

Abstract
The principal aim of the present study was to determine whether long-term treatment of human lung mast cells (HLMC) with the clinically-relevant β2-adrenoceptor agonists, salbutamol and terbutaline, leads to desensitization of β2-adrenoceptor-mediated responses in these cells.
The non-selective β-adrenoceptor agonist, isoprenaline, and the selective β2-adrenoceptor agonists, salbutamol and terbutaline, inhibited the IgE-mediated release of histamine from HLMC. Salbutamol (pD2; 7.7±0.3) and terbutaline (pD2; 7.3±0.2) were roughly equipotent as inhibitors of histamine release although both agonists were less potent than isoprenaline (pD2; 8.6±0.2).
Isoprenaline (10−5 M), salbutamol (10−5 M) and terbutaline (10−5 M) enhanced total cell cAMP levels in HLMC over basal by 361±90, 150±38 and 165±35%, respectively.
Long-term exposure (24 h) of HLMC to either salbutamol (10−7 M) or terbutaline (10−7 M) led to a subsequent reduction in the effectiveness of salbutamol and terbutaline (both 10−9–10−4 M) to inhibit histamine release. However, salbutamol was significantly (P<0.05) more effective than terbutaline at promoting the functional desensitization.
Radioligand binding studies, using iodinated cyanopindolol, were performed to determine β2-adrenoceptor density in cell membranes after pretreatment (24 h) of cells with either salbutamol (10−6 M) or terbutaline (10−6 M). Both agonists reduced β2-adrenoceptor density in membranes to about the same extent (∼25% reduction) but these changes in receptor density were not statistically significant (P>0.05).
These data indicate that long-term exposure of mast cells to salbutamol causes greater levels of desensitization to β2-adrenoceptor-mediated responses in HLMC than terbutaline. These findings may have wider clinical significance in the context of asthma treatment as compromised mast cell inhibition could result following long-term exposure of mast cells to short-acting bronchodilators.
Keywords: Mast cells, β2-adrenoceptor, salbutamol, terbutaline
Introduction
Bronchodilator β2-adrenoceptor agonists continue to be critically important in the management of asthma (Tattersfield, 1992; Barnes, 1999). The principal action of these drugs is to relax airway smooth muscle although additional effects may include the stabilization of inflammatory cell activity (Barnes, 1999). In this regard, pulmonary mast cells may serve as an important target for the actions of bronchodilators because β2-adrenoceptor agonists have been shown to inhibit mast cell responses both in vivo and in vitro (Assem & Schild, 1969; Orange et al., 1971; Butchers et al., 1980; Church & Hiroi, 1987; Peachell et al., 1988; Chong et al., 1998).
One potential problem that may disadvantage the continued use of bronchodilators is the development of tolerance. Several clinical studies have shown that tolerance to the mast cell-stabilizing effects of β2-adrenoceptor agonists occurs more readily than tolerance to bronchodilation (O'connor et al., 1992; Cockcroft et al., 1993). These studies are supported by in vitro comparative studies in guinea-pig (van der heijden et al., 1984) and man (Chong & Peachell, 1999) showing that airway smooth muscle is relatively resistant to the development of tolerance when compared to mast cells. In the clinical context, selective tolerance to mast cell stabilization could promote a potentially undesirable situation in which the release of spasmogenic and proinflammatory mediators from mast cells could occur unchecked and masked by the continued symptomatic relief that β2-adrenoceptor agonists would continue to provide by bronchodilation.
At the molecular level, tolerance to β2-adrenoceptor agonists may reflect β2-adrenoceptor desensitization (Ferguson, 2001; Lefkowitz, 1998). A large number of in vitro studies in tissue and cell systems has shown that exposure to β2-adrenoceptor agonists leads to receptor desensitization. Desensitization is an agonist-induced and time-dependent process that may involve, the rapid (within seconds) uncoupling of receptors, followed by sequestration of receptors into intracellular vesicles with longer exposures (hours) leading to receptor degradation (Ferguson, 2001). Phosphorylation of β2-adrenoceptors by cyclic AMP-dependent protein kinase (PKA) and β2-adrenoceptor kinase may contribute to these processes (Benovic et al., 1988; January et al., 1997). Other non-receptor related mechanisms that have been invoked to explain tolerance include the induction of phosphodiesterases (PDE), enzymes that could be involved in the catabolism of cAMP generated by the β2-adrenoceptor, and downregulation of Gsα, the G protein coupling the β2-adrenoceptor with adenylate cyclase (Giembycz, 1996; Finney et al., 2000; 2001).
We have previously reported that tolerance to β-adrenoceptor agonists may be readily induced in human lung mast cells (HLMC). Long-term incubation of HLMC with the non-selective β-adrenoceptor agonist, isoprenaline, leads to a situation in which isoprenaline is, consequently, less effective as an inhibitor of mediator release from these cells (Chong et al., 1997; Drury et al., 1998; Chong & Peachell, 1999). The aim of the present study was to extend these observations and to determine whether long-term treatment of HLMC with alternative, clinically-relevant, β2-adrenoceptor agonists would also induce tolerance in HLMC. In this study, the effects of salbutamol and terbutaline, the two most commonly-prescribed short-acting bronchodilators, have been compared.
Methods
Buffers
Tyrode's buffer contained (mM): NaCl 137, HEPES 1.2, KCl 2.7, NaH2PO4.H2O 0.04, glucose 5.6. Tyrode's-BSA was Tyrode's which additionally contained: CaCl2.2H2O 0.5 mM, MgCl2.6H2O 1 mM, bovine serum albumin (BSA) 1 mg ml−1, DNase 15 μg ml−1. Phosphate buffered saline (PBS) contained (mM): NaCl 137, Na2HPO4.12H2O 8, KCl 2.7, KH2PO4 1.5, CaCl2.2H2O 1, MgCl2.6H2O 1, glucose 5.6, human serum albumin (HSA) 30 μg ml−1. The pH of Tyrode's and PBS buffers was titrated to 7.3. Tris buffer contained (mM): Tris 50, NaCl 154, MgCl2.6H2O 10, EDTA 2. The pH of Tris buffer was titrated to 7.4.
Preparation of compounds
Stock solutions (10 mM) of (−)-isoprenaline bitartrate were prepared daily in 0.05% sodium metabisulphite (dissolved in 0.9% NaCl). Both (±)-salbutamol hemisulphate and (±)-terbutaline hemisulphate were prepared daily as stock solutions (10 mM) in buffer. The β-adrenoceptor antagonists, (±)-propranolol HCl and CGP20712A (2-hydroxy-5-(2-(hydroxy - 3 - (4((1 - methyl- 4-trifluoromethyl)-1-H-imidazol-2-yl)-phenoxy)-propyl)-aminoethoxyl)-benzamide) were prepared daily as stock solutions (10 mM) in buffer. Lyophilized polyclonal goat anti-human IgE antibody was reconstituted in distilled water and stored at 4°C.
Lung tissue
Macroscopically normal tissue from lung resections of patients was obtained following surgery. Most of the patients were undergoing surgery for carcinoma. The male to female split was 70 to 30% and 90% of the patients were white caucasians.
Isolation of mast cells
Mast cells were isolated from human lung tissue by a modification of the method described by Ali & Pearce (1985). The tissue was stripped of its pleura and chopped vigorously for 15 min with scissors in a small volume of Tyrode's buffer. The chopped tissue was washed over a nylon mesh (100 μm pore size; Cadisch and Sons, London, U.K.) with 0.5–1 l of Tyrode's buffer to remove lung macrophages. The tissue was reconstituted in Tyrode's-BSA (10 ml per g of tissue) containing collagenase Ia (350 Units per ml of Tyrode's-BSA) and agitated by using a water-driven magnetic stirrer immersed in a water bath set at 37°C. The supernatant (containing some HLMC) was separated from the tissue by filtration over nylon mesh. The collagenase-treated tissue was then reconstituted in a small volume of Tyrode's-BSA buffer and disrupted mechanically with a syringe. The disrupted tissue was then washed over nylon gauze with Tyrode's-BSA (300–600 ml). The pooled filtrates were sedimented (120×g, room temperature, 8 min), the supernatant discarded and the pellets reconstituted in Tyrode's-BSA (100 ml). The pellet was washed a further two times. The dispersion procedure generated 0.2 to 1×106 HLMC per g of lung tissue at 5 to 20% purity as assessed by alcian blue staining (Gilbert & Ornstein, 1975). These cell preparations were used in histamine release experiments. Mast cell-enriched preparations (43 to 67% purity) were generated by countercurrent elutriation (Beckman J6B centrifuge, JE-5.0 elutriator head) and these preparations were used in some of the radioligand binding studies. Further purification (⩾85%) was achieved by flotation of mast cell-enriched preparations over Percoll density gradients using slight modifications of the methods that have been described in detail elsewhere (Schulman et al., 1982; Ishizaka et al., 1983). Purified mast cells were used in cAMP assays and in some of the radioligand binding studies.
Histamine release
Isolated mast cells were resuspended in PBS buffer for histamine release experiments. Histamine release from HLMC was initiated immunologically with an optimal releasing concentration of anti-IgE (1 : 300). Secretion was allowed to proceed for 25 min at 37°C after which time the cells were pelleted by centrifugation (400×g, room temperature, 3 min). Histamine released into the supernatant was determined by a modification (Ennis, 1991) of the automated fluorometric method of Siraganian (1974). When an inhibitor was employed, cells were incubated with the inhibitor for 10 min at 37°C before the addition of stimulus and then samples were processed as indicated above. Total histamine content was determined by lysing aliquots of the cells with 1.6% perchloric acid. Cells incubated in buffer alone served as a measure of spontaneous histamine release (<6%). Histamine release was thus expressed as a percentage of the total histamine content after subtracting the spontaneous histamine release.
When long-term incubations were performed, RPMI 1640 buffer supplemented with penicillin/streptomycin (10 μg ml−1) and gentamicin (50 μg ml−1) was employed. Cells were incubated (24–48 h) at a density of 0.1×106 HLMC per ml in 6-well plates with, usually, 0.5×106 HLMC per condition with or without a β-adrenoceptor agonist. After completion of the incubations, the cells were washed three times with PBS and reconstituted in the same buffer for mediator release experiments. Incubations of HLMC with the β-adrenoceptor agonists had no effect on either the total number of HLMC recovered, the total histamine content or the spontaneous histamine release compared to HLMC incubated in buffer.
Assays for cyclic AMP
Total cell cAMP levels were monitored according to methods that have been described in more detail elsewhere (Chong et al., 1998). Cells were incubated (10 min) without or with a β-adrenoceptor agonist and the reaction terminated by the addition of ice-cold acidified ethanol and snap-freezing of samples in liquid nitrogen. After thawing, samples were pelleted by centrifugation, supernatants saved and the ethanol evaporated using a rotary evaporator. Samples were reconstituted in assay buffer and cAMP levels were determined using commercially-available EIA kits.
Radioligand binding
Membrane fractions were prepared from mast cell-enriched (54±5%) and purified (⩾90% purity) mast cell preparations (⩾3×106 cells per condition) after treatment (24 h) with buffer or a given β-adrenoceptor agonist. Membranes were prepared by homogenizing in ice-cold Tris buffer using an Ultra Turrax homogenizer for 20 s followed by four strokes (×4) of a Teflon homogenizer. The homogenate was centrifuged (500×g, 40 min), the supernatant was harvested and subjected to further centrifugation (40,000×g, 15 min) in an ultra-centrifuge (L80, Beckman). The pellet was washed and the high-speed centrifugation step repeated. The pellet was resuspended in Tris buffer and used in receptor binding assays. All procedures were carried out at 4°C. In saturation binding assays, the membrane preparations were assayed for β-adrenoceptor binding sites using [125I]-cyanopindolol ([125I]-CYP). Membrane suspensions (100 μl) were incubated (1 h, 37°C) using a range of radioligand concentrations (0.03125–2 nM) in a total volume of 250 μl. Non-specific binding was determined by displacement with propranolol (1 μM). Specific binding, expressed as a percentage of the total binding, ranged from 69 to 78% at a [125I]-CYP concentration of 0.0625 nM. Additions of ice-cold Tris buffer were used to terminate the reactions followed by rapid filtration through Whatman GF/B glass fibre filters. The filters were rapidly washed four times with 3 ml ice-cold buffer and the radioactivity remaining on filters measured in a Packard Cobra auto-gamma counter. All binding experiments were performed in duplicate. Protein content of the membranes was determined by the method of Lowry et al. (1951).
Materials
The following were purchased from the sources indicated; anti-human IgE, BSA, collagenase, DNase, HSA, Percoll, isoprenaline, salbutamol, terbutaline, propranolol (all Sigma, Poole, U.K.); [125I]-CYP (New England Nuclear, Stevenage, U.K.); cAMP EIA kits (Amersham, Little Chalfont, U.K.). CGP20712A was kindly supplied as a gift from Ciba-Geigy (Basel, Switzerland).
Data analysis
Maximal responses (Emax) and potencies (pD2) were determined by computer-assisted curve fitting (GraphPad Prism, version 2). The equation fitted was:
where Emin is the basal response, X is the logarithm of the agonist concentration and n is the Hill slope. Receptor densities (Bmax) and dissociation constants (KD) were determined by non-linear regression analysis (GraphPad Prism, version 2) using the following equation:
where X is the concentration of radioligand. To determine whether there was any difference in the responses of mast cells to drugs after treatments either paired t-tests or repeated measures ANOVA was performed.
Results
Effects of β-adrenoceptor agonists on mast cells
The effects of isoprenaline, terbutaline and salbutamol (all at 10−10–10−5 M) on the IgE-mediated release of histamine from HLMC were investigated (Figure 1). All three β-adrenoceptor agonists inhibited histamine release in a concentration-dependent manner. Terbutaline and salbutamol were essentially equipotent as inhibitors of histamine release but some 10 fold less potent than isoprenaline (see Table 1 for pD2 values). Both terbutaline and salbutamol displayed variable intrinsic activities among HLMC preparations acting as full agonists, relative to isoprenaline, in five (for salbutamol) and nine (for terbutaline) experiments out of 23. Overall, terbutaline was slightly more efficacious than salbutamol (see Table 1 for Emax val ues). There was a good correlation (r=0.78) between the intrinsic activities of terbutaline and salbutamol among HLMC preparations (Figure 2).
Figure 1.
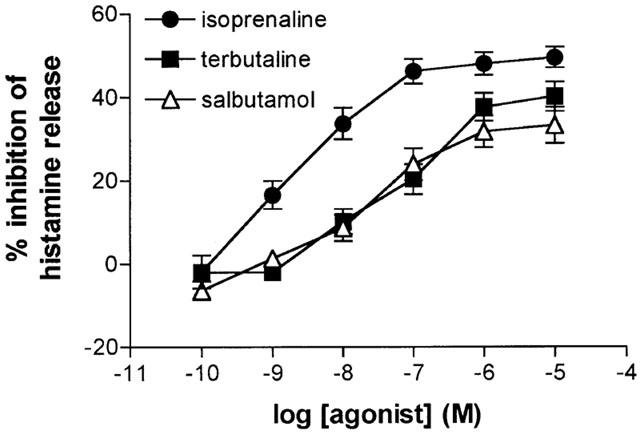
Inhibition of histamine release by β-adrenoceptor agonists. HLMC were incubated for 10 min without or with either isoprenaline, salbutamol or terbutaline before challenge with anti-IgE (1 : 300) for 25 min for histamine release. Values are expressed as the per cent inhibition of the control histamine release which was 30±3%. Values are means±s.e.mean from 23 experiments.
Table 1.
Emax and pD2 values for the inhibition of histamine release from human lung mast cells by β-adrenoceptor agonists

Figure 2.
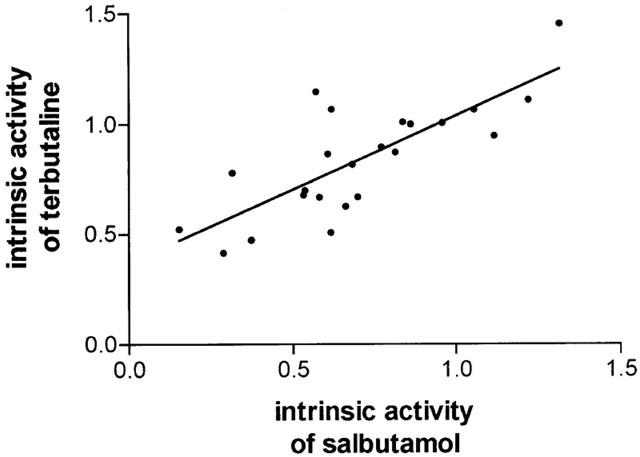
Correlation of the intrinsic activities of salbutamol and terbutaline. Intrinsic activities for these agonists were calculated relative to isoprenaline and by (Emax for agonist/Emax for isoprenaline) for each of the 23 experiments represented by Figure 1. Each point represents an individual experiment. The correlation coefficient is 0.78 (P<0.0001).
The effects of isoprenaline, salbutamol and terbutaline (all at 10−5 M) on cAMP generation in HLMC were determined (Figure 3). Terbutaline and salbutamol generated similar levels of total cAMP in these cells and about a half of the amount generated by isoprenaline.
Figure 3.
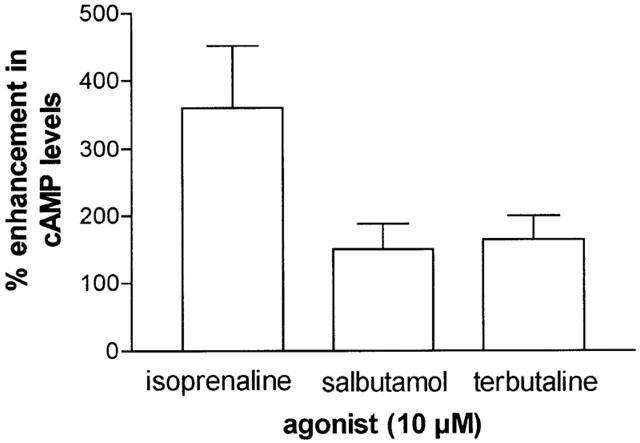
Elevations in cAMP induced by β-adrenoceptor agonists. HLMC (91±6% purity) were incubated without or with an agonist (10−5 M) for 10 min and total cell cAMP levels were determined. Values are expressed as the per cent enhancement in cAMP over basal levels which were 0.4±0.1 pmol per 106 cells. Isoprenaline caused statistically (P<0.05) larger increases in cAMP than both salbutamol and terbutaline. Values are means±s.e.mean from four experiments.
Desensitization induced by β-adrenoceptor agonists
We have previously reported that long-term (24 h) incubation of HLMC with isoprenaline (10−6 M) impairs the subsequent effectiveness of isoprenaline to inhibit histamine release (Chong et al., 1997; Drury et al., 1998; Chong & Peachell, 1999). In the present study, we determined the effects of long-term (24 h) treatment of HLMC with sub-maximal and equieffective concentrations of either salbutamol (10−7 M) or terbutaline (10−7 M) on the subsequent ability of salbutamol and terbutaline (10−9–10−4 M) to inhibit histamine release (Figure 4). Long-term treatment of HLMC with salbutamol and terbutaline reduced the maximal inhibitory effects (Emax) of these two agonists although only salbutamol caused statistically significant (P<0.01) changes (see Table 2). The extent of desensitization (calculated as [1–Emax after treatment/Emax with no treatment]×100) induced by exposure to salbutamol and terbutaline of the inhibition by salbutamol was 64±8% and 29±7%, respectively, and salbutamol caused significantly greater levels of desensitization than terbutaline (P<0.005). Similarly, the extent of desensitization induced by salbutamol and terbutaline of the inhibition by terbutaline was 45±10% and 19±6%, respectively, and salbutamol caused greater levels of desensitization than terbutaline (P<0.05).
Figure 4.
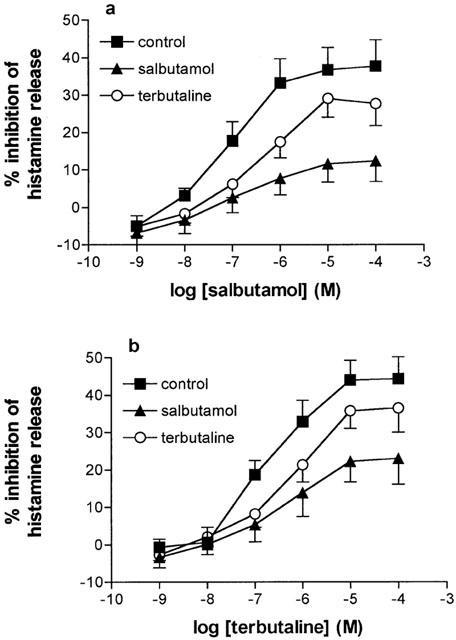
Functional desensitization of β-adrenoceptor agonist-mediated responses in mast cells. HLMC were incubated (24 h) without (control) or with either salbutamol (10−7 M) or terbutaline (10−7 M) and then washed extensively. Cells were then incubated without or with either salbutamol (a) or terbutaline (b) for 10 min before challenge with anti-IgE (1 : 300) for histamine release. Values are expressed as the per cent inhibition of the control histamine releases which were 34±4, 30±2 and 31±3% following 24 h treatments with buffer, salbutamol and terbutaline, respectively. Values are means±s.e.mean from 14 experiments.
Table 2.
Emax and pD2 values for salbutamol and terbutaline following desensitizing treatments

The effects of a longer incubation (48 h) with salbutamol (10−7 M) and terbutaline (10−7 M) and, for comparative purposes, isoprenaline (10−7 M), on the subsequent inhibitory effects of isoprenaline (10−6 M) were determined (Figure 5). There was a modest increase in the extent of desensitization induced by salbutamol and terbutaline at 48 h compared to 24 h and a slight reduction in the extent of desensitization observed with isoprenaline following the longer incubation although none of these time-dependent differences were significant (P>0.05).
Figure 5.
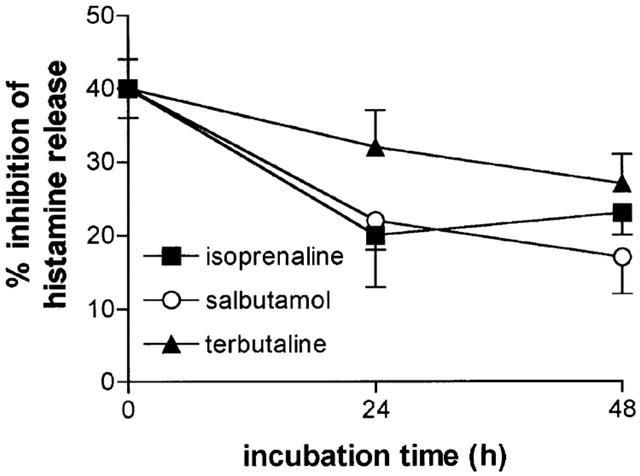
Time-dependence of the functional desensitization. Cells were incubated either in buffer or for 24 or 48 h with either isoprenaline (10−7 M), salbutamol (10−7 M) or terbutaline (10−7 M) before extensive washing. The cells were then incubated without or with isoprenaline (10−6 M) for 10 min before challenge with anti-IgE (1 : 300) for 25 min for histamine release. Values show the extent of isoprenaline (10−6 M) inhibition following the treatments. Values are per cent inhibition of the control histamine releases which ranged from 44±4 to 48±6%. Values are means±s.e.mean from 10 experiments except for isoprenaline which was studied in seven of these 10 experiments.
In order to establish whether long-term treatments affected the responses to alternative cAMP-active agents, HLMC were incubated (24 h) without (control) or with either salbutamol, terbutaline or isoprenaline (all at 10−7 M) and then the effects of prostaglandin E2 (PGE2), a receptor-mediated activator of adenylate cyclase, forskolin, a direct activator of adenylate cyclase, isobutyl-methylxanthine (IBMX), an inhibitor of phosphodiesterases, and the β-adrenoceptor agonists, isoprenaline and clenbuterol, evaluated (Table 3). Long-term exposure of HLMC to salbutamol reduced the inhibitory effects of forskolin and IBMX but did not affect the inhibition by PGE2. Of these agents, the effects of IBMX alone were reduced by long-term treatment with terbutaline (and isoprenaline). However, treatment with salbutamol was significantly (P<0.05) more effective at reducing the degree of inhibition seen with IBMX than terbutaline (and isoprenaline). Long-term treatment of HLMC with salbutamol (and isoprenaline) reduced the inhibitory effects of both clenbuterol and isoprenaline to a greater extent than long-term terbutaline.
Table 3.
Effects of desensitizing treatments of cAMP-active compounds

In order to establish whether the effects of β-adrenoceptor agonists on IBMX could be observed with alternative PDE inhibitors, theophylline was studied (n=4). Long-term treatment (24 h) with either buffer (control), salbutamol or terbutaline led to inhibition by theophylline (1 mM) of IgE-mediated histamine release of 61±6%, 32±5%, and 43±4%, respectively. Both agonists reduced the effectiveness of theophylline to inhibit but only salbutamol did to a significant extent (P<0.05).
To investigate further the potential interactions of β-adrenoceptor agonists with alternative cAMP-active agents, the effects of long-term treatment (24 h) of HLMC with increasing concentrations of salbutamol (10−8–10−6 M) on the inhibitory effects of salbutamol, PGE2, forskolin and IBMX were evaluated (Figure 6). Incubation (24 h) of HLMC with increasing concentrations of salbutamol led to a concentration-dependent reduction in the subsequent inhibitory effects of salbutamol with complete abrogation of inhibition following long-term incubation with 10−6 M salbutamol. By contrast, although the inhibitory effects of PGE2, forskolin and IBMX were attenuated to some degree by long-term treatment with the β-adrenoceptor agonist, these agents were not influenced to the same extent as salbutamol. Moreover, there was no concentration-dependent relationship following desensitizing treatments as the degree of attenuation of the inhibitory effects of PGE2, forskolin and IBMX was similar following long-term incubation with the lowest (10−8 M) and highest (10−6 M) concentrations of salbutamol.
Figure 6.
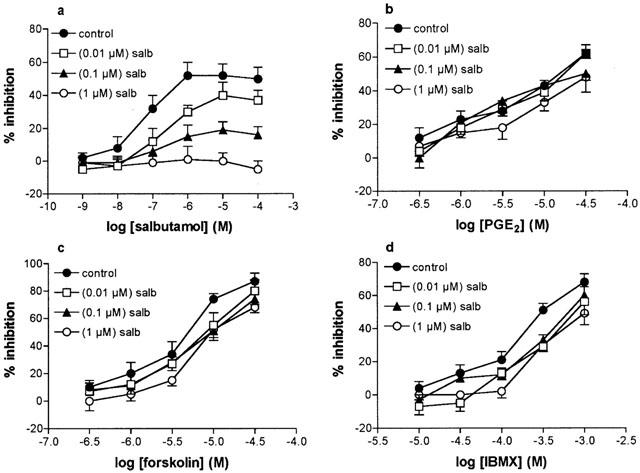
Effect of salbutamol treatment on cAMP-active compounds. Cells were incubated (24 h) in buffer (controls) or with increasing concentrations of salbutamol (salb) of 10−8 M, 10−7 M or 10−6 M and then extensively washed. Cells were then incubated for 10 min without or with either salbutamol (a), PGE2 (b), forskolin (c) or IBMX (d) for 10 min before challenge with anti-IgE for 25 min for histamine release. Values are expressed as the per cent inhibition of the control histamine releases which were 36±3, 37±4, 31±4 and 30±4% following 24 h treatments with buffer or salbutamol at 10−8, 10−7 or 10−6 M, respectively. Values are means±s.e.mean from five experiments.
Radioligand binding studies
In further experiments, we sought to determine the effects on β2-adrenoceptor density in membranes of mast cells after long-term treatment with β-adrenoceptor agonists. In the main, mast cell-enriched (43 to 67% purity) preparations were used in this study because the quantities of purified mast cells required for these studies could only be generated infrequently. Competition binding studies using [125I]-CYP as radioligand and the β1-selective antagonist, CGP20712A (Dooley et al., 1986), to discriminate between β-adrenoceptor subtypes indicate that mast cell-enriched preparations (n=5, data not shown) and HLMC (Chong et al., 2002) express a homogeneous population of β2-adrenoceptors. Preliminary studies of membranes of mast cell-enriched preparations treated (24 h) without or with isoprenaline (10−8–10−6 M) indicated that concentrations of ⩽10−7 M had little effect on receptor density (n=3, data not shown). In further studies, the effects of long-term exposure of mast cell-enriched preparations to isoprenaline (10−6 M), salbutamol (10−6 M) or terbutaline (10−6 M) on β2-adrenoceptor density were determined (Table 4). Our studies show that long-term exposure of cells to these three agonists led to similar (∼30% reduction) levels of reduction in β2-adrenoceptor density. Similar results were obtained for purified mast cells (purity⩾90%) as the β2-adrenoceptor density expressed by membranes generated from cells treated (24 h) with β-adrenoceptor agonists (10−6 M) was between 25–35% lower than that measured in membranes from untreated cells (data not shown, n=2).
Table 4.
Effects of long-term treatment of mast cell-enriched preparations with β-adrenoceptor agonists on β-adrenoceptor density (Bmax)

Discussion
Salbutamol is the most-commonly employed short-acting bronchodilator used in asthma and terbutaline is also used widely. As well as acting to relax airway smooth muscle and thereby providing symptomatic relief in asthma, additional effects of these drugs could include the stabilization of pulmonary inflammatory cells (Barnes, 1999). In this context, inhibition of mast cell responses could be important and the present study has shown that both terbutaline and salbutamol are effective inhibitors of the stimulated release of histamine from HLMC. Salbutamol and terbutaline were roughly equipotent and both showed similar intrinsic activities among HLMC preparations acting as full agonists in about a quarter of preparations and as partial agonists in the remainder. These data suggest that, in the clinical context, both salbutamol and terbutaline may stabilize mast cell responses to similar degrees but the extent of stabilization may show inter-individual variation.
Both salbutamol and terbutaline increased total cell cAMP consistent with the widely-held recognition that bronchodilators interact with β2-adrenoceptors to activate adenylate cyclase (Barnes, 1999). Salbutamol and terbutaline increased cAMP to similar degrees but to about a half of that seen with isoprenaline. In the context of cAMP generation, therefore, both drugs act as partial agonists. As the intrinsic activities, relative to isoprenaline, of these compounds are about 0.4 and 0.8 for cAMP generation and inhibition of histamine release, respectively, these data indicate that some intracellular amplification of the cAMP response in mast cells may occur if cAMP is involved in inhibiting histamine release.
These preliminary data suggest that both salbutamol and terbutaline mediate very similar quantitative responses in mast cells. In experiments to address the comparative effects of these two agonists on functional desensitization in mast cells, an expectation would be that both agonists would induce very similar levels of desensitization. However, in the present system, incubation (24 h) of mast cells with salbutamol induced far greater levels of desensitization than terbutaline to the inhibitory effects of a wide range of β-adrenoceptor agonists (salbutamol, terbutaline, isoprenaline and clenbuterol). This desensitizing capability remained stable over 48 h. This outcome was surprising given that studies in transfected cells demonstrate a correlation between the relative efficacy of agonists and the extent of desensitization (January et al., 1997). However, it should be borne in mind that the studies in cells transfected with the β2-adrenoceptor followed desensitization over relatively short time periods (5 to 30 min) so direct comparison with the present system, in which desensitization was followed over 24 h, may be inappropriate.
A factor that could contribute to the functional desensitization induced by β-adrenoceptor agonists is down-regulation of the β2-adrenoceptor. Studies in tissue and cell systems indicate that long-term treatments with β-adrenoceptor agonists leads to reductions in receptor density (Pittman et al., 1984; Nishikawa et al., 1996; Williams et al., 2000). However, incubation (24 h) of mast cells with salbutamol or terbutaline, even at a concentration (10−6 M) 10 fold higher than that which caused effective levels of functional desensitization, caused modest reductions in β2-adrenoceptor density that were not significant, statistically (P>0.05). Moreover, there was no difference in the extent of β2-adrenoceptor down-regulation promoted by either salbutamol or terbutaline. These data suggest that down-regulation of β2-adrenoceptors is unlikely to be a major mechanism involved in promoting the functional desensitization and, furthermore, that down-regulation is not the mechanism by which salbutamol induces greater levels of desensitization than terbutaline.
Recent studies have shown that exposure (7 days) of rats to salbutamol causes a loss in Gsα, the G protein coupling the β2-adrenoceptor to adenylate cyclase, and that this mechanism could be responsible for the development of tolerance to salbutamol (Finney et al., 2000). Although it is possible that levels of Gsα may change following long-term treatments of mast cells with β-adrenoceptor agonists, that the inhibitory effects of PGE2 remain largely unaffected by desensitizing treatments, suggest that this is unlikely to be a major mechanism to explain the functional desensitization in mast cells. An alternative explanation that has been proposed is that tolerance to β2-adrenoceptor agonists develops as a consequence of up-regulation of PDE the idea being that receptor-driven cAMP may be catabolized more readily by increased PDE content (Giembycz, 1996). Although a possibility, the data, on the whole, argue against an up-regulation of PDE especially as the inhibitory effects of the PDE inhibitors, IBMX and theophylline, were reduced, rather than increased, following desensitizing treatments with β-adrenoceptor agonists.
PDE inhibitors, such as IBMX and theophylline, cause increases in cAMP and inhibit mast cell responses without the need for ligand-induced activation of adenylate cyclase suggesting that a spontaneous and substantial generation of cAMP occurs in mast cells and that PDE inhibitors act to increase cAMP by preventing the catabolism of this spontaneous generation of cAMP (Peachell et al., 1988). It is of interest that long-term treatment of terbutaline and salbutamol reduced the inhibitory effects of IBMX and theophylline in mast cells but that salbutamol was more effective. Moreover, desensitizing treatment with salbutamol reduced the inhibitory effects on mast cells of the direct activator of adenylate cyclase, forskolin, to a greater degree than desensitizing treatments with terbutaline. Taken together, these data could suggest that the effects of desensitizing treatments with β-adrenoceptor agonists on the responses of IBMX and forskolin may reflect reduced levels of adenylate cyclase or its activation and that salbutamol is the more effective agonist in this regard. A reduction in adenylate cyclase would mean both fewer targets for forskolin and, a reduction in spontaneously generated cAMP, hence, PDE inhibitors might not work quite so effectively.
Notwithstanding these considerations, it still seems very probable that the functional desensitization observed to β-adrenoceptor agonists in response to long-term treatment with β-adrenoceptor agonists primarily involves mechanisms associated with the β2-adrenoceptor itself. This is because, although the effects of cAMP-active compounds may be affected by desensitizing treatments, the quite striking concentration-dependent desensitization of the inhibitory effects of salbutamol following long-term incubation with salbutamol, was not observed for the inhibitory effects of alternative cAMP-active compounds. These data suggest that desensitizing treatments affect the β2-adrenoceptor signalling pathway, specifically. Although our data suggest that receptor down-regulation is not a major mechanism to explain the functional desensitization, other considerations include the extent of receptor uncoupling and sequestration, the mechanisms that mediate these events and whether different β-adrenoceptor agonists promote these processes to varying degrees.
A further aspect that merits emphasis is the relative sensitivity of either salbutamol or terbutaline to desensitizing treatments. The inhibition by terbutaline of release is less susceptible to desensitizing conditions than the inhibition observed with salbutamol. Of particular interest is the considerably greater extent to which salbutamol causes desensitization to itself (∼64%, P<0.005) compared to that which terbutaline causes to itself (∼19%, P>0.05). Should similar processes be operative in vivo then terbutaline is far less likely to compromise the subsequent ability of terbutaline to stabilize mast cells than an equivalent concentration of salbutamol on the subsequent effectiveness of salbutamol to stabilize mast cells.
To summarise, salbutamol induces greater levels of desensitization than terbutaline even though both agonists are roughly equipotent as inhibitors of histamine release. This could be important in the clinical setting as it suggests that compromised stabilization of mast cell responses may occur more readily with continued use of salbutamol than terbutaline.
Acknowledgments
The authors are grateful to Mr N. Vaughan, Mr G. Rocco and Mr G. Cooper (Cardiothoracic Surgery) and Dr K. Suvarna and Dr A. Sherif (Histopathology) at the Northern General Hospital, Sheffield for their invaluable help in providing lung tissue specimens. The authors also thank C. Layton for technical support and Dr G.G.S. Collins for helpful comments and suggestions. This work was supported by the National Asthma Campaign
Abbreviations
- BSA
bovine serum albumin
- HLMC
human lung mast cells
- HSA
human serum albumin
- IBMX
isobutyl-methylxanthine
- [125I]-CYP
[125I]-cyanopindolol
- PBS
phosphate buffered saline
References
- ALI H., PEARCE F.L. Isolation and properties of cardiac and other mast cells from the rat and guinea-pig. Agents Actions. 1985;16:138–140. doi: 10.1007/BF01983121. [DOI] [PubMed] [Google Scholar]
- ASSEM E.S.K., SCHILD H.O. Beta-adrenergic receptors concerned with the anaphylactic mechanism. Int. Archs Allergy Appl. Immunol. 1969;45:62–69. doi: 10.1159/000231003. [DOI] [PubMed] [Google Scholar]
- BARNES P.J. Effect of β-agonists on inflammatory cells. J. Allergy Clin. Immunol. 1999;104:S10–17. doi: 10.1016/s0091-6749(99)70269-1. [DOI] [PubMed] [Google Scholar]
- BENOVIC J.L., STANISZWESKI C., MAYOR F., CARON M.G., LEFKOWITZ R.J. β-Adrenergic receptor kinase. Activity of partial agonists for stimulation of adenylate cyclase correlates with ability to promote receptor phosphorylation. J. Biol. Chem. 1988;263:3893–3897. [PubMed] [Google Scholar]
- BUTCHERS P.R., SKIDMORE I.F., VARDEY C.J., WHEELDON A. Characterisation of the receptor mediating the anti-anaphylactic activities of β-adrenoceptor agonists in human lung tissue in vitro. Br. J. Pharmacol. 1980;71:663–667. doi: 10.1111/j.1476-5381.1980.tb10987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHONG L.K., CHESS-WILLIAMS R., PEACHELL P.T. Pharmacological characterisation of the β-adrenoceptor expressed by human lung mast cells. Eur. J. Pharmacol. 2002;437:1–7. doi: 10.1016/s0014-2999(02)01263-3. [DOI] [PubMed] [Google Scholar]
- CHONG L.K., COOPER E., VARDEY C.J., PEACHELL P.T. Salmeterol inhibition of mediator release from human lung mast cells by β-adrenoceptor-dependent and independent mechanisms. Br. J. Pharmacol. 1998;123:1009–1015. doi: 10.1038/sj.bjp.0701703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHONG L.K., DRURY D.E.J., DUMMER J.F., GHAHRAMANI P., SCHLEIMER R.P., PEACHELL P.T. Protection by dexamethasone of the functional desensitization to β2-adrenoceptor-mediated responses in human lung mast cells. Br. J. Pharmacol. 1997;121:717–722. doi: 10.1038/sj.bjp.0701185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHONG L.K., PEACHELL P.T. β-Adrenoceptor reserve in human lung: a comparison between airway smooth muscle and mast cells. Eur. J. Pharmacol. 1999;378:115–122. doi: 10.1016/s0014-2999(99)00425-2. [DOI] [PubMed] [Google Scholar]
- CHURCH M.K., HIROI J. Inhibition of IgE-dependent histamine release from human dispersed lung mast cells by anti-allergic drugs and salbutamol. Br. J. Pharmacol. 1987;90:421–429. doi: 10.1111/j.1476-5381.1987.tb08972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COCKCROFT D.W., MCPARLAND C.P., BRITTO S.A., SWYSTUN V.A., RUTHERFORD B.C. Regular inhaled salbutamol and airway responsiveness to allergen. Lancet. 1993;342:833–837. doi: 10.1016/0140-6736(93)92695-p. [DOI] [PubMed] [Google Scholar]
- DOOLEY D.J., BITTIGER H., REYMANN N.C. CGP20712A: a useful tool for quantitating β1- and β2-adrenoceptors. Eur. J. Pharmacol. 1986;130:137–139. doi: 10.1016/0014-2999(86)90193-7. [DOI] [PubMed] [Google Scholar]
- DRURY D.E.J., CHONG L.K., GHAHRAMANI P., PEACHELL P.T. Influence of receptor reserve on β-adrenoceptor-mediated responses in human lung mast cells. Br. J. Pharmacol. 1998;124:711–718. doi: 10.1038/sj.bjp.0701897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENNIS M. Current techniques of histamine determination. Automated fluorometric assays. Handbook Exp. Pharmacol. 1991;97:31–38. [Google Scholar]
- FERGUSON S.S.G. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol. Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- FINNEY P.A., BELVISI M.G., DONNELLY L.E., CHUANG T.-T., MAK J.C.W., SCORER C., BARNES P.J., ADCOCK I.M., GIEMBYCZ M.A. Albuterol-induced down-regulation of Gsα accounts for pulmonary β2-adrenoceptor desensitization in vivo. J. Clin. Invest. 2000;106:125–135. doi: 10.1172/JCI8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FINNEY P.A., DONNELLY L.E., BELVISI M.G., CHUANG T.-T., BIRRELL M., HARRIS A., MAK J.C.W., SCORER C., BARNES P.J., ADCOCK I.M., GIEMBYCZ M.A. Chronic systemic administration of salmeterol to rats promotes pulmonary β2-adrenoceptor desensitization and down-regulation of Gsα. Br. J. Pharmacol. 2001;132:1261–1270. doi: 10.1038/sj.bjp.0703946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIEMBYCZ M.A. Phosphodiesterase 4 and tolerance to β2-adrenoceptor agonists in asthma. Trends Pharmacol. Sci. 1996;17:331–336. [PubMed] [Google Scholar]
- GILBERT H.S., ORNSTEIN L. Basophil counting with a new staining method using Alcian Blue. Blood. 1975;46:279–282. [PubMed] [Google Scholar]
- ISHIZAKA T., CONRAD D.H., SCHULMAN E.S., STERK A.R., ISHIZAKA K. Biochemical analysis of initial triggering events of IgE-mediated histamine release from human lung mast cells. J. Immunol. 1983;130:2357–2362. [PubMed] [Google Scholar]
- JANUARY B., SEIBOLD A., WHALEY B., HIPKIN R.W., LIN D., SCHONBRUNN A., BARBER R., CLARK R.B. β2-Adrenergic receptor desensitization, internalization, and phosphorylation in response to full and partial agonists. J. Biol. Chem. 1997;272:23871–23879. doi: 10.1074/jbc.272.38.23871. [DOI] [PubMed] [Google Scholar]
- LEFKOWITZ R.J. G protein-coupled receptors. III. New roles for receptor kinases and β-arrestins in receptor signaling and desensitization. J. Biol. Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- NISHIKAWA M., MAK J.C.W., BARNES P.J. Effect of short- and long-acting β2-adrenoceptor agonists on pulmonary β2-adrenoceptor expression in human lung. Eur. J. Pharmacol. 1996;318:123–129. doi: 10.1016/s0014-2999(96)00769-8. [DOI] [PubMed] [Google Scholar]
- O'CONNOR B.J., AIKMAN S.L., BARNES P.J. Tolerance to the nonbronchodilator effects of inhaled β2-agonists in asthma. N. Engl. J. Med. 1992;327:1204–1208. doi: 10.1056/NEJM199210223271704. [DOI] [PubMed] [Google Scholar]
- ORANGE R.P., AUSTEN W.G., AUSTEN K.F. Immunological release of histamine and slow reacting substance of anaphylaxis from human lung. I. Modulation by agents influencing cellular levels of cyclic 3′,5′-adenosine monophosphate. J. Exp. Med. 1971;134:136–148. [PMC free article] [PubMed] [Google Scholar]
- PEACHELL P.T., MACGLASHAN D.W., LICHTENSTEIN L.M., SCHLEIMER R.P. Regulation of human basophil and lung mast cell function by cAMP. J. Immunol. 1988;140:571–579. [PubMed] [Google Scholar]
- PITTMAN R.N., REYNOLDS E.E., MOLINOFF P.B. Relationship between intrinsic activities of agonists in normal and desensitized tissue and agonist-induced loss of beta adrenergic receptors. J. Pharmacol. Exp. Ther. 1984;230:614–618. [PubMed] [Google Scholar]
- SCHULMAN E.S., MACGLASHAN D.W., PETERS S.P., SCHLEIMER R.P., NEWBALL H.H., LICHTENSTEIN L.M. Human lung mast cells: purification and characterisation. J. Immunol. 1982;129:2662–2667. [PubMed] [Google Scholar]
- SIRAGANIAN R.P. An automated continuous-flow system for the extraction and fluorometric analysis of histamine. Anal. Biochem. 1974;57:283–287. doi: 10.1016/0003-2697(74)90093-1. [DOI] [PubMed] [Google Scholar]
- TATTERSFIELD A.E. Bronchodilators: new developments. Br. Med. Bull. 1992;48:51–64. doi: 10.1093/oxfordjournals.bmb.a072534. [DOI] [PubMed] [Google Scholar]
- VAN DER HEIJDEN P.J.C.M., VAN AMSTERDAM J.G.C., ZAAGSMA J. Desensitization of smooth muscle and mast cell β-adrenoceptors in the airways of the guinea pig. Eur. J. Resp. Dis. 1984;65 Suppl 135:128–134. [PubMed] [Google Scholar]
- WILLIAMS B.R., BARBER R., CLARK R.B. Kinetic analysis of agonist-induced down-regulation of the β2-adrenergic receptor in BEAS-2B cells reveals high and low-affinity components. Mol. Pharmacol. 2000;58:421–430. doi: 10.1124/mol.58.2.421. [DOI] [PubMed] [Google Scholar]