

Abstract
This study reports on the identification and characterization of a 1,4-dihydropyridine analogue, 9-(3,4-dichlorophenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydro-1,8(2H,5H)-acridinedione (A-184209) as a novel inhibitor of ATP-sensitive K+ channels.
A-184209 inhibited membrane potential changes evoked by the prototypical cyanoguanidine ATP-sensitive K+ channel opener (KCO) P1075 in both vascular (A10) and urinary bladder smooth muscle cells with IC50 values of 1.44 and 2.24 μM respectively.
P1075-evoked relaxation of 25 mM K+ stimulated aortic strips was inhibited by A-184209 in an apparently competitive fashion with a pA2 value of 6.34.
The potencies of A-184209 to inhibit P1075-evoked decreases in membrane potential responses in cardiac myocytes (IC50=0.53 μM) and to inhibit 2-deoxyglucose-evoked cation efflux pancreatic RINm5F cells (IC50=0.52 μM) were comparable to the values for inhibition of smooth muscle KATP channels.
On the other hand, a structural analogue of A-184209 that lacked the gem-dimethyl substituent, 9-(3,4-dichlorophenyl)-3,4,6,7,9,10-hexahydro-1,8(2H,5H)-acridinedione (A-184208), was found to be a KATP channel opener, evoking membrane potential responses in A10 smooth muscle cells (EC50=385 nM) and relaxing aortic smooth muscle strips (IC50=101 nM) in a glyburide-sensitive manner.
Radioligand binding studies demonstrated that A-184209 displaced SUR1 binding defined by [3H]glyburide binding to RINm5F cell membranes with a Ki value of 0.11 μM whereas A-184208 was ineffective. On the other hand, both A-184209 (Ki=1.34 μM) and A-184208 (Ki=1.14 μM) displaced binding of the KCO radioligand, [125I]A-312110 in guinea-pig bladder membranes with similar affinities.
These studies demonstrate that A-184209 is a novel and structurally distinct compound that inhibits KATP channels in smooth muscle with potencies comparable to glyburide. The structural overlap between DHP openers and blockers, together with their differential interaction with ligand binding sites, support the notion that both openers and blockers bind to similar or very closely coupled sites on the sulfonylurea receptor and that subtle changes in the pharmacophore itself could switch functional properties from KATP channel activation to inhibition.
Keywords: ATP sensitive potassium channel, dihydropyridine, A-184209, urinary bladder, vascular smooth muscle, KATP channel inhibitor, KATP channel opener
Introduction
ATP-sensitive potassium (KATP) channels belong to a family of weak inward rectifying K+ channels. These proteins couple cellular energy metabolism to membrane electrical activity by sensing changes in adenine nucleotide levels, particularly ATP and MgADP that respectively inhibits and activates the channel (Noma, 1983; Nichols & Lederer, 1991). KATP channels have the distinction of being the most widely explored of the K+ channels for their therapeutic potential with channel blockers used in the management of diabetes and structurally diverse openers evaluated in cardiovascular medicine as antihypertensives and cardioprotectants, and more recently, in the control of nonvascular smooth muscle disorders (Longman & Hamilton, 1992; Edwards & Weston, 1996; Coghlan et al., 2001).
Although sulfonylurea analogues such as glyburide and glipizide have proven useful both clinically and as valuable tools, KATP channel blockers are structurally not as diverse as the KATP channel openers. Sulfonylureas such as glyburide interact with high affinity at the sulfonylurea receptor SUR1, and hence are substantially more potent at pancreatic KATP channels compared to cardiac and vascular type channels (Bryan & Aguilar-Bryan, 1999). Besides sulfonylureas, other earlier known blockers such as phentolamine, tetraethylammonium (quaternary ammonium salt), 5-hydroxydecanoate, guanethidine and bretylium, albeit weak and nonselective, have served as pharmacophores for further structural optimization. Imidazoline-based KATP channel blockers including IMID-1M, IMID-26F and IMID-4F that functionally antagonized responses to cromakalim in vascular smooth muscle tissues have been described although these compounds do not displace [3H]-P1075 or [3H]-glyburide binding (Bell et al., 2000). The morpholinoguanidine, U-37783A, reportedly a vascular selective KATP channel blocker, has been shown to inhibit arterial smooth muscle channels more effectively than those from cardiac/skeletal muscle and to possess diuretic and natriuretic activity in animal models (Wellman et al., 1999). More recently, modifications of the sulfonylurea core have led to the identification of the sulfonylthiourea, HMR-1883, that inhibits KATP channels in cardiac muscle cells with 10–50 fold higher potency than at pancreatic KATP channels (Gögelein et al., 2000).
Although the binding sites for blockers that interact with the sulfonylurea receptor remains to be precisely defined, there is evidence to suggest that KATP channel opener and inhibitor sites may be similar or at least very closely coupled. The interaction domains of tolbutamide and glyburide are localized within the transmembrane (12–17) domains of the SUR1 subunit based on the identification of segments required for high affinity tolbutamide inhibition and [3H]-glyburide binding (Uhde et al., 1999; Babenko et al., 1999; Ashfield et al., 1999). Patch clamp studies showed that the transmembrane (12–17) domains of the SUR2 subunit also confer sensitivity to the benzopyran cromakalim and the cyanoguanidine, pinacidil. By analysis of various chimeras, Moreau et al. (2000) showed that a minimal set of two residues within the transmembrane helix 17, Thr1253 and to a lesser extent Leu1249, are determinants of opener sensitivity at SUR2 for structurally distinct KATP channel openers including benzopyran analogues such as SR 47063 and cyanoguandines. More recently, it has also been demonstrated that the enantiomers of a KATP channel modulator PNU-94750 affected channel activity and coupling to MgATP in opposite directions with the (R)-enantiomer inhibiting SUR2–Kir6.2 channels whereas the (S)-enantiomer behaving as a weak opener. Interestingly, these opposite effects were apparently mediated by binding to the same site on the sulfonylurea receptor (Lange et al., 2002).
Exhaustive exploration of the cyanoguanidine chemotype has led to the identification of both openers and blockers of KATP channels that show remarkable structural overlap. For example, PNU-99963 is a potent KATP channel blocker (IC50=18 nM; Khan et al., 1997) of the cyanoguandine class that is structurally related to the prototypical opener, P1075. Analogues belonging to the dihydropyridine series such as ZM-244085 (Li et al., 1996) and A-278637 (Gopalakrishnan et al., 2002b) have been shown to activate KATP channels and suppress smooth muscle contractility in vitro. In the present study, we describe the activator and inhibitory profiles of a pair of 1,4-dihydropyridine analogues at native smooth muscle KATP channels. These studies show that 9-(3,4-dichlorophenyl) -3,3,6,6 - tetramethyl - 3,4,6,7,9,10- hexahydro-1,8(2H,5H)-acridinedione (A-184209; Figure 1), is a KATP channel blocker, whereas its analogue, lacking the gemdimethyl substitutions, 9-(3,4-dichlorophenyl)-3,4,6,7,9,10-hexahydro-1,8(2H,5H)-acridinedione (A-184208), behaves as a KATP channel opener, the latter consistent with previous observations (Frank et al., 1993; Li et al., 1996). The structural overlap between openers and blockers belonging to the dihydropyridine and cyanoguanidine class support the notion that both openers and blockers bind to similar or very closely coupled sites on the sulfonylurea receptor and that subtle changes in the pharmacophore could switch the functional properties from channel activation to channel inhibition.
Figure 1.

Chemical structures of A-184209, A-184208 along with that of glyburide and P1075.
Methods
Materials
Cell culture reagents were purchased from Gibco Life Technologies (Gaithersburg, MD, U.S.A.). DiBAC4(3) was purchased from Molecular Probes (Eugene, OR, U.S.A.). P1075 was synthesized in-house. Other compounds, including glyburide (glibenclamide) were purchased from Research Biochemicals International/Sigma (St. Louis, MO, U.S.A.). Labelled radioligands were purchased from NEN Life Sciences or, in case of [125I]A-312110 [(9R)-9-(4 - fluoro -3-125iodophenyl)-2,3,5,9-tetrahydro - 4H - pyrano[3,4 - b]thieno[2,3-e]pyridin-8(7H)-one1,1-dioxide], custom synthesized in-house. Compounds were prepared as a 10 mM stock in dimethylsulphoxide and stored protected from light until use. Other chemicals and inorganic salts were from Fisher or Sigma.
Cell culture
A10 and RINm5F cell lines were propagated as previously described and plated in 96-well blackwall plates for FLIPR assays or in 24-well plates for cation efflux studies (Miller et al., 1999; Gopalakrishnan et al., 2000). Guinea-pig bladder smooth muscle cells and rat neonatal ventricular myocytes were isolated by enzymatic digestion and plated in 96-well black-wall plates as described (Gopalakrishnan et al., 1999; Whiteaker et al., 2001).
Membrane potential studies
Changes in membrane potential responses were assessed using the bisoxonol dye bis-(1,3-dibutylbarbituric acid) (DiBAC4(3)), an anionic potentiometric probe which partitions between cellular and extracellular fluids in a membrane potential-dependent manner in the Fluorometric Imaging Plate Reader (FLIPR; Sunnyvale, CA, U.S.A.) as previously described (Gopalakrishnan et al., 1999; Miller et al., 1999). Cells were rinsed twice with 200 μl assay buffer (mM: HEPES 20, NaCl 120, KCl 2, CaCl2 2, MgCl2 1, glucose 5, 5 μM DiBAC4(3), pH 7.4, at 25°C) and then incubated in 180 μl of assay buffer in a cell incubator for 1 h to ensure homogeneous dye distribution across the cell membrane. Assays were performed at 37°C and initiated upon addition of varying concentrations of the test compounds. Changes in fluorescence were monitored for 25 min with sampling done at 30 s intervals simultaneously in all 96-wells at excitation and emission wavelengths of 488 and 520 nm respectively. Generally, a second addition of 5 μM glyburide was made after the 25 min period to observe its effects on activatorevoked fluorescence changes and data was collected for an additional 15 min. In studies where the effect of sulfonylureas or other inhibitors were assessed, both the opener and inhibitor were added simultaneously.
Tissue bath studies
The entire thoracic aorta from male Sprague Dawley rats (200–350 g) was removed and immediately placed into Krebs Ringer bicarbonate solution (Composition (mM): NaCl 120, NaHCO3 18.0, dextrose 11.0, KCl 4.7, CaCl2 2.5, MgSO4 1.5, KH2PO4 1.2, K2EDTA 0.01 equilibrated with 5% CO2: 95% O2; pH=7.4 at 37°C adjusted with NaHCO3). The aorta was cleaned of extraneous tissue; endothelium removed, cut into 3–4 mm rings and mounted in 10 ml isolated tissue baths at 37°C. One end was fixed to a stationary glass rod and the other to a Grass FT03 transducer at a basal preload of 1.0 g. Tissues were rinsed every 10 min for a total of 45–60 min. Absence of functional endothelium was confirmed by loss of acetylcholine (10 μM)-evoked relaxation. After an additional 60 min equilibration period, tension was established using 25 mM K+ and a cumulative concentration relaxation response curve was generated for the KATP channel opener, P1075, in the presence of varying concentrations of the inhibitor.
Radioligand binding studies
Binding to RINm5F membranes were performed in Tris HCl buffer with 0.3 nM [3H]-glyburide in an assay volume of 0.5 ml at 22°C as previously described (Gopalakrishnan et al., 1991). Nonspecific binding was determined by the addition of 10 μM unlabelled glyburide. Voltage-dependent L-type calcium channel binding to rat brain membranes was performed using 80 pM [3H]-isradipine and 100 nM unlabelled isradipine to define nonspecific binding. Binding of the KCO radioligand, [125I]A-312110 to guinea-pig bladder membranes was performed in a total 250 μl volume of HEPES buffer containing (in mM) NaCl 139, KCl 5, MgCl2 25, CaCl2 1.25, HEPES 20 (pH 7.4 at 25°C) containing an ATP regenerating system (20 mM creatine phosphate, 50 U creatine phosphokinase and 1 mM Na2ATP) with [125I]A-312110 (1 nM) at 37°C for 90 min (Gopalakrishnan et al., 2002a). Incubations were terminated by rapid vacuum filtration and bound radioactivity was assessed by liquid scintillation or gamma counting.
Data analysis
The concentration-dependence of maximal steady-state changes in fluorescence or changes in tension responses was fitted by nonlinear regression analysis (GraphPad Prism, San Diego, CA, U.S.A.) to obtain EC50 or IC50 values. The Ki values for the compounds were determined by using the Cheng–Prusoff equation, Ki=IC50/(1+[ligand]/KD). Data are expressed as means±standard error.
Results
Inhibition of smooth muscle KATP channels by A-184209
During the course of evaluation of diverse dihydropyridine analogues as potential KATP channel openers, several analogues were found to be inactive in decreasing membrane potential responses, as assessed by DiBAC4(3) fluorescence in the vascular smooth muscle A10 cell line. Closer examination of these agents, to evaluate whether any structural modifications may have converted the openers to blockers, led to the identification of the KATP channel blocker, A-184209 (Figure 1).
A-184209 inhibits KCO-evoked membrane potential effects in smooth muscle cells
The effect of A-184209 on changes in membrane potential responses evoked by a prototypical cyanoguanidine KCO, P1075 was assessed in the vascular smooth muscle A10 cell line. A-184209 inhibited P1075-evoked DiBAC4(3) fluorescence responses in a concentration-dependent manner (Figure 2A). The pD2 value of A-184209 for inhibition of P1075 responses was 5.95±0.22 (IC50=1.44±0.70 μM; n=3). Similarly, in smooth muscle cells isolated from the guinea-pig urinary bladder, A-184209 inhibited P1075-evoked responses, with potency, pD2=5.72±0.26 (IC50=2.24±0.69 μM; n=4) similar to that observed in the A10 cell line. A-184209 is about 3 fold less potent than glyburide in both A10 and bladder smooth muscle cells. The potencies of A-184209 in the membrane potential assays together with those of the prototypical sulfonylurea blocker, glyburide are summarized in Table 1.
Figure 2.

Effects of A-184209 and A-184208 in the A10 vascular smooth muscle cell line. (A) Concentration-dependence of inhibition of P1075-evoked decreases in membrane potential responses (DiBAC4(3) fluorescence) by A-184209. (B) Concentration-dependent decreases in membrane potential responses evoked by A-184208. Data in both cases were normalized to the fluorescence response evoked by 10 μM P1075. The potencies are summarized in Table 1.
Table 1.
Inhibition of KATP channels by A-184209 and glyburide

A-184209 inhibits KCO-evoked relaxation of smooth muscle strips
Further confirmation of the inhibitory properties of A-184209 was assessed using rat aortic smooth muscle strips, a model widely employed to evaluate KATP channel function. As shown in Figure 3, P1075-evoked relaxation of 25 mM K+-stimulated aortic strips was shifted to the right by increasing concentrations of A-184209 in a parallel fashion suggestive of an apparent competitive interaction. Schild analysis of the data to determine the potency of A-184209 yielded a pA2 value of 6.34±0.20 and a slope of 0.87±0.07 (r2=0.94) close to unity. Under similar conditions, glyburide exhibited a pA2 value (7.02±0.14; slope=1.13±0.08; r2=0.92; Table 1).
Figure 3.

Inhibition by A-184209 of P1075 evoked relaxation of aortic smooth muscle strips. Shown are concentration-dependent relaxation of 25 mM K+ stimulated strips by P1075 in the absence (empty symbols) and presence of varying concentrations (0.3–30 μM) of A-184209 (filled symbols). Values depicted are means±s.e.mean of at least three separate determinations. Inset: Schild analysis of the concentration-response data in presence of varying concentrations of A-184209.
Selectivity of A-184209 vs pancreatic and cardiacKATP channels
The observation that A-184209 inhibits smooth muscle KATP channels prompted us to examine its profile in some detail. To assess interactions of A-184209 with other KATP channels, assays utilizing neonatal cardiac myocytes and rat insulinoma RINm5F cells were employed.
In neonatal rat cardiac ventricular myocytes, A-184209 inhibited sarcolemmal KATP channels as assessed by its sensitivity to inhibit P1075-evoked decreases in DiBAC4(3) fluorescence. The potency of A-184209 (IC50 value = 0.53±0.11 μM; n=5) to inhibit cardiac KATP channels is about 12 fold lower than that of glyburide in this assay (IC50=0.045 μM) (Table 1).
Assessment of cation efflux in RINm5F cells that expresses SUR1-containing KATP channels showed that A-184209 inhibited 2-deoxyglucose-oligomycin stimulated 86Rb+ efflux with an IC50 value of 0.52±0.08 μM (n=3), which is some 290 fold lower than that of glyburide (1.8 nM; Gopalakrishnan et al., 2000).
Activation of smooth muscle KATP channels by A-184208
The analogue of A-184209 lacking the gem-dimethyl substitution, A-184208, was found to be an activator of KATP channels (Figure 2B). The −log EC50 value of A-184208 for decreasing membrane potential in A10 cells, as assessed by glyburide-sensitive changes in DiBAC4(3) fluorescence, was 6.46±0.13 (385.7±103 nM; n=4) and the maximal efficacy was comparable to that of P1075. Pretreatment of cells with 5 μM glyburide inhibited the responses of A-184208, indicating that the effects are indeed mediated by opening KATP channels. Consistent with its KATP-channel opening properties, A-184208 also relaxed 25 mM K+ precontracted rat aorta strips with an −log IC50 value of 7.02±0.09 (101±9.5 nM; n=4) and with maximal efficacy (97%) comparable to P1075 (−log IC50=7.72±0.15; n=7). The effects were reversed upon addition of 10 μM glyburide. On the other hand, when tissues were precontracted with 80 mM K+, A-184208 was weakly effective in relaxing aortic strips (Figure 4). This observation is consistent with the K+ channel opening property of this compound and distinct from the profile of a prototypical calcium channel blocker such as nifedipine.
Figure 4.
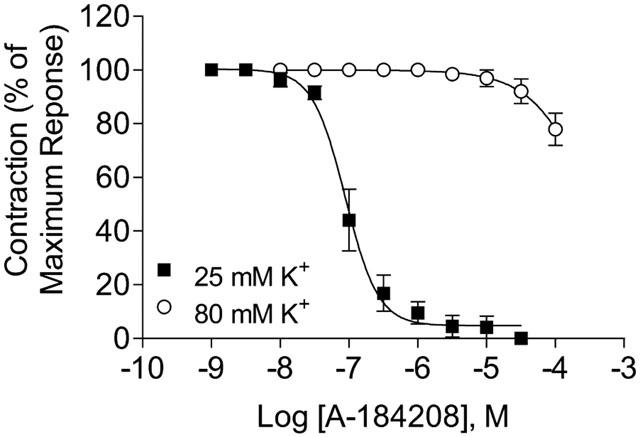
Concentration-dependent inhibition of K+ evoked contractions of aortic smooth muscle strips. Consistent with its KATP channel opening profile, A-184208 suppressed contractions evoked by 25 mM K+ in a glyburide reversible manner, but was ineffective against contractions generated by 80 mM K+.
Displacement of radioligand binding
The observation that A-184209 inhibited smooth muscle, cardiac and pancreatic KATP channels with similar potencies prompted us to examine the nature of KATP channel interactions. KATP channel inhibition could, in principle, be at the level of the sulfonylurea receptor (SUR) itself as in the case of sulfonylurea analogues or via direct blockade of the pore-forming Kir6.2 subunit as suggested for certain imidazoline antagonists (Bell et al., 2000). Since Kir6.2 is a constituent of various KATP channels, it may be possible that the lack of tissue selectivity might potentially be due to its interaction with the inward rectifier alone.
[3H]-Glyburide binding
To determine whether the inhibition of KATP channels involves an interaction with the sulfonylurea receptor, we carried out displacement studies using [3H]-glyburide, a radioligand with high affinity for the SUR1 subunit. A-184209 was found to displace [3H]-glyburide binding to RINm5F membranes with a Ki value of 0.11±0.03 μM (nH=0.95±0.02; n=3). On the other hand, high affinity [3H]-glyburide binding was not displaced (<20% at 10 μM) by the KATP channel opener, A-184208 (Figure 5A).
Figure 5.
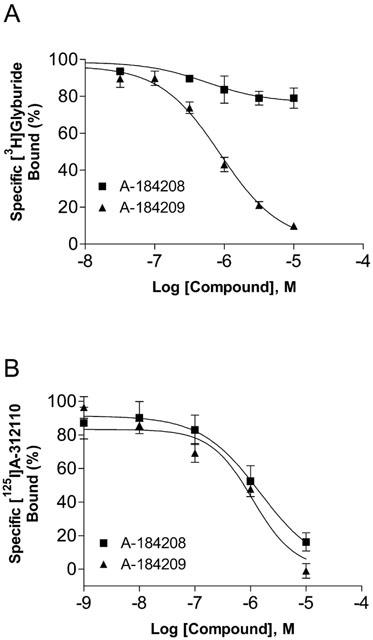
Displacement of [3H]-glyburide (A) and [125I]A-312110 (B) binding by the KATP channel blocker, A-184209 and the opener, A-184208. Assays were carried out as described in Methods and mean Ki values are summarized in Table 1.
[125I]A-312110 binding
To further evaluate interactions with opener binding sites, studies were conducted to assess displacement of binding of a dihydropyridine KATP channel opener, [125I]A-312110 (Gopalakrishnan et al., 2002a). [125I]A-312110 binding was displaced by both A-184209 and A-184208 with similar Ki values of 1.34±0.19 (nH=0.98±0.16) and 1.16±0.04 μM (nH=0.93±0.15) respectively with Hill slopes close to unity (Figure 5B; Table 1).
[3H]-isradipine binding
Since A-184209 and A-184208 are structurally related to the 1,4-dihydropyridine calcium channel antagonists, we assessed interactions with L-type calcium channels by [3H]-isradipine binding. A-184209 and A-184208 did not significantly inhibit [3H]-isradipine binding to rat brain membranes (<30% at 10 μM), consistent with the lack of effect on contractions evoked by 80 mM K+ (Figure 4).
Discussion
The present study describes the characterization of A-184209 belonging to the 1,4-dihydropyridine chemotype as a novel KATP channel blocker. A-184209 inhibited KATP channels in rat aorta and guinea-pig bladder smooth muscle cells with potencies about 3 fold lower than that of glyburide. A-184209 also shifted P1075-evoked relaxation of low K+-stimulated aortic strips and displaced high affinity antagonist ([3H]-glyburide) and opener ([125I]A-312110) binding. A-184209 also inhibited cardiac and pancreatic KATP channels with similar potencies, unlike glyburide which is about 10 and 300 fold more potent at inhibiting cardiac and pancreatic type KATP channels respectively compared to smooth muscle. On the other hand, the analogue lacking the gem-dimethyl substituent, A-184208, behaved as a typical KATP channel opener, decreasing membrane potential responses and relaxing aortic smooth muscle strips in a glyburide-sensitive manner.
Frank et al. (1993) and, subsequently, Li et al. (1996) reported dihydropyridines such as ZM244085 and the corresponding 3-nitro analogue, as openers of KATP channels, devoid of L-type calcium channel activity. More recently, other structurally related KCOs such as A-278637 with improved potencies have been reported (Gopalakrishnan et al., 2002b). During the process of evaluating compounds for KATP channel opening activities, it was revealed that several inactive analogues were, in fact, blockers of KATP channels. A-184209 is a prototypical example, that was ineffective in decreasing membrane potential responses or relaxing tissue strips per se, but did inhibit membrane potential decreases and relaxation responses evoked by the cyanoguanidine KCO, P1075, in smooth muscles. The potencies of A-184209 were similar in both vascular and bladder smooth muscle cells (IC50 ∼1–2 μM), comparable to that of glyburide in A10 (0.6 μM; Miller et al., 1999) and guinea-pig bladder smooth muscle cells (0.9 μM; Gopalakrishnan et al., 2000). Tissue reactivity studies in aortic smooth muscle strips confirmed the KATP channel inhibitory properties of A-184209. As depicted in Figure 3, A-184209 shifted the concentration response curve of P1075 to the right in an apparently competitive manner with a pA2 value of 6.34. By this comparison, A-184209 is about 7 fold less potent than glyburide.
On the other hand, a close analogue of A-184209 that lacks the gem dimethyl-substitution on the cyclohexane ring, A-184208, behaved as a prototypical activator, decreasing membrane potential responses assessed in FLIPR based assays and relaxing low, but not high, K+ stimulated rat aortic strips in a glyburide reversible manner. A-184208 (IC50= 385 nM) is 11 fold more potent than ZM244085 (IC50=4.2 μM), but less potent compared to P1075 (IC50=95 nM) in relaxing rat aortic strips.
To assess whether the interaction of A-184209 shows selectivity vs pancreatic and cardiac KATP channels, we examined its effects on KCO mediated functional responses in pancreatic RINm5F insulinoma and cardiomyocytes. A-184209 was also found to be 12 fold less potent in inhibiting KCO-evoked responses in cardiac myocytes and about 290 fold less potent in pancreatic RINm5F cells compared to glyburide (Gopalakrishnan et al., 2000). Thus, A-184209 exhibits similar potencies for KATP channel inhibition across pancreatic, cardiac and vascular smooth muscle KATP channels, which is distinct from the profile of glyburide.
In principle, the inhibition of KATP channels by A-184209 could occur at the level of the SUR protein or via direct blockade of the inward rectifier K+ channel subunit. Radioligand binding studies showed that A-184209 displaced high affinity [3H]-glyburide binding with Ki values that compare well with their KATP channel inhibitory potencies. In addition, A-184209 also displaced the binding of [125I]A-312110, a high affinity KCO radioligand, with Ki values about 10 fold lower than that for displacement of [3H]-glyburide. These studies collectively demonstrate that A-184209 interacts with both antagonist and opener binding sites on the sulfonylurea receptor, unlike certain other blockers such as phentolamine and cibenzoline that directly block the channel pore by interacting with the inward rectifier alone. The opener, A-184208 effectively displaced [125I]A-312110 binding sites, but not high affinity [3H]-glyburide binding sites. The structural overlap between DHP openers and blockers, together with their differential interaction with ligand binding sites, support the notion that both openers and blockers bind to similar or very closely coupled sites on the sulfonylurea receptor. Our studies also show that subtle changes in the dihydropyridine pharmacophore could switch functional properties from KATP channel activation to inhibition. A recent study of a set of cyanoguanidine analogues have demonstrated that the enantiomers of PNU-94750 altered KATP channel activity and coupling to MgATP in opposite directions with the (R)-enantiomer inhibiting SUR2–Kir6.2 channels whereas the (S)-enantiomer behaved as a weak opener; these opposing effects were apparently mediated by binding to the same site on the sulfonylurea receptor (Lange et al., 2002).
Structural modifications of the 1,4-dihydropyridine pharmacophore have been well recognized as activators or antagonists at the L-type voltage-dependent Ca2+ channel. The differentiation of the activity derives from the nature of the functional groups substituted on the 1,4-dihydropyridine moiety, the stereochemistry at the C-4 position and the voltage-dependence of interaction with the channel (Triggle et al., 1989; 1991). While precise structural determinants of antagonist sensitivities of dihydropyridine class of KATP channel modulators remain to be elucidated, the identification of KATP channel openers and blockers from this series (present study) and the cyanoguanidine series (Khan et al., 1997) represents parallel developments in the medicinal chemistry of KATP channel modulators. In particular, this also illustrates that openers and blockers of both L-type voltage-dependent calcium channels and KATP channels can be identified within the dihydropyridine pharmacophore, with remarkable degrees of selectivity.
Abbreviations
- DHP
dihydropyridine
- FLIPR
Fluorometric Imaging Plate Reader
- KATP channels
ATP-sensitive K+ channels
- KCO
KATP channel opener
- P1075
(N-cyano-N′-(1,1-dimethylpropyl)-N′′-3-pyridylguanidine)
- SUR
sulfonylurea receptor
References
- ASHFIELD R., GRIBBLE F.M., ASHCROFT S.J., ASHCROFT F.M. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the K(ATP)channel. Diabetes. 1999;48:1341–1347. doi: 10.2337/diabetes.48.6.1341. [DOI] [PubMed] [Google Scholar]
- BABENKO A.P., GONZALEZ G., BRYAN J. The tolbutamide site of SUR1 and a mechanism for its functional coupling to K(ATP) channel closure. FEBS Lett. 1999;459:367–376. doi: 10.1016/s0014-5793(99)01215-6. [DOI] [PubMed] [Google Scholar]
- BELL K., FAVALORO J., KHALIL V., ISKANDER M.M., MCPHERSON G.A. The identification of a potent imidazoline-based vascular KATP channel antagonist. Naunyn Schmiedebergs Arch. Pharmacol. 2000;362:145–151. doi: 10.1007/s002100000261. [DOI] [PubMed] [Google Scholar]
- BRYAN J., AGUILAR-BRYAN L. Sulfonylurea receptors: ABC transporters that regulate ATP-sensitive K+ channels. Biochim. Biophys. Acta. 1999;1461:285–303. doi: 10.1016/s0005-2736(99)00164-9. [DOI] [PubMed] [Google Scholar]
- COGHLAN M.J., CARROLL W.A., GOPALAKRISHNAN M. Recent developments in the biology and medicinal chemistry of potassium channel modulators: Update from a decade of progress. J. Med. Chem. 2001;44:1627–1653. doi: 10.1021/jm000484+. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. Recent advances in the pharmacology and therapeutic potential of potassium channel openers. Expert Opin. Invest. Drugs. 1996;5:1453–1464. [Google Scholar]
- FRANK C.A., FORST J.M., GRANT T., HARRIS R.J., KAU S.T., LI J.H., OHNMACHT C.J., SMITH R.W., TRAINOR D.A., TRIVEDI S. Dihydropyridine KATP channel openers. Biorg. Med Chem Lett. 1993;3:2725–2726. [Google Scholar]
- GÖGELEIN H., ENGLERT H.C., KOTZAN A., HACK R., LEHR K.-H., SEIZ W., BECKER R.H.A., SULTAN E., SCHOLKENS B.A., BUSCH A.E. HMR 1098: An inhibitor of cardiac ATP-sensitive potassium channels. Cardiovasc. Drug Rev. 2000;18:157–174. [Google Scholar]
- GOPALAKRISHNAN M., BUCKNER S.A., WHITEAKER K.L., MOLINARI E.J., SHIEH C.-C., SCOTT V.E.S., DAZA A., MILICIC R., DAVIS-TABER I., LIU Y., LIU X., SELLERS D., CHESSWILLIAMS R., CHAPPLE C., WILLIAMS M., SULLIVAN J.P., COGHLAN M.J. A-278637: A Novel ATP-sensitive potassium channel opener with potential for the treatment of overactive bladder I. In vitro characterization. J. Pharmacol. Exp. Ther. 2002b;303:379–386. doi: 10.1124/jpet.102.034538. [DOI] [PubMed] [Google Scholar]
- GOPALAKRISHNAN M., DAVIS-TABER R., MOLINARI E.J., WHITEAKER K.L., BUCKNER S.A., SHIEH C.-C., SCOTT V.E.S., ROTERT G.A., ALTENBACH R.A., COGHLAN M.J., CARROLL W.A. [125I]-A-312110: A novel high affinity 1,4-dihydropyridine ATP-sensitive K+ channel opener: Characterization and pharmacology of binding. FASEB J. 2002a;16:A171.1. doi: 10.1124/mol.64.1.143. [DOI] [PubMed] [Google Scholar]
- GOPALAKRISHNAN M., JOHNSON D., JANIS R.A., TRIGGLE D.J. Characterization of binding of the ATP-sensitive potassium channel ligand, [3H]glyburide, to neuronal and muscle preparations. J. Pharmacol. Exp. Ther. 1991;257:1162–1171. [PubMed] [Google Scholar]
- GOPALAKRISHNAN M., MOLINARI E.J., SHIEH C.C., MONTEGGIA L.M., ROCH J.-M., SCOTT V.E.S., ANDERSON K.L., SULLIVAN J.P., BRIONI J.D. Pharmacological characterization of the human sulfonylurea receptor SUR1 and inward rectifier Kir6.2 stably expressed in HEK-293 cells. British J. Pharmacol. 2000;129:1323–1332. doi: 10.1038/sj.bjp.0703181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOPALAKRISHNAN M., WHITEAKER K.L., MOLINARI E.J., DAVIS-TABER R., SCOTT V.E.S., SHIEH C.-C., BUCKNER S.A., MILICIC I., CAIN J.C., POSTL S., SULLIVAN J.P., BRIONI J.D. Characterization of the ATP-sensitive potassium channels (KATP) expressed in guinea pig bladder smooth muscle cells. J. Pharmacol. Exp. Ther. 1999;289:551–558. [PubMed] [Google Scholar]
- KHAN S.A., HIGDON N.R., HESTER J.B., MEISHERI K.D. Pharmacological characterization of novel cyanoguanidines as vascular KATP channel blockers. J. Pharmacol. Exp. Ther. 1997;283:1207–1213. [PubMed] [Google Scholar]
- LANGE U., LOFFLER-WALZ C., ENGLERT H.C., HAMBROCK A., RUSS U., QUAST U. The stereoenantiomers of a pinacidil analogue open or close cloned ATP-sensitive K+ channels. J. Biol. Chem. 2002;277:40196–40205. doi: 10.1074/jbc.M206685200. [DOI] [PubMed] [Google Scholar]
- LI J.H., YASAY G.D., KAU S.T., OHNMACHT C.J., TRAINOR D.A., BONEV A.D., HEPPNER T.J., NELSON M.T. Studies of the KATP channel opening activity of the new dihydropyridine compound 9-(3-cyanophenyl)-3,4,6,7,9,10-hexahydro-1,8-(2H,5H)- acridinedione in bladder detrusor in vitro. Arzneim-Forsch. 1996;46:525–530. [PubMed] [Google Scholar]
- LONGMAN S.D., HAMILTON T.C. Potassium channel activator drugs: mechanism of action, pharmacological properties, and therapeutic potential. Med. Res. Rev. 1992;12:73–148. doi: 10.1002/med.2610120202. [DOI] [PubMed] [Google Scholar]
- MILLER T.R., DAVIS-TABER R., MOLINARI E.J., WHITEAKER K.L., MONTEGGIA L.M., SCOTT V.E.S., BRIONI J.D., SULLIVAN J.P., GOPALAKRISHNAN M. Pharmacological and molecular characterization of ATP-sensitive K+ channels in the TE671 human medulloblastoma cell line. Eur. J. Pharmacol. 1999;370:179–185. doi: 10.1016/s0014-2999(99)00128-4. [DOI] [PubMed] [Google Scholar]
- MOREAU C., JACQUET H., PROST A.-L., D'HAHAN N., VIVAUDOU M. The molecular basis of the specificity of action of KATP channel openers. EMBO J. 2000;19:6644–6651. doi: 10.1093/emboj/19.24.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLS C.G., LEDERER W.J. Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am. J. Physiol. 1991;261:H1675–H1686. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- NOMA A. ATP-regulated potassium channels in cardiac muscle. Nature (London) 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- TRIGGLE D.J., HAWTHORN M., GOPALAKRISHNAN M., MINARINI A., AVERY S., RUTLEDGE A., BANGALORE R., ZHENG W. Synthetic organic ligands active at voltage-gated calcium channels. Ann. N.Y. Acad. Sci. 1991;635:123–138. doi: 10.1111/j.1749-6632.1991.tb36487.x. [DOI] [PubMed] [Google Scholar]
- TRIGGLE D.J., LANGS D.A., JANIS R.A. Calcium channel ligands: structure-function relationships of the 1,4-dihydropyridines. Med. Res. Rev. 1989;9:123–180. doi: 10.1002/med.2610090203. [DOI] [PubMed] [Google Scholar]
- UHDE I., TOMAN A., GROSS I., SCHWANSTECHER C., SCHWANSTECHER M. Identification of the potassium channel opener site on sulfonylurea receptors. J. Biol. Chem. 1999;274:28079–28082. doi: 10.1074/jbc.274.40.28079. [DOI] [PubMed] [Google Scholar]
- WELLMAN G.C., BARRETT-JOLLEY R., KOPPEL H., EVERITT D., QUAYLE J.M. Inhibition of vascular KATP channels by U-37883A: a comparison with cardiac and skeletal muscle. Br. J. Pharmacol. 1999;128:909–916. doi: 10.1038/sj.bjp.0702868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITEAKER K.L., DAVIS-TABER R., SCOTT V.E.S., GOPALAKRISHNAN M. Fluorescence-based functional assay for sarcolemmal ATP-sensitive potassium channel activation in cultured neonatal rat ventricular myocytes. J. Pharmacol. Toxicol. Methods. 2001;46:45–50. doi: 10.1016/s1056-8719(02)00160-0. [DOI] [PubMed] [Google Scholar]