

Abstract
G protein-linked P2Y nucleotide receptors are known commonly to stimulate the phosphoinositide signalling pathway. However, we have previously demonstrated that the cloned P2Y2, P2Y6 and P2Y1 receptors couple to neuronal N-type Ca2+ channels and to M-type K+ channels. Here we investigate the coupling of recombinant, neuronally expressed rat- and human P2Y4 receptors (rP2Y4, hP2Y4) to those channels.
Rat sympathetic neurones were nuclear-injected with a P2Y4 cDNA plasmid. A subsequent activation of rP2Y4 or hP2Y4 by UTP (100 μM) in whole-cell (ruptured-patch) mode produced only about 12% inhibition of the N-type Ca2+ current (ICa(N)). Surprisingly, in perforated patch mode, UTP produced much more inhibition of ICa(N) (maximally 51%), with an IC50 value of 273 nM. This inhibition was voltage-dependent and was blocked by co-expression of the βγ-binding transducin Gα-subunit. Pertussis toxin (PTX) pretreatment also suppressed ICa(N) inhibition.
UTP inhibited the M-current, recorded in perforated patch mode, by (maximally) 52%, with IC50 values of 21 nM for rP2Y4 and 28 nM for hP2Y4. This inhibition was not affected by PTX pretreatment.
With rP2Y4, ATP inhibited the M-current (IC50 524 nM, 26 times weaker than UTP), whereas ATP had no agonist activity at hP2Y4. This suggests a difference in agonist binding site between rP2Y4 and hP2Y4.
We conclude that, in contrast to other nucleotide receptors studied, the P2Y4 receptor couples much more effectively to M-type K+ channels than to Ca2+ channels. Coupling to the Ca2+ channels involves the βγ-subunits of Gi/o-proteins and requires a diffusible intracellular component that is lost in ruptured-patch recording.
Keywords: Nucleotide receptors, P2Y receptors, P2Y4 receptor, UTP, ion channels, calcium channels, potassium channels, M-current
Introduction
The P2Y receptors are G-protein-linked nucleotide receptors (North & Barnard, 1997). The family of vertebrate P2Y receptors has nine or so members (depending on the assignment made of species orthologues) cloned and recognized (Barnard & Simon, 2001). At the great majority of these, including the four discussed here, the P2Y1, P2Y2, P2Y4 and P2Y6 receptors, their agonists stimulate inositol trisphosphate (IP3) production and mobilization of intracellular Ca2+. P2Y receptor activity has been frequently observed in non-neuronal cells and in recombinant-transfected cell lines (reviewed by Ralevic & Burnstock, 1998), but transduction pathways have not been studied for molecularly identified P2Y receptors in neurones.
An exception to this is that P2Y1, P2Y2 and P2Y6 receptors, when expressed in rat superior cervical ganglion (SCG) neurones by nuclear injection of each cDNA, couple with high agonist potency to both N-type Ca2+ channels and M-type K+ channels (Filippov et al., 1997; 1998; 1999; 2000). Those studies showed that P2Y receptors inhibit the Ca2+ channel current using apparently distinct G-proteins which are either sensitive or insensitive to Pertussis toxin (PTX), while they inhibit the M-channel current using only a PTX-insensitive G-protein. Since these ion channels can control neurotransmitter release and excitability, this modulation of ion channels by nucleotides is likely to be of physiological importance in the brain if these receptors are expressed there. Indeed, P2Y1 receptors are expressed in high abundance in many brain regions and their neuronal localization has been demonstrated (Barnard et al., 1997; Webb et al., 1998a; Moore et al., 2000; Moran-Jimenez & Matute, 2000). In contrast, no evidence was found for P2Y4 expression in brain neurones, although it occurs in some other cell types in the brain (Webb et al., 1998b), including neonatal cortical astrocytes (Lenz et al., 2000). It is therefore of interest to know if it lacks the ability to couple to those neuronal ion channels, a question investigated here in the SCG expression system.
The P2Y4 receptor was cloned first from human placenta (Communi et al., 1995) and from genomic human DNA (Nguyen et al., 1995); a similar sequence was demonstrated in rat heart (Webb et al., 1996; Bogdanov et al., 1998) and was also cloned and expressed from rat brain (Webb et al., 1998b). The rat brain P2Y4 receptor has 84% amino acid sequence identity with human P2Y4 (90% in the combined transmembrane regions and extracellular loops) and 50% homology with the rat P2Y2 receptor. As with the P2Y2 receptor, inositol phosphate accumulation stimulated by P2Y4 was partially sensitive to Pertussis toxin, suggesting a parallel involvement of two distinct G-proteins (Communi et al., 1996). We find now that channel interactions of the P2Y4 receptor provide further information on multiple G-protein pathways for this receptor.
Another interesting feature here is that ATP and UTP are equipotent agonists in the Ca2+ mobilization response of the rat P2Y4 receptor expressed in transfected cell lines, but at the human P2Y4 receptor, ATP is not an agonist and can even be an antagonist (Webb et al., 1998b; Kennedy et al., 2000). The mouse P2Y4 receptor has also been described, and in this respect behaves as the rat receptor (Lazarowski et al., 2001; Suarez-Huerta et al., 2001). We have investigated also whether that agonist specificity applies in the channel coupling of the rat and human P2Y4 receptors.
Methods
cDNA Plasmids
The rat P2Y4 receptor (rP2Y4) cDNA was as described previously (Webb et al., 1998b); the human P2Y4 receptor (hP2Y4; Communi et al., 1995) was a gift from J.M. Boeynaems and B. Robaye (Brussels). Each was cloned into the pcDNA3.1 vector (Stratagene) and checked by re-sequencing. Plasmids were stored at −20°C for injection in sterile 10 mM Tris /1 mM EDTA, pH 8 (TE solution).
Cell isolation and DNA injection
Isolation and injection procedures were similar to those described previously in detail (Filippov et al., 1997; 1998). Briefly, single SCG neurones were isolated from 15–19-day-old rats. Cells were plated on glass coverslips coated with laminin/poly-L-lysine and incubated at 37°C for 4–6 h prior to DNA injection. The plasmid, in sterile TE solution (90–118 ng μl−1), was microinjected into the nucleus. The enhanced green fluorescent (mutant S65T) protein (GFP) cDNA in pcDNA3 (Clontech) was co-injected (10–50 ng μl−1) to identify later the cells with successful expression. Transducin Gα subunit cDNA cloned into pcDNA3 (200 ng μl−1) was also co-injected where stated. These cells were incubated at 37°C for 14–24 h prior to electrophysiological recording.
Electrophysiology and data analysis
Membrane currents were recorded from GFP-labelled neurones at room temperature (20–22°C) in Krebs' solution continuously-flowing at 20–25 ml min−1, using a discontinuous (‘switching') amplifier (Axoclamp 2B) sampling voltage at 6–8 kHz. Drugs were applied in this same perfusing solution (bath exchange rate ⩽1 s); this avoids any change in nucleotide composition due to enzymic action at the surface of the cells. Voltage commands were generated and currents digitized and analysed using ‘pClamp 8' software (Axon Instruments, Foster City, U.S.A.).
Currents through voltage-gated Ca2+ channels (ICa), which in these cells were largely N-type (ICa(N); Filippov et al., 1997), were recorded using the whole-cell (disrupted-patch) method or a perforated patch method as described in detail previously (Filippov et al., 1999). The bathing solution for ICa(N) recordings consisted of (mM): tetraethylammonium (TEA) chloride 120, KCl 3, MgCl2 1.5, BaCl2 5, HEPES 10, glucose 11.1 and 0.5 μM tetrodotoxin (pH adjusted to 7.35 with NaOH). Patch electrodes (2–3 MΩ) were filled with a solution containing (in mM) CsCl 110, MgCl2 3, HEPES 40, EGTA 3, Na2ATP 2, Na2GTP 0.5 (pH adjusted to 7.4 with CsOH). Amphotericin B, 0.125 mg ml−1, was added to the pipette solution for perforated-patch recordings. Currents were routinely evoked every 20 s with a 50–100 ms depolarizing rectangular test pulse to 0 mV from a holding potential of −90 mV. Current amplitudes were measured isochronally 10 ms from the onset of the rectangular test pulse, i.e., near to the peak of the control current. To eliminate leak currents, Co2+ was substituted for Ba2+ in the external solution at the end of each experiment to block all Ca2+ channel currents and the residual current was digitally subtracted from the corresponding currents in Ba2+ solution.
M-type potassium currents, IK(M), were recorded as described in detail previously (Filippov et al., 1998) using a perforated-patch method. Patch pipettes (2–3 MΩ) were filled with a solution containing (mM) potassium acetate 90, KCl 20, MgCl2 3, HEPES 40, BAPTA 0.1, 0.125 mg ml−1 amphotericin B (adjusted to pH 7.4 by KOH). The bathing solution contained (mM) NaCl 120, KCl 3, MgCl2 1.5, CaCl2 2.5, HEPES 10, glucose 11.1 (adjusted to pH 7.3 with NaOH). Neurones were voltage-clamped at −20 mV or −30 mV and M-currents deactivated with 1 s hyperpolarizing steps at 5 s intervals. Current/voltage (I/V) relationships were obtained using incremental voltage steps of 10 mV between −10 and −100 mV; currents were measured at the end of each hyperpolarizing step. For dose/response curves, currents were measured at −30 mV from a steady-state I/V relationship obtained using a ramp voltage command of 20 s from −20 to −90 mV. Currents measured were leak subtracted. The leak component of current was estimated in both cases by extrapolating a linear fit to the I/V relationship from the negative potential region, where only ohmic currents were observed.
Data are presented as means±s.e.mean. Statistical significance was verified by Student's test (P⩽0.05). Dose–response curves were determined using concentrations added cumulatively, with 1 min exposure times. Curves were fitted (using Origin 5.0 software) to pooled data points using the equation y=ymaxxnH/(xnH+KnH) where y=observed % inhibition, ymax=extrapolated maximal % inhibition, x=nucleotide concentration (μM), K=IC50 (μM) and nH=Hill coefficient.
Chemicals
Uridine 5′-triphosphate (UTP) was from Pharmacia Biotech, molecular biology grade; it was 99.5% pure and free from adenine nucleotides. ATP was from Boehringer Mannheim GmbH (Germany) and was freed from nucleotide diphosphates by pre-treatment with creatine kinase/creatine phosphate (Filippov et al., 2000). Purity was verified on samples by HPLC analysis of nucleotides (Sak et al., 2000). Guanosine 5′-triphosphate (GTP), (−)-norepinephrine (noradrenaline) bitartrate, EGTA, BAPTA, amphotericin B were all from Sigma. Oxotremorine-M (OxoM) was from RBI-Sigma, tetrodotoxin from Tocris, Pertussis toxin (PTX) from Porton Products (U.K.), CoCl2 (AnalaR grade) from BDH (U.K.) and BaCl2 and CsCl from Aldrich.
Results
P2Y4 coupling to Ca2+ channels
In contrast to P2Y1, P2Y2 and P2Y6 receptors, in whole-cell mode activation of the rat- or human P2Y4 receptor by the agonist UTP produced only a marginal inhibition of Ca2+ channel current, ICa(N), up to 12.5±3.2% at 100 μM (Figure 1A,C). Expression of P2Y4 receptors did not prevent modulation of ICa(N) by endogenous G-protein linked receptors, since noradrenaline (10 μM) inhibited ICa via α2 receptors by 41.8±4.1% on the same cells.
Figure 1.
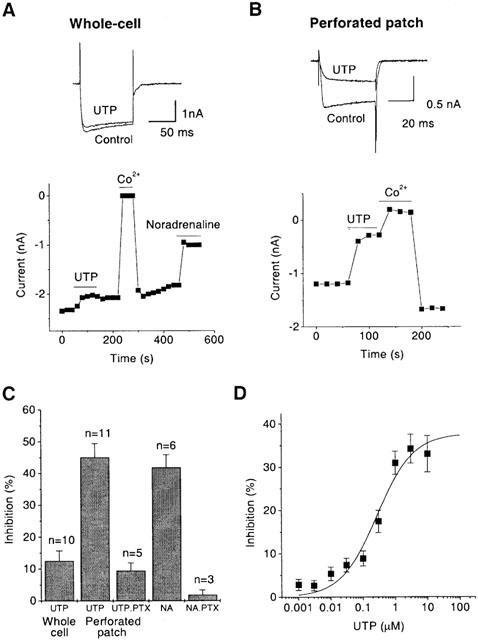
P2Y4 nucleotide receptors expressed in SCG neurones after cDNA injection couple to Ca2+ channels in perforated patch mode but do not couple in whole-cell mode. Ca2+ channel currents (ICa(N)) were recorded by stepping for 50–100 ms every 20 s from −90 mV to 0 mV and leak-corrected by subtracting currents remaining after substituting 5 mM Co2+ for Ba2+. Current amplitude was measured 10 ms from the onset of the test pulse. Records show superimposed leak-subtracted currents in the absence and presence of 100 μM UTP; plots show the time course of changes in current amplitude in whole-cell (ruptured patch) mode (A) and in perforated patch mode (B). Solid bars indicate time of exposure to UTP, Co2+ or 10 μM noradrenaline. (C) Bar charts show the mean inhibition of ICa(N) amplitude by 100 μM UTP and by 10 μM noradrenaline (NA) in neurones pretreated with Pertussis toxin (PTX) and in untreated neurones in whole-cell mode and in perforated patch mode. Bars show s.e.mean; n=number of cells. (D) Concentration-dependence of ICa(N) inhibition by UTP in perforated patch mode. Points show means±s.e.mean of measurements in three cells, concentrations were added cumulatively, with 1 min exposure times. Curves were fitted to pooled data points using Origin 5.0 software to the Hill equation y=ymax.xnH/(xnH+KnH) where y=observed % inhibition, ymax=extrapolated maximal % inhibition, x=UTP concentration (μM), K=IC50 (μM) and nH=Hill coefficient. Values of Hill constants (mean±s.e.mean) were as follows: ymax=37.8±4.3%; K=272.6±129.6 nM; nH=0.8±0.2.
Surprisingly, in perforated patch mode, UTP (100 μM) significantly inhibited the ICa(N) current by 45.1±4.3% (Figure 1B,C). As with the three other P2Y receptors studied before (Filippov et al., 1997; 1999; 2000), the decrease of current was accompanied by a slowed current onset (Figure 1B), suggesting that the inhibition was voltage-dependent. Voltage-dependence was directly confirmed, since a strong depolarizing pre-pulse restored current onset and decreased inhibition from 47.4±4.5% to 21.5±1.5% (Figure 2).
Figure 2.
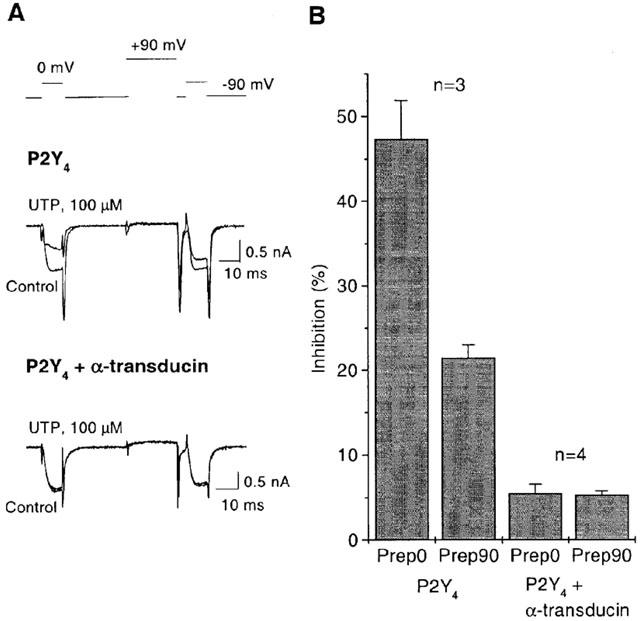
βγ-subunits are responsible for P2Y4 mediated inhibition of Ca2+ channel current. (A) records show superimposed ICa(N) obtained with a double-pulse voltage protocol (top trace) in the absence (control) and presence of 100 μM UTP in cells injected with P2Y4 cDNA alone (upper records) or together with cDNA for transducin (lower records). (B) The bar-charts show the mean per cent current inhibition (measured after 10 ms at 0 mV command potential) produced by UTP before (Prep. 0) and after (Prep. 90) the +90 mV prepulse in cells injected with P2Y4 cDNA alone (two first columns) or together with transducin cDNA (two last columns). Note that the prepulse significantly decreased P2Y4 mediated inhibition of ICa(N) and eliminated the slowing of the kinetics demonstrating voltage-dependence of the effect in cells expressing P2Y4 alone. However practically no ICa(N) inhibition and no voltage-dependence can be seen in cells where βγ-subunit buffering transducin was co-expressed with P2Y4 receptor.
Pertussis toxin (PTX) pretreatment (0.5 μg ml−1, overnight) dramatically decreased ICa(N) inhibition in perforated patch mode, to only 9.4±2.4% (Figure 1C). This effect was almost as great as the abolition of α2-adrenergic inhibition of Ca2+ current by PTX in the same cells (Figure 1C).
Together, the voltage-dependence of P2Y4 mediated ICa(N) inhibition and the effect of PTX suggested the involvement of βγ-subunits of mainly PTX-sensitive Gi/Go proteins (Caulfield et al., 1994; Hille, 1994; Ikeda, 1996; Herlitze et al., 1996; Delmas et al., 1998a, b). To confirm βγ-subunit involvement, we introduced the transducin Gα subunit known to bind βγ-subunits and confirmed to do so to completion when expressed in SCG neurones (Delmas et al., 1999). Co-expression of transducin here almost completely prevented the inhibition of ICa(N) by UTP (Figure 2). Hence, all of the evidence here demonstrated that ICa(N) inhibition via P2Y4 receptors is mediated by Gβγ subunits.
UTP inhibited ICa(N) with IC50 272.6±129.6 nM (Figure 1D). Thus, the potency of P2Y4 coupling to N-type Ca2+ channels is (for native nucleotide agonists) similar to that of the P2Y2 receptor but of the order of 50 times less than that of P2Y1 or P2Y6 (reviewed by Brown et al., 2000).
P2Y4 coupling to M-type K+ channels
As found with the P2Y1, P2Y2 and P2Y6 receptors, activation of the rat- or human P2Y4 receptor by UTP inhibited the M-current (as recorded in perforated patch mode) (Figure 3). In recordings of the M-current (Figure 3A), this inhibition was evident as a decrease of the holding current pre-activated at −20 mV and as a decrease in the amplitude of the deactivation tails after stepping to hyperpolarizing voltages (see Methods). Also, the current measured at the end of the applied hyperpolarizing steps was decreased at all voltages within the M-current activation range (Figure 3B). No voltage dependence of the inhibition occurs. The leak-subtracted current (see Methods) measured at −30 mV was decreased by 52.0±4.1% (Figure 3C). This inhibition was similar to that seen with oxotremorine M (OxoM) acting via endogenous M1 muscarinic receptors. The P2Y4-mediated inhibition was not affected by PTX pretreatment (Figure 3C).
Figure 3.
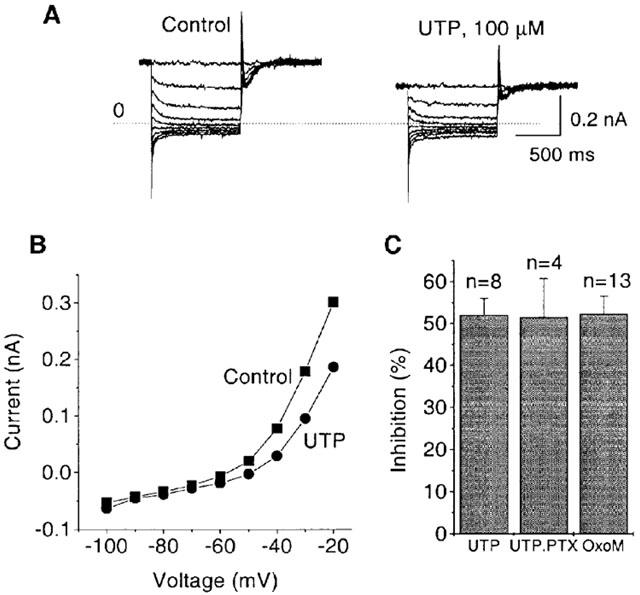
Expressed P2Y4 nucleotide receptors couple to the M-type K+ channels in SCG neurones. (A) traces show the superimposed membrane currents recorded in perforated-patch mode using a standard voltage protocol for M-type K+ current (M-current) measurements (see Methods). M-current was pre-activated by holding the membrane potential at −20 mV, then deactivated with a series of 1 s hyperpolarizing steps in increments of 10 mV at 5 s intervals. The dotted line indicates the zero current. Note that UTP decreased an outward current at the holding potential and reduced the amplitude of the deactivation tails during the steps. (B) The graph shows the current amplitude at the end of each 1 s step measured as change from zero current from records in A. Note that UTP reduced the outward rectification of the current–voltage curve positive to −70 mV. (C) Bar charts show mean inhibition of M-current at −30 mV by 100 μM UTP and by 10 μM oxotremorine-M (OxoM) in P2Y4 expressing neurones. The effect of UTP is shown in neurones pretreated with PTX and in PTX-untreated neurones. The effect of OxoM in PTX-untreated neurones is shown for comparison. M-current was leak-subtracted (see Methods). Bars show s.e.mean; n=number of cells. Note that PTX pretreatment did not affect P2Y4 mediated inhibition of M-current.
The inhibition of the M-current by UTP action showed similar concentration dependence for the rat and human P2Y4 receptors (Figure 4A). IC50 values were 21.5±4.3 nM (rat receptor) and 27.8±4.7 nM (human receptor). ATP was much less effective than UTP on the rat receptor in M-current inhibition (Figure 4A), with an IC50 of 524±273 nM. The greater variability between cells with ATP was unavoidable due to some interference from the response to ATP levels above 1 μM from low but variable levels of endogenous P2X receptors in SCG neurones (Cloues et al., 1993). On the human P2Y4 receptor ATP was inactive as an agonist. For example, 1 μM ATP inhibited the M-current by 17.8±4.7% for rat P2Y4 but by 3.97±2.1% for human P2Y4 (Figure 4B), these values being significantly different (rat) and not significantly different (human) from controls respectively. Although ATP has been reported to be an antagonist (KB 708 nM) at human P2Y4 receptors expressed by transfection into astrocytoma cells (Communi et al., 1996; Kennedy et al., 2000), in the neurones here any antagonist activity of ATP on the human receptor was found to be too weak to measure. Thus, the effect of 1 μM UTP in the presence of 1 μM ATP was indistinguishable from that without ATP (data not shown). A KB value of the order of 10 μM or greater for ATP would not be seen in our cells due to the above-mentioned effect of endogenous P2X receptors.
Figure 4.
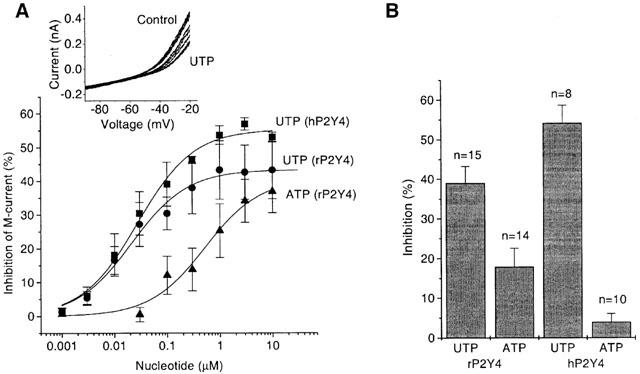
Comparison of UTP- and ATP effects on M-current in neurones expressing rP2Y4- and hP2Y4 receptors. (A) Concentration–dependence of M-current inhibition by UTP in neurones pre-injected with rP2Y4 cDNA or with hP2Y4 cDNA. Also effect of ATP on rP2Y4 is shown. M-current was recorded using a ramp-voltage protocol, leak-subtracted and measured at −30 mV (see Methods). Representative original current records before and after adding increasing concentrations of UTP at hP2Y4 are shown in the inset. Points show means±s.e.mean of measurements in 3–5 cells; concentrations were added cumulatively, with 1 min exposure times. Curves were fitted to pooled data points using Origin 5.0 software to the Hill equation (see legend to the Figure 1D). Values of constants (mean±s.e.mean) were as follows: UTP (hP2Y4): ymax=55.3±1.7%; K=27.8±4.7 nM; nH=0.8±0.1; UTP (rP2Y4): ymax=43.3±1.6%; K=21.5±4.3 nM; nH=0.8±0.1; ATP(rP2Y4): ymax=40.6±5.9%; K=523.7±276.5 nM; nH=0.8±0.2. (B) Bar charts show mean inhibition of M-current at −30 mV by 1 μM UTP and 1 μM ATP in neurones pre-injected with hP2Y4 cDNA, or with rP2Y4 cDNA. Bars show s.e.mean; n=number of cells.
Comparing the actions of preferred agonists on other P2Y receptors in coupling to the M-type K+ channel (reviewed by Brown et al., 2000), the potency at P2Y4 is about three times less than that for the P2Y1 receptor, but it is comparable to that for the P2Y6 receptor and 75 times greater than that for the P2Y2 receptor.
Discussion
The results demonstrate that the P2Y4 receptor can, under appropriate conditions, couple both to N-type Ca2+ channels and to M-type K+ channels. On the basis of these and our previous results on other P2Y receptors (P2Y1, P2Y2 and P2Y6) we can conclude that the ability to couple to these neuronal ion channels and to inhibit them is a common attribute of nucleotide P2Y receptors. However, there is a significant difference between the channel coupling of the P2Y4 receptor and that of the other P2Y receptors studied: the P2Y4 receptor, alone, couples much more effectively to M-type K+ channels than to Ca2+ channels. M-channels are 13 times more sensitive than Ca2+ channels in this case, whereas at other P2Y receptors there was, with a given agonist, either no difference or it was small and in the converse direction (Table 1). Further, the coupling of P2Y4 (but not of the other P2Y receptors ) to the Ca2+ channels is so vulnerable that it can be observed only in perforated patch recording mode, where intracellular composition is preserved, in contrast to the losses by diffusion occurring in whole-cell mode. This indicates that the P2Y4 receptor action to close the Ca2+ channel requires the presence of some intracellular component(s).
Table 1.
Sensitivities of M-type K+ currents (M-current) and N-type Ca2+ currents (ICa(N)) in rat sympathetic neurones to agonists activating expressed P2Y receptors
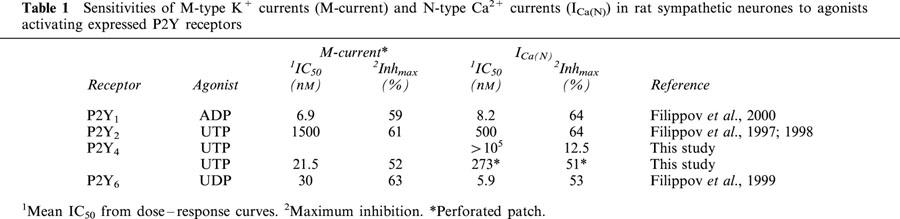
It can also be noted that this contrast between the coupling of the other P2Y receptors to the N-type Ca2+ channel and the lack of it with the P2Y4 receptor, when all are compared in whole cell patch-clamping, confirms that the channel couplings seen in this neuronal expression system are not some artefact of an overloading of a foreign receptor, since those effects are seen to differ greatly between P2Y subtypes. That difference occurs despite the approximately similar levels of P2Y expression in each case, as judged by the rather similar maximum extents of inhibition of the M-channel or Ca2+ channel in all cases (Table 1). Further, both here and with the other P2Y receptors, that maximum is well below 100% and is similar or less than that attainable by activation of the native α2-adrenergic or M1 muscarinic receptors in the same cells (Figures 1C, 3C).
In other cases of rat receptor inhibitory coupling to M-channels, only a voltage-independent effect through a PTX-insensitive G-protein is involved (Brown et al., 1989; Ikeda et al., 1995; Jones et al., 1995). This has been identified as the α-subunit of Gq or G11 (Jones et al., 1995; Haley et al., 1998; 2000). This fits the observations on M-channel inhibition by the P2Y4 receptor and by the other PLC-linked P2Y receptors we have previously studied. In contrast, almost all of the P2Y4 modulation of Ca2+ channels is sensitive to PTX. This, plus its voltage-dependence and its abolition by transducin Gα co-expression, show the involvement of G protein βγ-subunits, in a direct membrane-delimited action on the Ca2+ channel. The parent trimeric G protein is presumed to be one or more of the Go or Gi subtypes, on the basis of the identifications made in parallel behaviour found in rat SCG neurones with several other receptor types (Delmas et al., 1999; Jeong & Ikeda, 2000).
When the P2Y4 receptor was expressed by transfection into astrocytoma cells (Communi et al., 1996) it was reported that its activation (via UTP) of PLC to form IP3 comprises a PTX-sensitive (∼60% ) and a PTX-insensitive component. For the PTX-insensitive component of the same response (due to native P2Y receptors in rabbit smooth muscle cells, activatable by either UTP or ATP ), evidence was obtained to assign it to PLC-β1 coupling via Gαq/11, and to assign the PTX-sensitive component to PLC-β3 coupling via Gβγi3 (Murthy & Makhlouf, 1998). This illustrates how, in our case, the activated P2Y4 receptor could use G-protein βγ subunits in an IP3 pathway and in PTX-sensitive closure of a Ca2+ channel. It has not been proven that the PLC stimulation actually mediates the latter event, although evidence has been provided to suggest that activation of the PLC/IP3/Ca2+ pathway may be involved in the PTX-insensitive inhibition of M current by UTP via native P2Y receptors that is seen in long-term cultures of ganglion cells from newborn rats (Bofill-Cardona et al., 2000). These two responses – PTX-insensitive inhibition of ICa(N) and IK(M) – frequently operate in parallel but may, or may not, use similar downstream signalling pathways (Hille, 1994). However, the initial transduction stages for the P2Y4 receptor must be more complex than has hitherto been described, since they show the first example of a requirement for a diffusible intracellular component as a co-factor of a βγ-subunit effect on Ca2+ channels. Such βγ action would be expected, instead, to be direct and entirely membrane-delimited (reviewed by Hille, 1994; Ikeda, 1996; Kaneko et al., 1999). A broad family of diffusible regulators of G-protein signalling (RGS) is now known (Hepler, 1999), but thus far their actions on channel responses are known only as negative regulations. The identity of the positively-acting factor here will be of interest.
We have observed that, at the rat P2Y4 receptor, both UTP and ATP are agonists in inhibiting the M-current whereas, at the human P2Y4 receptor, UTP is an agonist but ATP is not. This unusual specificity difference is not due to the linkage to this ion channel, since it was found earlier with other transductions, i.e. the mobilization of intracellular Ca2+ in P2Y4-transfected cells (Webb et al., 1998b; Kennedy et al., 2000) and Xenopus oocyte P2Y4 expression (Bogdanov et al., 1998). This indicates a difference within the nucleotide binding site of the rat and human P2Y4 receptors.
With expressed rat P2Y4 receptors UTP was 26 fold more effective than ATP in inhibiting the M-current, but only about 2 fold more effective in mobilization of intracellular Ca2+ (Webb et al., 1998b; Kennedy et al., 2000). The lower ATP potency here cannot be explained by ATP metabolism since we applied fresh nucleotide solutions for each measurement, in continuous and fast superfusion over cells at a low density. Also, UTP was 10–30 times more effective in inhibiting the M-current than in its intracellular Ca2+ response in rat P2Y4-transfected cells, a confirmation that there was no more nucleotide triphosphate degradation in our experiments than in the latter case. The marked difference in the agonist potency ratio for the two actions does not favour PLC-β activation being the direct precursor of the M-channel closure for this receptor (cf. Bofill-Cardona et al., 2000), and further tests of the mechanism of the latter are indicated.
As noted in the Introduction, the P2Y4 receptor has not so far been shown to occur in central neurones, unlike P2Y1 and P2Y6, but is prominent in the brain ventricular system, cardiac and skeletal muscles, some smooth muscles and some other peripheral sites. It is, therefore, not surprising that its coupling to the neurone-specific N-type Ca2+ channel via G-protein βγ-subunits, imposed on it here, is far less efficient than with those other two P2Y receptors, as cited in Results. Its coupling to the M-type K+ channel is, exceptionally, far stronger than to the Ca2+ channel. The M-type channel is now known to be constituted of KCNQ subunits (Wang et al., 1998; Selyanko et al., 1999; Schroeder et al., 2000). Members of that subunit family are prominent in other channels in, e.g., the cardiac sites of P2Y4 receptors. Since all members of this family can be inhibited by Gq/11-coupled receptors (Selyanko et al., 2000; Schroeder et al., 2000), M-channel closure may be a manifestation of the normal non-neuronal role of the P2Y4 receptor; this may explain its ability to couple strongly to the M-channel when introduced in a neurone. This proposal can be tested after co-expressing P2Y4 and each of a series of KCNQ subunits or their combinations in a null host cell line.
Acknowledgments
We thank Christian Lowson for tissue culture. This work was supported by the Wellcome Trust.
Abbreviations
- GFP
green fluorescent protein
- GTP
guanosine 5′-triphosphate
- ICa(N)
N-type Ca2+ current
- IP3
inositol trisphosphate
- I/V
current/voltage
- OxoM
oxotremorine M
- PTX
Pertussis toxin
- hP2Y4
human P2Y4 receptor
- rP2Y4
rat P2Y4 receptor SCG, superior cervical sympathetic ganglion
- UTP
uridine 5′-triphosphate
References
- BARNARD E.A., SIMON J. An elusive receptor is finally caught: P2Y12, an important drug target in platelets. Trends Pharmacol. Sci. 2001;22:388–391. doi: 10.1016/s0165-6147(00)01759-4. [DOI] [PubMed] [Google Scholar]
- BARNARD E.A., SIMON J., WEBB T.E. Nucleotide receptors in the nervous system – an abundant component using diverse transduction mechanisms. Mol. Neurobiol. 1997;15:103–129. doi: 10.1007/BF02740631. [DOI] [PubMed] [Google Scholar]
- BOGDANOV Y.D., WILDMAN S.S., CLEMENTS M.P., KING B.F., BURNSTOCK G. Molecular cloning and characterization of rat P2Y4 nucleotide receptor. Br. J. Pharmacol. 1998;124:428–430. doi: 10.1038/sj.bjp.0701880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOFILL-CARDONA E., VARTIAN N., NANOFF C., FREISSMUTH M., BOEHM S. Two different signaling mechanisms involved in the excitation of rat sympathetic neurons by uridine nucleotides. Mol. Pharmacol. 2000;57:1165–1172. [PubMed] [Google Scholar]
- BROWN D.A., FILIPPOV A.K., BARNARD E.A. Inhibition of potassium and calcium currents in neurones by molecularly-defined P2Y receptors. J. Autonomic Nervous System. 2000;81:31–36. doi: 10.1016/s0165-1838(00)00150-8. [DOI] [PubMed] [Google Scholar]
- BROWN D.A., MARRION N.V., SMART T.G. On the transduction mechanism for muscarine-induced inhibition of M-current in cultured rat sympathetic neurones. J. Physiol. 1989;413:469–488. doi: 10.1113/jphysiol.1989.sp017664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAULFIELD M.P., JONES S., VALLIS Y., BUCKLEY N.J., KIM G.-D., MILLIGAN G., BROWN D.A. Muscarinic M-current inhibition via Gαq/11 and α-adrenoceptor inhibition of Ca2+ current via Gαo in rat sympathetic neurones. J. Physiol. 1994;477:415–422. doi: 10.1113/jphysiol.1994.sp020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLOUES R., JONES S., BROWN D.A. Zn2+ potentiates ATP-activated currents in rat sympathetic neurons. Pflug. Arch. 1993;424:152–158. doi: 10.1007/BF00374606. [DOI] [PubMed] [Google Scholar]
- COMMUNI D., MOTTE S., BOEYNAEMS J.M., PIROTTON S. Pharmacological characterization of the human P2Y4 receptor. Eur. J. Pharmacol. 1996;317:383–389. doi: 10.1016/s0014-2999(96)00740-6. [DOI] [PubMed] [Google Scholar]
- COMMUNI D., PIROTTON S., PARMENTIER M., BOEYNAEMS J.M. Cloning and functional expression of a human uridine nucleotide receptor. J. Biol. Chem. 1995;270:30849–30852. doi: 10.1074/jbc.270.52.30849. [DOI] [PubMed] [Google Scholar]
- DELMAS P., ABOGADIE F.C., DAYRELL M., HALEY J.E., MILLIGAN G., CAULFIELD M.P., BROWN D.A., BUCKLEY N.J. G-proteins and G-protein subunits mediating cholinergic inhibition of N-type calcium currents. Eur. J. Neurosci. 1998a;10:1654–1666. doi: 10.1046/j.1460-9568.1998.00170.x. [DOI] [PubMed] [Google Scholar]
- DELMAS P., ABOGADIE F.C., MILLIGAN G., BUCKLEY N.J., BROWN D.A. βγ dimers derived from Go and Gi proteins contribute different components of adrenergic inhibition of Ca2+ channels in rat sympathetic neurones. J. Physiol. 1999;518:23–36. doi: 10.1111/j.1469-7793.1999.0023r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DELMAS P., BROWN D.A., DAYRELL M., ABOGADIE F.C., CAULFIELD M.P., BUCKLEY N.J. On the role of endogenous G-protein βγ subunits in N-type Ca2+ current inhibition by neurotransmitters in rat sympathetic neurones. J. Physiol. 1998b;495:353–366. doi: 10.1111/j.1469-7793.1998.319bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FILIPPOV A.K., BROWN D.A., BARNARD E.A. The P2Y1 receptor closes the N-type Ca2+ channel in neurones, with both adenosine triphosphates and diphosphates as potent agonists. Br. J. Pharmacol. 2000;129:1063–1066. doi: 10.1038/sj.bjp.0703185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FILIPPOV A.K., WEBB T.E., BARNARD E.A., BROWN D.A. Inhibition by heterologously-expressed P2Y2 nucleotide receptors of N-type calcium currents in rat sympathetic neurones. Br. J. Pharmacol. 1997;121:849–851. doi: 10.1038/sj.bjp.0701270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FILIPPOV A.K., WEBB T.E., BARNARD E.A., BROWN D.A. P2Y2 nucleotide receptors heterologously-expressed in rat sympathetic neurons inhibit both Ca2+ and K+ currents. J. Neurosci. 1998;18:5170–5179. doi: 10.1523/JNEUROSCI.18-14-05170.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FILIPPOV A.K., WEBB T.E., BARNARD E.A., BROWN D.A. Dual coupling of heterologously-expressed rat P2Y6 nucleotide receptors to N-type Ca2+ and M-type K+ currents in rat sympathetic neurones. Br. J.Pharmacol. 1999;126:1009–1017. doi: 10.1038/sj.bjp.0702356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALEY J.E., ABOGADIE F.C., DELMAS P., DAYRELL M., VALLIS Y., MILLIGAN G., CAULFIELD M.P., BUCKLEY N.J., BROWN D.A. The α subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons. J. Neurosci. 1998;18:4521–4531. doi: 10.1523/JNEUROSCI.18-12-04521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALEY J.E., DELMAS P., OFFERMANNS S., ABOGADIE F.C., SIMON M.I., BUCKLEY N.J., BROWN D.A. Muscarinic inhibition of calcium current and M current in Galpha q deficient mice. J. Neurosci. 2000;20:3973–3979. doi: 10.1523/JNEUROSCI.20-11-03973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEPLER J.R. Emerging roles for RGS proteins in cell signalling. Trends Pharmacol. Sci. 1999;20:376–382. doi: 10.1016/s0165-6147(99)01369-3. [DOI] [PubMed] [Google Scholar]
- HERLITZE S., GARCIA D.E., MACKIE K., HILLE B., SCHEUER T., CATTERALL W.A. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;105:113–119. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- HILLE B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- IKEDA S.R. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- IKEDA S.R., LOVINGER D.M., MCCOOL B.A., LEWIS D.L. Heterologous expression of metabotropic glutamate receptors in adult rat sympathetic neurons: subtype-specific coupling to ion channels. Neuron. 1995;14:1029–1038. doi: 10.1016/0896-6273(95)90341-0. [DOI] [PubMed] [Google Scholar]
- JEONG S.W., IKEDA S.R. Effect of G protein heterotrimer composition on coupling of neurotransmitter receptors to N-type Ca2+ channel modulation in sympathetic neurons. Proc. Natl. Acad. Sci. U.S.A. 2000;97:907–912. doi: 10.1073/pnas.97.2.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONES S., BROWN D.A., MILLIGAN G., WILLER E., BUCKLEY N.J., CAULFIELD M.P. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and Gαq/11. Neuron. 1995;14:399–405. doi: 10.1016/0896-6273(95)90295-3. [DOI] [PubMed] [Google Scholar]
- KANEKO S., AKAIKE A., SATOH M. Receptor-mediated modulation of voltage-dependent Ca2+ channels via heterotrimeric G-proteins in neurons. Jpn. J. Pharmacol. 1999;81:324–331. doi: 10.1254/jjp.81.324. [DOI] [PubMed] [Google Scholar]
- KENNEDY C., QI A.D., HEROLD C.L., HARDEN T.K., NICHOLAS R.A. ATP, an agonist at the rat P2Y4 receptor, is an antagonist at the human P2Y4 receptor. Mol. Pharmacol. 2000;57:926–931. [PubMed] [Google Scholar]
- LAZAROWSKI E.R., ROCHELLE L.G., O'NEAL W.K., RIBEIRO C.M., GRUBB B.R., ZHANG V., HARDEN T.K., BOUCHER R.C. Cloning and functional characterization of two murine uridine nucleotide receptors reveal a potential target for correcting ion transport deficiency in cystic fibrosis gallbladder. J. Pharmacol. Exp. Ther. 2001;297:43–49. [PubMed] [Google Scholar]
- LENZ G., GOTTFRIED C., LUO Z., AVRUCH J., RODNIGHT R., NIE W.J., KANG Y., NEARY J.T. P2Y purinoceptor subtypes recruit different Mek activators in astrocytes. Br. J. Pharmacol. 2000;129:927–936. doi: 10.1038/sj.bjp.0703138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORAN-JIMENEZ M.J., MATUTE C. Immunohistochemical localization of the P2Y1 purinergic receptor in neurons and glial cells of the central nervous system. Mol. Brain Res. 2000;78:50–58. doi: 10.1016/s0169-328x(00)00067-x. [DOI] [PubMed] [Google Scholar]
- MOORE D., CHAMBERS J., WALDVOGEL H., FAULL R., EMSON P. Regional and cellular distribution of the P2Y1 purinergic receptor in the human brain: striking neuronal localisation. J. Comp. Neurol. 2000;421:374–384. doi: 10.1002/(sici)1096-9861(20000605)421:3<374::aid-cne6>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- MURTHY K.S., MAKHLOUF G.M. Coexpression of ligand-gated P2X and G protein-coupled P2Y receptors in smooth muscle. Preferential activation of P2Y receptors coupled to phospholipase C (PLC)-β1 via Gαq/11 and to PLC-β3 via Gβγi3. J. Biol. Chem. 1998;273:4695–4704. doi: 10.1074/jbc.273.8.4695. [DOI] [PubMed] [Google Scholar]
- NGUYEN T., ERB L., WEISMAN G.A., MARCHESE A., HENG H.H., GARRAD R.C., GEORGE S.R., TURNER J.T., O'DOWD B.F. Cloning, expression, and chromosomal localization of the human uridine nucleotide receptor gene. J. Biol. Chem. 1995;270:30845–30848. doi: 10.1074/jbc.270.52.30845. [DOI] [PubMed] [Google Scholar]
- NORTH R.A., BARNARD E.A. Nucleotide receptors. Current Opin. Neurobiol. 1997;7:346–357. doi: 10.1016/s0959-4388(97)80062-1. [DOI] [PubMed] [Google Scholar]
- RALEVIC V., BURNSTOCK G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- SAK K., BARNARD E.A., JARV J. Dual effect of nucleotides on P2Y receptors. IUBMB Life. 2000;50:99–103. doi: 10.1080/713803703. [DOI] [PubMed] [Google Scholar]
- SCHROEDER B.C., HECHENBERGER M., WEINREICH F., KUBISCH C., JENTSCH T.J. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- SELYANKO A.A., HADLEY J.K., WOOD I.C., ABOGADIE F.C., DELMAS P., BUCKLEY N.J., LONDON B., BROWN D.A. Two types of K(+) channel subunit, Erg1 and KCNQ2/3, contribute to the M-like current in a mammalian neuronal cell. J. Neurosci. 1999;9:7742–7756. doi: 10.1523/JNEUROSCI.19-18-07742.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SELYANKO A.A., HADLEY J.K., WOOD I.C., ABOGADIE F.C., JENTSCH T.J., BROWN D.A. Inhibition of KCNQ1-4 channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J.Physiol. 2000;522:349–355. doi: 10.1111/j.1469-7793.2000.t01-2-00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUAREZ-HUERTA N., POUILLON V., BOEYNAEMS J., ROBAYE B. Molecular cloning and characterization of the mouse P2Y4 nucleotide receptor. Eur. J. Pharmacol. 2001;416:197–202. doi: 10.1016/s0014-2999(01)00875-5. [DOI] [PubMed] [Google Scholar]
- WANG H.S., PAN Z., SHI W., BROWN B.S., WYMORE R.S., COHEN I.S., DIXON J.E., MCKINNON D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- WEBB T.E., BOLUYT M.O., BARNARD E.A. Molecular biology of P2Y purinoceptors: Expression in rat heart. J. Autonom. Pharmacol. 1996;16:303–307. doi: 10.1111/j.1474-8673.1996.tb00040.x. [DOI] [PubMed] [Google Scholar]
- WEBB T.E., SIMON J., BARNARD E.A. Regional distribution of [35S] 2′-deoxy 5′-O-(1-thio) ATP binding sites and the P2Y1 mRNA within the chick brain. Neuroscience. 1998a;84:825–837. doi: 10.1016/s0306-4522(97)00478-8. [DOI] [PubMed] [Google Scholar]
- WEBB T.E., HENDERSON D.J., ROBERTS J.A., BARNARD E.A. Molecular cloning and characterization of the rat P2Y4 receptor. J. Neurochem. 1998b;71:1348–1357. doi: 10.1046/j.1471-4159.1998.71041348.x. [DOI] [PubMed] [Google Scholar]