1998.tb06867.x. [DOI] [PubMed] [Google Scholar]
- CHALOTHORN D., MCCUNE D.F., EDELMANN S.E., GARCIA-CAZARÍN M.L., TSUJIMOTO G., PIASCIK M.T. Differences in the cellular localization and agonist-mediated internalization properties of the alpha 1-adrenoceptor subtypes. Mol. Pharmacol. 2002;61:1008–1016. doi: 10.1124/mol.61.5.1008. [DOI] [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CHULIÁ S., IVORRA M.D., LUGNIER C., VILA E., NOGUERA M.A., D'OCON P. Mechanism of the cardiovascular activity of laudanosine: comparison with papaverine and other benzylisoquinolines. Br. J. Pharmacol. 1994;113:1377–1385. doi: 10.1111/j.1476-5381.1994.tb17150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRAIG D.A. The Cheng-Prusoff relationship: something lost in the translation. Trends Pharmacol. Sci. 1993;14:89–91. doi: 10.1016/0165-6147(93)90070-z. [DOI] [PubMed] [Google Scholar]
- FAGURA M.S., LYDFORD S.J., DOUGALL I.G. Pharmacological classification of α1-adrenoceptors mediating contractions of rabbit isolated ear artery: comparison with rat isolated thoracic aorta. Br. J. Pharmacol. 1997;120:247–258. doi: 10.1038/sj.bjp.0700917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORD A.P.D.W., DANIELS D.V., CHANG D.J., GEVER J.R., JASPER J.R., LESNICK J.D., CLARKE D.E. Pharmacological pleitropime of the human recombinant α1A-adrenoceptor: implications for α1-adrenoceptors classification. Br. J. Pharmacol. 1997;121:1127–1135. doi: 10.1038/sj.bjp.0701207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARCÍA SÁINZ J.A., TORRES-PADILLA M.E. Modulation of basal intracellular calcium by inverse agonists and phorbol myristate acetate in rat-1 fibroblasts stably expressing α1d-adrenoceptors. FEBS Lett. 1999;443:277–281. doi: 10.1016/s0014-5793(98)01738-4. [DOI] [PubMed] [Google Scholar]
- GISBERT R., NOGUERA M.A., IVORRA M.D., D'OCON P. Functional evidence of a constitutively active population of α1D adrenoceptors in rat aorta. J. Pharmacol. Exp. Ther. 2000;295:810–817. [PubMed] [Google Scholar]
- GISBERT R., ZIANI K., MIQUEL R., NOGUERA M.A., IVORRA M.D., ANSELMI E., D'OCON P. Pathological role of a constitutively active population of α1D adrenoceptors in arteries of spontaneously hypertensive rats. Br. J. Pharmacol. 2002;135:206–216. doi: 10.1038/sj.bjp.0704447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOETZ A.S., KING H.K., WARD S.D., TRUE T.A., RIMELE T.J., SAUSSY D.L. BMY 7378 is a selective antagonist of the D subtype of α1-adrenoceptors. Eur. J. Pharmacol. 1995;272:R5–R6. doi: 10.1016/0014-2999(94)00751-r. [DOI] [PubMed] [Google Scholar]
- HROMETZ S.L., EDELMANN S.E., MCCUNE D.F., OLGES J.R., HADLEY R.W., PEREZ D.M., PIASCIK M.T. Expression of multiple α1-adrenoceptors on vascular smooth muscle: correlation with the regulation of contraction. J. Pharmacol. Exp. Ther. 1999;290:452–463. [PubMed] [Google Scholar]
- HUSSAIN M.B., MARSHALL I. Characterization of α1-adrenoceptor subtypes mediating contractions to phenylephrine in rat thoracic aorta, mesenteric artery and pulmonary artery. Br. J. Pharmacol. 1997;122:849–858. doi: 10.1038/sj.bjp.0701461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IVORRA M.D., D'OCON P., CASSELS B.K., SCHWINN D.A.Structure activity relationships in a series of aporphine derivatives showing selective affinity for human cloned α1A-adrenoceptors Naunyn-Schmiedeberg's Arch. Pharmacol. 1998358Suppl. 2P 6.61 [Google Scholar]
- IVORRA M.D., LUGNIER C., SCHOTT C., CATRET M., NOGUERA M.A., ANSELMI E., D'OCON P. Multiple actions of gluacine on cyclic nucleotide phosphodiesterases, α1-adrenoceptor and benzothiazepine binding site at the calcium channel. Br. J. Pharmacol. 1992;106:387–394. doi: 10.1111/j.1476-5381.1992.tb14345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNY B.A., CHALMERS D.H., PHILPOTT P.C., NAYLOR A.M. Characterization of an α1D-adrenoceptor mediating the contractile response of rat aorta to noradrenaline. Br. J. Pharmacol. 1995;115:981–986. doi: 10.1111/j.1476-5381.1995.tb15907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LACHNIT W.G., TRAN A.M., CLARKE D.E., FORD A.P.D.W. Pharmacological characterization of an α1A-adrenoceptor mediating contractile responses to noradrenaline in isolated caudal artery of the rat. Br. J. Pharmacol. 1997;120:819–826. doi: 10.1038/sj.bjp.0700983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAZARENO S., BIRDSALL N.J.M. Estimation of competitive antagonist affinity from functional inhibition curves using the Gaddum, Schild and Cheng-Prusoff equations. Br. J. Pharmacol. 1993;109:1110–1119. doi: 10.1111/j.1476-5381.1993.tb13737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEFF P., DOUGLAS I.G. Further concerns over Cheng-Prusoff analysis. Trends Pharmacol. Sci. 1993;14:110–112. doi: 10.1016/0165-6147(93)90080-4. [DOI] [PubMed] [Google Scholar]
- MICHEL M.C., KENNY B., SCHWINN D.A. Classification of alpha1-adrenoceptor subtypes. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995;352:1–10. doi: 10.1007/BF00169183. [DOI] [PubMed] [Google Scholar]
- MURAMATSU I., MURATA S., ISAKA M., PIAO H.-L., ZHU J., SUZUKI F., MIYAMOTO S., OSHITA M., WATANABE Y., TANIGUCHI T. α1-adrenoceptor subtypes and two receptor systems in vascular tissues. Life Sciences. 1998;62:1461–1465. doi: 10.1016/s0024-3205(98)00090-3. [DOI] [PubMed] [Google Scholar]
- MURATA S., TANIGUCHI T., MURAMATSU I. Pharmacological analysis of the novel, selective α1-adrenoceptor antagonist, KMD-3213, and its suitability as a tritiated radioligand. Br. J. Pharmacol. 1999;127:19–26. doi: 10.1038/sj.bjp.0702489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCUNE D.F., EDELMANN S.E., OLGES J.R., POST G.R., WALDROP B.A., WAUGH D.J.J., PEREZ D.M., PIASCIK M.T. Regulation of the cellular localization and signaling properties of the α1B and α1D-adrenoceptors by agonists and inverse agonists. Mol. Pharmacol. 2000;57:659–666. doi: 10.1124/mol.57.4.659. [DOI] [PubMed] [Google Scholar]
- NOGUERA M.A., D'OCON M.P. Evidence that depletion of internal calcium stores sensitive to noradrenaline elicits a contractile response dependent on extracellular calcium in rat aorta. Br. J. Pharmacol. 1993;110:861–867. doi: 10.1111/j.1476-5381.1993.tb13892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOGUERA M.A., IVORRA M.D., CHULIA S., D'OCON P. Capacitative Ca2+ entry associated with α1-adrenoceptors in rat aorta. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:83–89. doi: 10.1007/pl00005033. [DOI] [PubMed] [Google Scholar]
- NOGUERA M.A., IVORRA M.D., D'OCON P. Functional evidence of inverse agonism in vascular smooth muscle. Br. J. Pharmacol. 1996;119:158–164. doi: 10.1111/j.1476-5381.1996.tb15689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIASCIK M.T., GUARINO R.D., SMITH M.S., SOLTIS E.E., SAUSSY D.L., JR, PEREZ D.M. The specific contribution of the novel α1D adrenoceptor to the contraction of vascular smooth muscle. J. Pharmacol. Exp. Ther. 1995;275:1583–1589. [PubMed] [Google Scholar]
- PIASCIK M.T., PEREZ D.M. α1-Adrenergic receptors: new insights and directions. J. Pharmacol. Exp. Ther. 2001;298:403–410. [PubMed] [Google Scholar]
- SAUSSY D.L., JR, GOETZ A.S., QUEEN K.L., KING H.K., LUTZ M.W., RIMELE T.J. Structure activity relationships of a series of buspirone analogs at alpha-1 adrenoceptors; further evidence that rat aorta alpha-1 adrenoceptors are of the alpha-1d-subtype. J. Pharmacol. Exp. Ther. 1996;272:134–142. [PubMed] [Google Scholar]
- SCHWINN D.A., JOHNSTON G.I., PAGE S.O., MOSLEY M.J., WILSON K.H., WORMAN N.P., CAMPBELL S., FIDOCK M.D., FURNESS L.M., PARRY-SMITH D.J., PETER B., BAILEY D.S. Cloning and pharmacological characterization of human alpha-1 adrenergic receptors; sequence corrections and direct comparison with other species homologues. J. Pharmacol. Exp. Ther. 1995;272:134–142. [PubMed] [Google Scholar]
- TAGUCHI K., YANG M., GOEPEL M., MICHEL M.C. Comparison of human α1-adrenoceptor subtype coupling to protein kinase C activation and related signaling pathways. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;357:100–110. doi: 10.1007/pl00005143. [DOI] [PubMed] [Google Scholar]
- TESTA R., GUARNERI L., TADDEI C., POGGESI E., ANGELICO P., SARTANI A., LEONARDI A., GOFRIT O.F., MERETYK S., VAINE M. Functional antagonistic activity of Rec 15/2739, a novel alpha-1 antagonist selective for the lower urinary tract, on noradrenaline-induced contraction of human prostate and mesenteric artery. J. Pharmacol. Exp. Ther. 1996;277:1237–1246. [PubMed] [Google Scholar]
- THEROUX T.L., ESBENSHADE T.A., PEAVY R.D., MINNEMAN K.P. Coupling efficiencies of human α1-adrenergic receptor subtypes: titration of receptor density and responsiveness with inducible and repressible expression vectors. Mol. Pharmacol. 1996;50:1376–1587. [PubMed] [Google Scholar]
- VALIENTE M., MARTÍNEZ S., IVORRA M.D., CASSELS B.K., D'OCON P.Functional study on rat aorta and tail artery of a series of aporphine derivatives showing selective affinity for α1A-adrenoceptors Naunyn-Schmiedeberg's Arch. Pharmacol. 1998358Suppl. 2P 6.58 [Google Scholar]
- VAN DER GRAAF P.H., SHANKLEY N.P., BLACK J.W. Analysis of the activity of α1-adrenoceptor antagonists in rat aorta. Br. J. Pharmacol. 1996;118:299–310. doi: 10.1111/j.1476-5381.1996.tb15403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG M., VERFÜRTH F., BÜSCHER R., MICHEL M.C. Is α1D-adrenoceptor protein detectable in rat tissues. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;355:438–446. doi: 10.1007/pl00004966. [DOI] [PubMed] [Google Scholar]
- ZIANI K., GISBERT R., NOGUERA M.A., IVORRA M.D., D'OCON P. Modulatory role of a constitutively active population of α1D-adrenoceptors in conductance arteries. Am. J. Physiol. Heart. Circ. Physiol. 2002;282:H475–H481. doi: 10.1152/ajpheart.00411.2001. [DOI] [PubMed] [Google Scholar]