

Abstract
Presynaptic muscarinic receptors modulate sympathetic transmitter release. The goal of the present study was to identify the muscarinic receptor subtype(s) mediating inhibition of sympathetic transmitter release in mouse atria, urinary bladder and vas deferens. To address this question, electrically evoked noradrenaline release was assessed using tissue preparations from NMRI, M2- and M4-knockout, and the corresponding M2- and M4-wildtype mice, after preincubation with 3H-noradrenaline.
The muscarinic agonist carbachol decreased evoked tritium overflow (20 pulses/50 Hz) in each tissue and strain investigated. After deletion of the M2-receptor the maximal inhibition by carbachol was significantly reduced (by 41–72%), but not abolished, in all tissues. After deletion of the M4-receptor a moderate and significant reduction of the maximal inhibition by carbachol (by 28%) was observed only in the vas deferens.
Experiments with the muscarinic antagonists methoctramine and pirenzepine confirmed that the presynaptic muscarinic receptors were predominantly M2 in atria and bladder and probably a mixture of M2 and M4 in the vas deferens.
Experiments in the urinary bladder with the cholinesterase inhibitor physostigmine and the muscarinic antagonist ipratropium demonstrated that endogenously released acetylcholine predominantly acted through M2-receptors to inhibit noradrenaline release. However, the results do not exclude a minor contribution of M4-receptors to this endogenous inhibition.
In conclusion, our results clearly indicate that the release-inhibiting muscarinic receptors on postganglionic sympathetic axons in mouse atria, bladder and vas deferens represent mixtures of M2- and non-M2-receptors. The non-M2-receptors remain unknown in atria and the bladder, and may represent primarily M4-receptors in the vas deferens. These results reveal an unexpected heterogeneity among the muscarinic receptors mediating inhibition of noradrenaline release.
Keywords: Presynaptic muscarinic receptors, muscarinic receptor subtypes, sympathetic axon terminals, 3H-noradrenaline, M2-knockout, M4-knockout, mouse atria, mouse urinary bladder, mouse vas deferens, heterogeneity
Introduction
Presynaptic release-modulating muscarinic receptors on postganglionic sympathetic axon terminals, first described in 1967 (Löffelholz, 1967; Lindmar et al., 1968), contribute to the interplay between the sympathetic and parasympathetic nervous systems (reviewed by Fuder & Muscholl, 1995). In general, these receptors mediate an inhibition of sympathetic transmitter release, but enhancement has also been reported, at least in some tissues (Fuder & Muscholl, 1995).
There are five molecularly distinct subtypes of muscarinic receptors, M1 to M5 (Caulfield & Birdsall, 1998; Alexander et al., 2001). Probably due to the lack of highly subtype-selective drugs, pharmacological studies aimed at identifying the presynaptic muscarinic receptors at sympathetic axons have often led to conflicting results. Release-facilitating receptors were suggested to be M1-receptors across tissues and species (e.g. mouse atria, Costa & Majewski, 1991; see Fuder & Muscholl, 1995). The more prominent release-inhibiting receptors, on the other hand, were proposed to represent M2-receptors in the majority of tissues and species (e.g. mouse atria, Costa & Majewski, 1991). However, the existence of release-inhibiting M1-receptors (e.g. rabbit vas deferens; Grimm et al., 1994) and M3-receptors (e.g. guinea-pig atria; Olmez et al., 1995) has also been reported, and in a substantial number of studies the identity of the presynaptic muscarinic heteroreceptors has remained unclear (e.g. rat vas deferens; Miranda et al., 1994).
A new approach to receptor classification that circumvents the difficulties associated with pharmacological tools of limited subtype selectivity involves the use of mutant mice in which specific receptor genes have been disrupted by molecular genetic techniques. This strategy has shown, for example, that postganglionic sympathetic neurons and central noradrenergic neurons possess release-inhibiting presynaptic α2A-(which predominate) and α2C-adrenoceptors (autoreceptors; Altman et al., 1999; Hein et al., 1999; Trendelenburg et al., 1999; 2001). The use of mutant mice has also shown that parasympathetic cholinergic neurons possess release-inhibiting muscarinic autoreceptors which are exclusively M4 in the urinary bladder but M4 plus non-M4 (probably M2) in atria (Zhou et al., 2002).
In the present study, we used mice lacking either the M2-receptor (M2-knockout; Gomeza et al., 1999a) or the M4-receptor (M4-knockout; Gomeza et al., 1999b) to investigate presynaptic muscarinic receptors mediating inhibition of noradrenaline release. The corresponding M2-wildtype and M4-wildtype mice served as controls. In addition, NMRI mice were included as a commonly used laboratory strain. In the first part of the study, we identified and characterized presynaptic muscarinic receptors in heart atria, urinary bladder, and vas deferens. The agonist carbachol and the antagonists ipratropium (high-affinity, non-selective), methoctramine and pirenzepine were used as pharmacological tools. The latter two antagonists were chosen because they allow a limited distinction between muscarinic receptor subtypes: the rank order methoctramine>pirenzepine is typical for M2-receptors, whereas pirenzepine⩾methoctramine is typical for M3-, M4- and M5-receptors and pirenzepine>methoctramine for M1-receptors (Lazareno et al., 1990; Dörje et al., 1991; Lazareno & Birdsall, 1993; Eglen et al., 1996; Caulfield & Birdsall, 1998). In the second part, we examined through which receptor subtype(s) endogenously released acetylcholine modulates noradrenaline release in the urinary bladder. Neurotransmitter release experiments were carried out using isolated tissue segments after labelling with 3H-noradrenaline.
Methods
Tissues and superfusion
The generation of M2-knockout (genetic background: 129J1×CF1) and M4-knockout (genetic background: 129SvEv×CF1) mice has been described previously (Gomeza et al., 1999a, b). In all experiments, aged-matched wildtype mice of the corresponding genetic background were used as controls (M2-wildtype, 129J1×CF1; M4-wildtype, 129SvEv×CF1). Mouse genotyping was carried out by PCR analysis of mouse tail DNA. Male NMRI (Naval Medical Research Institute; bred in the local animal facility), M2-wildtype, M4-wildtype, M2-knockout and M4-knockout mice aged >2 months were killed by cervical dislocation. From each animal six to eight pieces of the atria, 12 to 15 pieces of the urinary bladder or eight to 12 pieces of the vas deferens were obtained. Tissue pieces were preincubated in 1 ml medium (see below) containing 0.2 μM 3H-noradrenaline for 30 min at 37°C and then placed in 12 superfusion chambers between platinum electrodes, one piece per chamber, where they were superfused with 3H-noradrenaline-free medium at a rate of 1.2 ml min−1. Successive 2-min samples of the superfusate were collected from t=50 min onwards (t=0 min being the start of superfusion). At the end of experiments, tissues were dissolved and tritium was determined in superfusate samples and tissues.
The superfusion medium contained (mM): NaCl 118, KCl 4.8, CaCl2 2.5, MgSO4 1.2, NaHCO3 25, KH2PO4 1.2, glucose 11, ascorbic acid 0.57, disodium EDTA 0.03 and desipramine 0.001. The medium for preincubation with 3H-noradrenaline contained no desipramine and only 0.2 mM CaCl2 (Limberger et al., 1992).
Protocols
There were seven periods of electrical stimulation. Each stimulation period consisted of rectangular pulses of 1 ms width and 47 V cm−1 voltage drop between the electrodes of each chamber, yielding a current strength of 80 mA. The first stimulation period (180 pulses/3 Hz) was delivered at t=30 min and was not used for determination of tritium overflow. The subsequent stimulation periods (S1 to S6) were applied at t=54, 72, 90, 108, 126 and 144 min and differed, depending on the type of experiment.
In experiments designed to detect and characterize presynaptic muscarinic receptors in atria, the urinary bladder and the vas deferens by means of carbachol and different muscarinic antagonists, S1 to S6 each consisted of 20 pulses/50 Hz. Carbachol was introduced at increasing concentrations after S1, 12 min before S2, S3, S4, S5 and S6. Antagonists were present throughout superfusion at a fixed concentration.
In experiments designed to study the potential modulation of noradrenaline release by endogenously released acetylcholine in the urinary bladder, the following stimulations were applied: S1 and S4 consisted of 20 pulses/50 Hz; S2 and S5 consisted of 120 pulses/3 Hz, and S3 and S6 consisted of 360 pulses/3 Hz. Drugs were present either throughout superfusion or introduced after S3, 12 min before S4.
Evaluation
The outflow of tritium was calculated as a fraction of the tritium content of the tissue at the onset of the respective collection period (fractional rate; min−1). The overflow elicited by electrical stimulation was calculated as the difference ‘total tritium outflow during and after stimulation' minus ‘basal outflow', and was then expressed as a percentage of the tritium content of the tissue at the time of stimulation (see Trendelenburg et al., 1997).
For further evaluation, overflow ratios were calculated: Sn/S1 in experiments with identical stimulation patterns at S1 to S6, and Sn/Sn-3 in experiments with different stimulation patterns (note that stimulation patterns at Sn and Sn-3 were identical). Overflow ratios obtained in the presence of drugs added after S1 or S3 were also calculated as a percentage of the corresponding ratio in controls in which no drug was added after S1 or S3. Effects of drugs on basal tritium outflow were evaluated similarly (Trendelenburg et al., 1997).
Concentration-response data for carbachol given alone were evaluated by sigmoid curve fitting (eq. 25 of Waud, 1976). This yielded the Emax (maximal effect) of carbachol and its EC50 (concentration causing a half-maximal effect) in the absence of antagonist. In most cases sigmoid curves could not be fitted to concentration-response data for carbachol in the presence of antagonists. Therefore, for determination of apparent antagonist pKd values (negative logarithms of the apparent Kd), carbachol EC50 values in the presence of antagonist were interpolated from the nearest points of the respective concentration-response curves, assuming that the Emax of the agonist had not changed; the pKd value was then calculated from the increase in EC50 values (equation no. 4 of Furchgott, 1972). The pKd values are apparent because only one antagonist concentration was used and the competitive character of the interaction was not verified.
Results are expressed as arithmetic means±s.e.mean (estimates±s.e. defined by Waud, 1976, in the case of Emax and EC50 values of carbachol; s.e. of pKd values were calculated by means of the Gaussian law of error propagation). Groups were tested for significant differences with the Mann-Whitney test with Bonferroni correction. P<0.05 was taken as limit of statistical difference. n represents the number of tissue pieces.
Drugs
Drugs were (−)-[ring-2,5,6-3H]-noradrenaline, specific activity 51.8–70.7 Ci/mmol (NEN, Köln, Germany), carbachol chloride, desipramine HCl, physostigmine hemisulphate, ipratropium bromide, methoctramine 4 HCl, pirenzepine 2 HCl, rauwolscine HCl (Sigma, Deisenhofen, Germany) and phentolamine methanesulfonate (Ciba-Geigy, Basel, Switzerland). Drugs were dissolved in distilled water.
Results
All experiments were done with tissue segments prepared from atria, urinary bladder and vas deferens from either NMRI, M2-wildtype, M4-wildtype, M2-knockout or M4-knockout mice after preincubation with 3H-noradrenaline to label vesicular noradrenaline pools. Electrical stimulation was applied to elicit the release of 3H-noradrenaline measured as tritium overflow.
Detection and characterization of presynaptic muscarinic heteroreceptors in atria, urinary bladder and vas deferens
In this series of experiments, tissues were stimulated by short bursts of 20 pulses/50 Hz. As shown in Figure 1 for the urinary bladder, which has not been studied in this manner previously, electrical stimulation led to clear peaks of tritium overflow. In control experiments without carbachol the magnitude of these peaks was similar from S1 to S6, giving Sn/S1 ratios close to unity (Figure 1). Similar observations were made in atria and the vas deferens (not shown; see also atria: Wahl et al., 1996; vas deferens: Trendelenburg et al., 1999). The overflow of tritium evoked by S1 amounted to 0.39±0.03% of tissue tritium in atria, 0.21±0.02% in bladder, and 0.30±0.01% in vas deferens from NMRI mice (n=16–29). Similar values were observed in tissues from M2-wildtype, M4-wildtype, M2-knockout and M4-knockout mice (data not shown). The overflow values in atria and vas deferens from NMRI mice were similar to previous studies (Trendelenburg et al., 1999; 2000).
Figure 1.
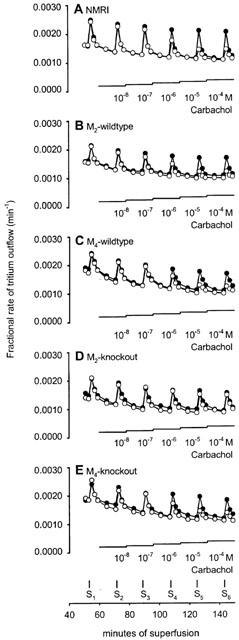
Outflow of tritium from urinary bladder pieces taken from NMRI (A), M2-wildtype (B), M4-wildtype (C), M2-knockout (D) or M4-knockout mice (E): effects of electrical stimulation and carbachol. After preincubation with 3H-noradrenaline tissues were superfused and stimulated electrically six times by 20 pulses/50 Hz (S1 to S6). Filled circles, controls (no carbachol). Empty circles, carbachol was added at increasing concentrations as indicated. Each line represents the mean of three to 13 bladder pieces.
We have shown previously that short bursts of 20 pulses/50 Hz led to little, if any, α2-autoinhibition of noradrenaline release in mouse atria and vas deferens (Trendelenburg et al., 1999; 2000). The same stimulation pattern (i.e. 20 pulses/50 Hz), also led to little α2-autoinhibition in the mouse urinary bladder as indicated by an only small facilitatory effect of the α-adrenoceptor antagonists phentolamine (1 μM) and rauwolscine (1 μM) on evoked tritium overflow (see section below on ‘Inhibition of noradrenaline release by endogenous acetylcholine in the urinary bladder'). These stimulation conditions with no or little α2-autoinhibition were chosen in order to provide optimal conditions for the detection and characterization of presynaptic modulation of noradrenaline release by exogenous muscarinic agonists (see Starke, 1987; Schlicker & Göthert, 1998).
The muscarinic agonist carbachol (10 nM to 100 μM) reduced the evoked overflow of tritium in all three tissues of all five mouse strains studied. This is shown as efflux-versus-time curves in Figure 1 and as concentration-response curves in Figure 2 (NMRI), Figure 3 (M2-wildtype and M2-knockout) and Figure 4 (M4-wildtype and M4-knockout). The Emax of carbachol (expressed as per cent inhibition of stimulation-evoked tritium overflow compared to controls without carbachol), obtained from logistic curve fitting, amounted to 65±1% in NMRI heart atria, 56±1% in NMRI urinary bladder, and 43±1% in NMRI vas deferens. The carbachol EC50 values were 1.9±0.4 μM in NMRI atria, 1.0±0.6 μM in NMRI bladder and 1.1±1.3 μM in NMRI vas deferens. The concentration-response curves of carbachol in M2-wildtype tissues (Figure 3) and M4-wildtype tissues (Figure 4) did not differ from the NMRI curves (Figure 2).
Figure 2.
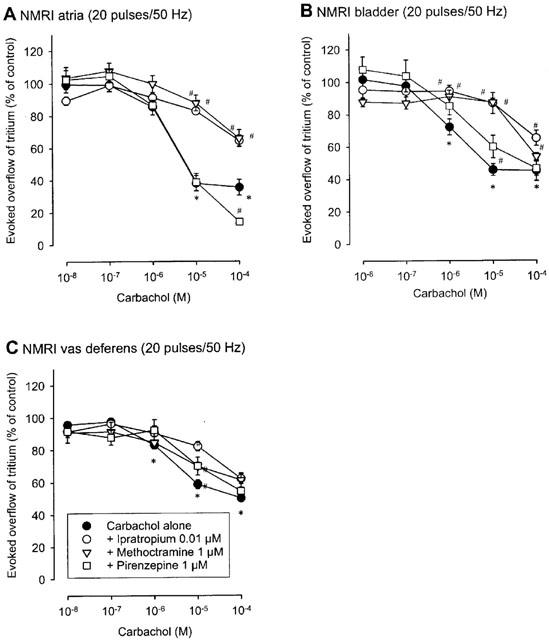
Interaction of muscarinic antagonists with carbachol on the evoked overflow of tritium from atria (A), urinary bladder (B) and vas deferens (C) of NMRI mice. After preincubation with 3H-noradrenaline tissues were superfused and stimulated electrically six times by 20 pulses/50 Hz (S1 to S6). Carbachol was added at increasing concentrations (abscissae) before S2 to S6. Carbachol was given either alone or combined with the indicated antagonists which were present throughout superfusion. Ordinates, evoked overflow of tritium, calculated from Sn/S1 ratios and expressed as a percentage of the corresponding control (no carbachol). Means±s.e.mean from n=6 to 16 tissue pieces. Significant differences from corresponding control (no carbachol): *P<0.05. Significant differences from carbachol alone: #P<0.05.
Figure 3.
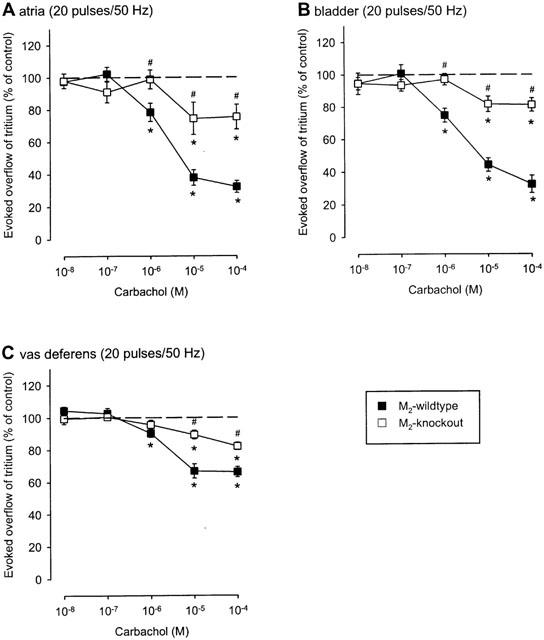
Effect of carbachol on the evoked overflow of tritium from atria (A), bladder (B) and vas deferens (C) of M2-wildtype or M2-knockout mice. After preincubation with 3H-noradrenaline tissues were superfused and stimulated electrically six times by 20 pulses/50 Hz (S1 to S6). Carbachol was added at increasing concentrations (abscissae) before S2 to S6. Ordinates, evoked overflow of tritium, calculated from Sn/S1 ratios and expressed as a percentage of the corresponding control (no carbachol). Means±s.e.mean from n=5 to 16 tissue pieces. Significant differences from corresponding control (no carbachol): *P<0.05. Significant differences from M2-wildtype: #P<0.05.
Figure 4.
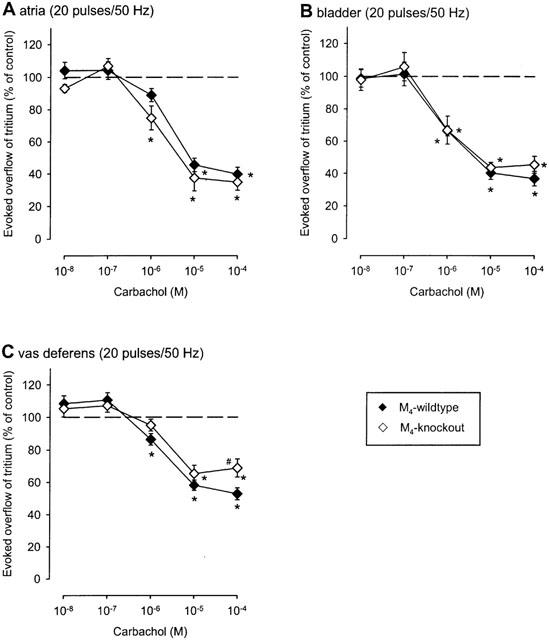
Effect of carbachol on the evoked overflow of tritium from atria (A), bladder (B) and vas deferens (C) of M4-wildtype or M4-knockout mice. After preincubation with 3H-noradrenaline tissues were superfused and stimulated electrically six times by 20 pulses/50 Hz (S1 to S6). Carbachol was added at increasing concentrations (abscissae) before S2 to S6. Ordinates, evoked overflow of tritium, calculated from Sn/S1 ratios and expressed as a percentage of the corresponding control (no carbachol). Means±s.e.mean from n=5 to 16 tissue pieces. Significant differences from corresponding control (no carbachol): *P<0.05. Significant differences from M4-wildtype: #P<0.05.
Strikingly, deletion of the M2-receptor led to a marked reduction (P<0.05) in the maximum inhibition of stimulation-evoked overflow of tritium in all three tissues investigated. Logistic curve fitting showed that the Emax of carbachol was reduced from 68±4% (M2-wildtype) to 26±10% (M2-knockout) in atria (Figure 3A), from 69±8% (M2-wildtype) to 19±4% (M2-knockout) in bladder (Figure 3B) and from 34±4% (M2-wildtype) to 20±3% (M2-knockout) in vas deferens (Figure 3C). In all three tissues, statistically significant inhibitory carbachol responses remained in the absence of M2-receptors (Figure 3). Lack of M4-receptors, in contrast, had no significant effect on the inhibition by carbachol in atria and urinary bladder (Figure 4A,B) although it moderately reduced the maximum inhibition in the vas deferens: the fitted Emax values for carbachol were 46±9% (M4-wildtype) and 33±6% (M4-knockout), respectively.
The receptors mediating the inhibition of noradrenaline release were characterized further by means of the antagonists ipratropium (high-affinity, non-selective; used in NMRI tissues only) as well as methoctramine and pirenzepine which allow a limited distinction between muscarinic receptor subtypes (see Introduction). Neither ipratropium (0.01 μM) nor pirenzepine (1 μM), when present throughout superfusion, changed the stimulation-evoked overflow of tritium (S1), whereas methoctramine (1 μM) increased the evoked overflow of tritium (S1) from atria, bladder and vas deferens of several mouse strains by up to 35% (data not shown; compare Casado et al., 1992). In control experiments without carbachol, Sn/S1 ratios were close to unity also in the presence of the antagonists. None of the muscarinic receptor ligands used had any effect on basal tritium outflow (data not shown).
In all three tissues from NMRI mice, ipratropium (0.01 μM), methoctramine (1 μM) and pirenzepine (1 μM) shifted the carbachol concentration-response curve to the right (Figure 2), except for pirenzepine in atria (Figure 2A). The apparent antagonist pKd values calculated from the shifts are given in Table 1. The rank order of antagonist potency was ipratropium>methoctramine>pirenzepine in atria and bladder but ipratropium>pirenzepine⩾methoctramine in vas deferens (Figure 2; Table 1).
Table 1.
pKd values of antagonists at presynaptic muscarinic receptors in NMRI, M2-wildtype, M4-wildtype and M4-knockout mice

Methoctramine (1 μM) and pirenzepine (1 μM) were also studied as antagonists against carbachol in preparations from M2- and M4-knockout mice and the two corresponding wildtype control strains. As indicated by the pKd values summarized in Table 1, the results obtained in M2- and M4-wildtype tissues agreed well with those obtained in NMRI tissues: in M2- and M4- wildtype atria and bladder, the antagonist potency order again was methoctramine>pirenzepine, whereas in M2- and M4-wildtype vas deferens, the potency order again was pirenzepine⩾methoctramine. The interaction of methoctramine and pirenzepine with carbachol in the M4-wildtype vas deferens is shown in Figure 5A. Comparison with Figure 2C shows the similarity to the NMRI vas deferens.
Figure 5.
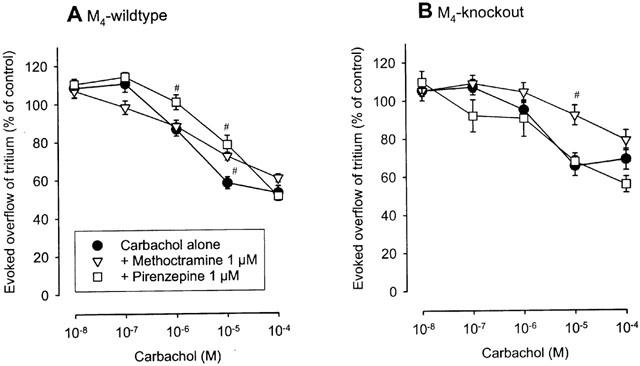
Interaction of muscarinic antagonists with carbachol on the evoked overflow of tritium from vas deferens of M4-wildtype (A) and M4-knockout (B) mice. After preincubation with 3H-noradrenaline tissues were superfused and stimulated electrically six times by 20 pulses/50 Hz (S1 to S6). Carbachol was added at increasing concentrations (abscissae) before S2 to S6. Carbachol was given either alone or combined with the indicated antagonists which were present throughout superfusion. Ordinates, evoked overflow of tritium, calculated from Sn/S1 ratios and expressed as a percentage of the corresponding control (no carbachol). Means±s.e.mean from n=5 to 16 tissue pieces. The concentration-inhibition curves of carbachol given alone in both M4-wildtype and M4-knockout are identical with those in Figure 4C. Significant differences from carbachol alone: #P<0.05.
In M2-knockout atria, bladder and vas deferens, the concentration-response curves of carbachol were flat, with a small maximum (Figure 3). For this reason, although antagonism by methoctramine and pirenzepine was evident, antagonist pKd values could not be quantified with a sufficient degree of certainty (data not shown).
In M4-knockout tissues, the concentration-response curves of carbachol were sufficiently steep (Figure 4) for quantification of antagonist pKd values for methoctramine and pirenzepine. The pKd values are summarized in Table 1. In M4-knockout atria and bladder, the values were very close to the corresponding NMRI, M2-wildtype and M4-wildtype values, yielding again the potency order methoctramine>pirenzepine. In contrast, a different pattern was observed in the vas deferens. Whereas the order of antagonist potencies was pirenzepine⩾methoctramine in vasa deferentia from NMRI mice and the two wildtype strains, the order was reversed to methoctramine>pirenzepine in the M4-knockout vas deferens (Table 1 and Figure 5).
Inhibition of noradrenaline release by endogenous acetylcholine in the urinary bladder
In the next series of experiments, we investigated whether the presynaptic muscarinic receptors mediating inhibition of noradrenaline release could also be activated by endogenously released acetylcholine, rather than being exclusively sites of action of exogenous drugs. These experiments were carried out using the mouse urinary bladder as a model system. Each bladder preparation was stimulated by 20 pulses/50 Hz (S1 and S4), 120 pulses/3 Hz (S2 and S5) and 360 pulses/3 Hz (S3 and S6). The long pulse trains were used to increase the chance that endogenous muscarinic inhibition of noradrenaline release developed. We noted that a considerable α2-adrenergic autoinhibition of noradrenaline release occurred during the long pulse trains but not during the short bursts in bladder segments of NMRI mice, as indicated by the effects of a combined administration of phentolamine (1 μM) and rauwolscine (1 μM) after S3: the overflow elicited by 20 pulses/50 Hz was increased only by 28±12%, whereas the overflow elicited by 120 pulses/3 Hz and 360 pulses/3 Hz was markedly increased by 244±25 and 212±19%, respectively (n=6). Thus, in order to prevent α2-adrenergic autoinhibition of noradrenaline release, α2-autoreceptors were blocked by adding phentolamine (1 μM) and rauwolscine (1 μM) throughout superfusion in all subsequent experiments. In addition, the cholinesterase inhibitor, physostigmine (1 μM), was added throughout superfusion in some experiments in order to raise synaptic acetylcholine levels.
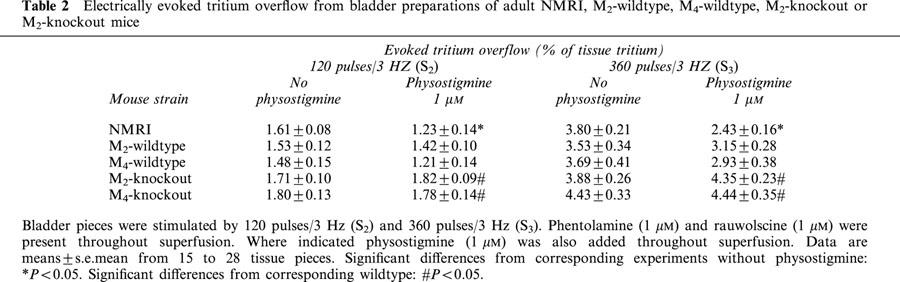
The overflow of tritium elicited by stimulation with 20 pulses/50 Hz was not significantly changed by deletion of either the M2- or the M4-receptor gene or by addition of physostigmine (1 μM) or ipratropium (1 μM; data not shown). Thus, only the results obtained with 120 pulses and 360 pulses/3 Hz are presented in the following. The stimulation-evoked overflow of tritium obtained under these conditions (S2 and S3) is shown in Table 2. Physostigmine (1 μM), when present throughout superfusion, significantly reduced (by 24 and 36%; respectively) the overflow elicited by 120 pulses/3 Hz (S2) and 360 pulses/3 Hz (S3) in bladder preparations from NMRI mice and tended to reduce the overflow in bladder segments from M2- and M4-wildtype mice (Table 2). Strikingly, deletion of either M2- or M4-receptors led to a significant increase (by 28 to 52%) of tritium overflow elicited by 120 (S2) or 360 pulses/3 Hz (S3) in the presence of physostigmine, as compared to the values obtained with the corresponding wildtype control mice (Table 2).
Table 2.
Electrically evoked tritium overflow from bladder preparations of adult NMRI, M2-wildtype, M4-wildtype, M2-knockout or M2-knockout mice
To further investigate muscarinic inhibition of noradrenaline release by endogenous acetylcholine in the mouse bladder, we next administered physostigmine (1 μM) and ipratropium (1 μM) after S3. Physostigmine, when administered after S3 to bladder preparations from NMRI mice not previously treated with physostigmine, reduced the evoked overflow of tritium by 24 to 38% (Figure 6). Under the same conditions, ipratropium had no significant effect on transmitter release (Figure 6). However, ipratropium increased the evoked overflow of tritium by 40 to 52% when added to bladder preparations superfused throughout the experiment with physostigmine (Figure 6).
Figure 6.
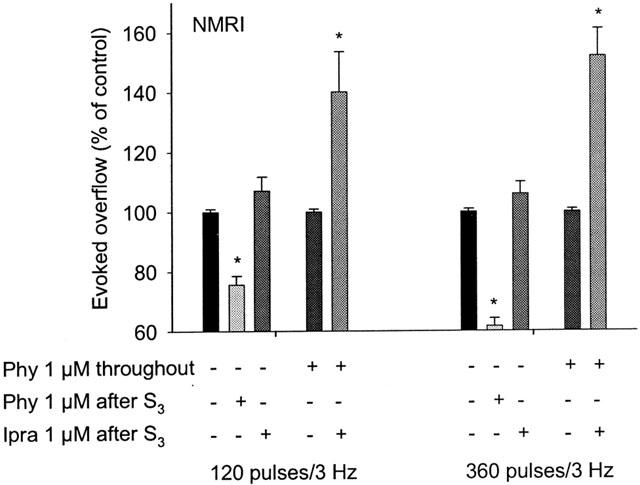
Effects of physostigmine and ipratropium on the evoked overflow of tritium from NMRI bladder. After preincubation with 3H-noradrenaline tissues were superfused and stimulated electrically six times (S1 to S6). S1 and S4 consisted of 20 pulses/50 Hz (results not shown), S2 and S5 of 120 pulses/3 Hz, and S3, and S6 of 360 pulses/3 Hz. Phentolamine (1 μM) and rauwolscine (1 μM) were present throughout superfusion. Physostigmine (Phy) was present either throughout superfusion or was added after S3, and ipratropium (Ipra) was added after S3, as indicated. Ordinates, evoked overflow of tritium, calculated from Sn/Sn-3 ratios and expressed as a percentage of the corresponding control (no drug added after S3). Means±s.e.mean from n=4 to 8 tissue pieces. Significant differences from corresponding control (no drug added after S3): *P<0.05.
Finally, analogous experiments were carried out using bladder preparations from M2- and M4-knockout mice and the two corresponding wildtype control strains to detect the muscarinic receptor subtype(s) responsible for the inhibition of noradrenaline release by endogenously released acetylcholine. The responses observed with bladder preparations from M2- and M4-wildtype mice closely resembled those found with NMRI mice (Figure 7A,B). Physostigmine (1 μM), when administered after S3, reduced the evoked overflow by 15 to 26% (Figure 7A,B). On the other hand, ipratropium (1 μM), when added to bladder preparations pre-exposed to physostigmine, increased the evoked overflow by 22 to 38% (Figure 7A,B).
Figure 7.
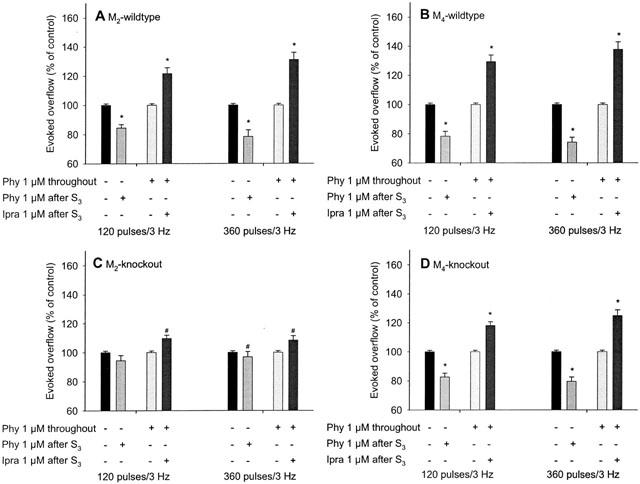
Effects of physostigmine (Phy) and ipratropium (Ipra) on the evoked overflow of tritium from M2-wildtype (A), M4-wildtype (B), M2-knockout (C) and M4-knockout bladder (D). For details, see legend to Figure 6. Means±s.e.mean from n=5 to 13 tissue pieces. Significant differences from corresponding control (no drug added after S3): *P<0.05. Significant differences from corresponding wildtype: #P<0.05. The tendencies of an inhibitory effect of physostigmine and a facilitatory effect of ipratropium, added after S3, are not significant (C).
Deletion of M2-receptors abolished both the inhibitory effect of physostigmine in previously physostigmine-free tissues and the facilitatory effect of ipratropium in tissues pre-exposed to physostigmine (Figure 7C). The small remaining effects in M2-knockout preparations were not statistically significant (Figure 7C). Deletion of the M4-receptor, in contrast, did not significantly change the effects of physostigmine and ipratropium. However, there was a trend towards reduced physostigmine and ipratropium effects, as compared to the corresponding values from M4-wildtype mice (compare Figure 7D with 7B).
Discussion
In this study, we initially demonstrated that mouse atria, urinary bladder, and vas deferens possess presynaptic release-inhibiting muscarinic receptors at their sympathetic fibres. In the mouse, muscarinic heteroreceptors on sympathetic axons have been described previously in atria (Costa & Majewski, 1991) and cultured sympathetic neurons (Göbel et al., 2000). This is the first report demonstrating their presence in mouse urinary bladder and vas deferens.
Identification of muscarinic receptor subtypes inhibiting noradrenaline release
Previous work has shown that presynaptic muscarinic receptors at postganglionic sympathetic axons are not homogeneous (Fuder & Muscholl, 1995). It is well known that these receptors can mediate inhibition as well as facilitation of noradrenaline release (Fuder & Muscholl, 1995). The release-facilitating receptors are likely to represent M1 receptors across tissues and species (Muscholl et al., 1989; Vizi et al., 1989; Habermeier-Muth et al., 1990; Costa & Majewski, 1991; Casado et al., 1992; Somogyi et al., 1996). In contrast, the majority of release-inhibiting receptors were suggested to be M2. However, the existence of release-inhibiting M1- and M3-receptors has also been proposed in certain tissues, and in some studies the identity of the presynaptic muscarinic receptors remained unclear (see Introduction). In most cases, these classifications were based on the use of muscarinic antagonists with a limited degree of subtype selectivity, which may be responsible for at least some of the discrepant results (see Introduction; Fuder & Muscholl, 1995).
Our knockout approach confirms the heterogeneity of presynaptic muscarinic heteroreceptors at postganglionic sympathetic axons and reveals an additional, hitherto unsuspected heterogeneity: in all three tissues, the sympathetic nerve terminals are endowed with at least two distinct muscarinic receptor subtypes mediating inhibition of transmitter release. The carbachol responses and antagonist potencies determined with preparations from M2-wildtype and M4-wildtype mice were generally similar to those obtained with NMRI mice (Figures 2 to 5; Table 1). Therefore, the presynaptic muscarinic heteroreceptors are essentially the same in the three wildtype mouse strains.
Deletion of M2-receptors significantly reduced the maximal inhibitory effect of carbachol on noradrenaline release in all tissues: by 62% in atria, by 72% in the urinary bladder, and by 41% in the vas deferens (Figure 3). These observations show that M2-receptors play a key role as release-inhibiting heteroreceptors in each of the three tissues. However, in all tissues lacking M2-receptors, a significant inhibition by carbachol remained, clearly indicating the additional involvement of non-M2-receptors in each of the three tissues. This complexity remained undetected in functional studies using different muscarinic antagonists in wildtype mice, probably due to the limited subtype selectivity of the antagonists.
What, then, is the identity of the non-M2-heteroreceptors? We observed that deletion of the M4-receptors did not significantly affect the release-inhibiting effect of carbachol in atria and urinary bladder (Figure 4A,B) but moderately reduced (by 28%; P<0.05) the maximal inhibitory effect of carbachol in the vas deferens (Figure 4C). This observation may support the view that M4-heteroreceptors are also present in the vas deferens. It also suggests that the non-M2-receptors in atria and bladder may not be M4. However, the possibility exists that a contribution of presynaptic M4-receptors to inhibition of noradrenaline release in atria and bladder remained undetected due to the predominance of the M2 pathway which may be sufficient to allow maximal inhibition. In fact, such a pattern has been found previously for α2A- and α2C-autoreceptors: although both co-exist on sympathetic fibres, only deletion of the α2A-adrenoceptor but not deletion of the α2C-adrenoceptor reduced the release-inhibiting effect of α2-adrenoceptor agonists; a contribution of the α2C-autoreceptor became manifest only as inhibition remaining in the α2A-receptor knockout mice (Altman et al., 1999; Hein et al., 1999; Trendelenburg et al., 1999).
In order to gain additional insight into the identity of the muscarinic heteroreceptors mediating inhibition of noradrenaline release, we also tried to determine antagonist potencies of methoctramine and pirenzepine against carbachol in all tissues and mouse strains. In atria and bladder from NMRI, M2-wildtype, M4-wildtype and M4-knockout mice, the potency order was methoctramine>pirenzepine, which is the M2 order (see Introduction), in accord with the view that in both tissues of the four strains the M2-heteroreceptor detected by the M2-knockout experiment determined the pharmacological properties of the overall M2-plus non-M2-heteroreceptor population. In contrast, in the vas deferens of NMRI, M2-wildtype and M4-wildtype mice the potency order was pirenzepine⩾methoctramine, the M3/4/5 order: thus, it seems that the non-M2-heteroreceptors determined the pharmacological properties of the overall heteroreceptor population in the vas deferens of wildtype mice.
Unfortunately, the residual effect of carbachol in tissues from M2-knockout was too small in size (Figure 3) to permit an acceptable quantification of antagonist potencies. The antagonist experiments are therefore not useful to classify the non-M2-heteroreceptors in the atria and bladder. In contrast, the antagonist potencies determined in M4-knockout preparations support the idea that non-M2-heteroreceptors in the vas deferens may be M4, as mentioned above: the potency order in the vas deferens of wildtype mice was pirenzepine⩾methoctramine, in accord with a pharmacological predominance of non-M2-receptors. Deletion of the M4-receptor reversed the order to methoctramine>pirenzepine, indicating that the remaining M2-heteroreceptors became dominant in the absence of M4-receptors (Figure 5). These findings support the concept that M4-heteroreceptors co-exist with M2-heteroreceptors in the vas deferens.
Activation of muscarinic heteroreceptors by released acetylcholine in the urinary bladder
An early study in rabbit atria (Löffelholz & Muscholl, 1970) showed that electrical stimulation of the vagus nerves reduced the release of noradrenaline elicited by stimulation of the accelerans nerves, suggesting that presynaptic muscarinic receptors were operative physiologically. Consistent with this observation, subsequent studies with simultaneous field stimulation of sympathetic and parasympathetic axons showed, for example, that atropine increased the release of noradrenaline in mouse atria and rat urinary bladder, at least under certain experimental conditions (Somogyi & de Groat, 1990; Costa & Majewski, 1991). To study whether endogenously released acetylcholine could trigger muscarinic receptor-mediated inhibition of noradrenaline release in the urinary bladder, and if so through which subtype(s), we carried out systematic release studies using bladder preparations from NMRI as well as M2- and M4-knockout mice and the two corresponding wildtype control strains. In preparations from NMRI and M2- and M4-wildtype mice, physostigmine (1 μM) reduced, whereas ipratropium (1 μM), if added in the presence of physostigmine, increased the stimulated release of noradrenaline, consistent with the concept that endogenous acetylcholine activates release-inhibiting presynaptic muscarinic receptors. To reveal this effect, the pulse trains applied to stimulate transmitter release had to be relatively long, presumably to release sufficient amounts of acetylcholine and to leave time for muscarinic inhibition to develop. When trains of 120 or 360 pulses/3 Hz were used, physostigmine reduced the release of noradrenaline both when present throughout superfusion (Table 2; this effect was significant only in bladder pieces from NMRI mice) and when added after S3 (Figures 6 and 7A,B). The release-facilitating effect of ipratropium became evident only in the presence of physostigmine, suggesting that high synaptic levels of acetylcholine were required to reveal this effect (Figure 6).
The release-inhibiting effects of endogenously released acetylcholine in mouse urinary bladder are mediated mainly by M2-receptors. This notion is supported primarily by the following two observations. First, in bladder preparations from M2-knockout mice, noradrenaline release in the presence of physostigmine throughout superfusion was increased, as compared to the corresponding responses in M2-wildtype mice (Table 2), suggesting that activation of M2-receptors by endogenously released acetylcholine caused inhibition of noradrenaline release. Second, both the release-inhibitory effect of physostigmine and the release-facilitatory effect of ipratropium, when added in the presence of physostigmine after S3, were virtually abolished in bladder preparations from M2-knockout mice (Figure 7C). As shown in Table 2, deletion of the M4-receptor also led to a significant increase in the release of noradrenaline in physostigmine-treated bladder preparations. This observation is consistent with a minor contribution of M4-receptors, besides the predominant M2-receptors, to the release-inhibitory effects of endogenously released acetylcholine in mouse urinary bladder.
In conclusion, presynaptic release-inhibiting muscarinic receptors occur on the sympathetic axons innervating the heart atria, urinary bladder and vas deferens of mice. In all three tissues, these receptors represent mixtures of M2- and non-M2-receptors. The non-M2-receptors remain unknown in atria and the bladder, and appear to represent primarily M4-receptors in the vas deferens. This is the first report demonstrating the co-existence of multiple inhibitory presynaptic muscarinic receptor subtypes in one and the same tissue. Our results underscore the usefulness of muscarinic receptor knockout mice to reveal the molecular identity of muscarinic heteroreceptors mediating inhibition of noradrenaline release. These results should contribute to a better understanding of the molecular mechanisms governing the interplay between the sympathetic and parasympathetic nervous systems under physiological and pathophysiological conditions.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft Grant SFB 505 (to A.-U. Trendelenburg) and by a CRADA between the NIDDK (to J. Wess) and the Eli Lilly Research Laboratories. The authors gratefully acknowledge the support and advice of Professor Klaus Starke.
Abbreviations
- NMRI
Naval Medical Research Institute
- PCR
polymerase chain reaction
References
- ALEXANDER S.P.H., MATHIE A., PETERS J.A. TiPS Receptor and Ion Channel Nomenclature. Cambridge: Elsevier Trends Journals; 2001. 2001. [Google Scholar]
- ALTMAN J.D., TRENDELENBURG A.U., MACMILLAN L., BERNSTEIN D., LIMBIRD L., STARKE K., KOBILKA B.K., HEIN L. Abnormal regulation of the sympathetic nervous system in α2A-adrenergic receptor knockout mice. Mol. Pharmacol. 1999;56:154–161. doi: 10.1124/mol.56.1.154. [DOI] [PubMed] [Google Scholar]
- CASADO M.A., MARIN J., SALAICES M. Evidence for M1 muscarinic cholinoceptors mediating facilitation of noradrenaline release in guinea-pig carotid artery. Naunyn-Schmiedeberg's Arch. Pharmacol. 1992;346:391–394. doi: 10.1007/BF00171079. [DOI] [PubMed] [Google Scholar]
- CAULFIELD M.P., BIRDSALL N.J.M. International union of pharmacology. XVII. classification of muscarinic acetylcholine receptors. Pharmacol. Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- COSTA M., MAJEWSKI H. Evidence for facilitatory and inhibitory muscarinic receptors on postganglionic sympathetic nerves in mouse isolated atria. Br. J. Pharmacol. 1991;102:855–860. doi: 10.1111/j.1476-5381.1991.tb12266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DÖRJE M., WESS J., LAMBRECHT G., TACKE R., MUTSCHLER E., BRANN M.R. Antagonist binding profiles of five cloned human muscarinic receptors. J. Pharmacol. Exp. Ther. 1991;256:727–733. [PubMed] [Google Scholar]
- EGLEN R.M., HEGDE S.S., WATSON N. Muscarinic receptor subtypes and smooth muscle function. Pharmacol. Rev. 1996;48:531–565. [PubMed] [Google Scholar]
- FUDER H., MUSCHOLL E. Heteroreceptor-mediated modulation of noradrenaline and acetylcholine release from peripheral nerves. Rev. Physiol. Biochem. Pharmacol. 1995;126:265–412. doi: 10.1007/BFb0049778. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F.The classification of adrenoceptors (adrenergic receptors). An evaluation from the standpoint of receptor theory Catecholamines. Handbook of experimental pharmacology 1972Vol. 33Berlin, Heidelberg, New York: Springer; 283–335.eds. Blaschko, H. & Muscholl, E. [Google Scholar]
- GÖBEL I., TRENDELENBURG A.U., COX S.L., MEYER A., STARKE K. Electrically evoked release of [3H]noradrenaline from mouse cultured sympathetic neurons: release-modulating heteroreceptors. J. Neurochem. 2000;75:2087–2094. doi: 10.1046/j.1471-4159.2000.0752087.x. [DOI] [PubMed] [Google Scholar]
- GOMEZA J., SHANNON H., KOSTENIS E., FELDER C., ZHANG L., BRODKIN J., GRINBERG A., SHENG H., WESS J. Pronounced pharmacologic deficits in M2 muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 1999a;96:1692–1697. doi: 10.1073/pnas.96.4.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOMEZA J., ZHANG L., KOSTENIS E., FELDER C., BYMASTER F., BRODKIN J., SHANNON H., XIA B., DENG C.X., WESS J. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M4 muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 1999b;96:10483–10488. doi: 10.1073/pnas.96.18.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIMM U., FUDER H., MOSER U., BÄUMERT H.G., MUTSCHLER E., LAMBRECHT G. Characterization of the prejunctional muscarinic receptors mediating inhibition of evoked release of endogenous noradrenaline in rabbit isolated vas deferens. Naunyn-Schmiedeberg's Arch. Pharmacol. 1994;349:1–10. doi: 10.1007/BF00178199. [DOI] [PubMed] [Google Scholar]
- HABERMEIER-MUTH A., ALTES U., FORSYTH K.M., MUSCHOLL E. A presynaptic excitatory M1 muscarine receptor at postganglionic cardiac noradrenergic nerve fibres that is activated by endogenous acetylcholine. Naunyn-Schmiedeberg's Arch. Pharmacol. 1990;342:483–489. doi: 10.1007/BF00169033. [DOI] [PubMed] [Google Scholar]
- HEIN L., ALTMAN J.D., KOBILKA B.K. Two functionally distinct α2-adrenergic receptors regulate sympathetic neurotransmission. Nature. 1999;402:181–184. doi: 10.1038/46040. [DOI] [PubMed] [Google Scholar]
- LAZARENO S., BIRDSALL N.J.M. Pharmacological characterization of acetylcholine-stimulated [35S]-GTPγS binding mediated by human muscarinic m1-m4 receptors: antagonist studies. Br. J. Pharmacol. 1993;109:1120–1127. doi: 10.1111/j.1476-5381.1993.tb13738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAZARENO S., BUCKLEY N.J., ROBERTS F.F. Characterization of muscarinic M4 binding sites in rabbit lung, chicken heart, and NG108-15 cells. Mol. Pharmacol. 1990;38:805–815. [PubMed] [Google Scholar]
- LIMBERGER N., TRENDELENBURG A.U., STARKE K. Pharmacological characterization of presynaptic α2-autoreceptors in rat submaxillary gland and heart atrium. Br. J. Pharmacol. 1992;107:246–255. doi: 10.1111/j.1476-5381.1992.tb14494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LINDMAR R., LÖFFELHOLZ K., MUSCHOLL E. A muscarinic mechanism inhibiting the release of noradrenaline from peripheral adrenergic nerve fibres by nicotinic agents. Br. J. Pharmacol. Chemother. 1968;32:280–294. doi: 10.1111/j.1476-5381.1968.tb00972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LÖFFELHOLZ K. On the release of noradrenaline from the perfused rabbit heart caused by acetylcholine. Naunyn-Schmiedeberg's Arch. Pharmacol. 1967;258:108–122. [PubMed] [Google Scholar]
- LÖFFELHOLZ K., MUSCHOLL E. The effect of dexamphetamine on the output of noradrenaline from isolated rabbit hearts. Naunyn-Schmiedeberg's Arch. Pharmacol. 1970;266:393–394. [PubMed] [Google Scholar]
- MIRANDA H.F., DURAN E., BUSTAMANTE D., PAEILE C., PINARDI G. Pre- and postjunctional muscarinic receptor subtypes in the vas deferens of rat. Gen. Pharmacol. 1994;8:1643–1647. doi: 10.1016/0306-3623(94)90366-2. [DOI] [PubMed] [Google Scholar]
- MUSCHOLL E., FORSYTH K.M., HABERMEIER-MUTH A. A presynaptic excitatory M1 muscarine receptor at postganglionic cardiac adrenergic fibres that is activated by endogenous acetylcholine. Naunyn-Schmiedebergs Arch. Pharmacol. 1989;339:R88. doi: 10.1007/BF00169033. [DOI] [PubMed] [Google Scholar]
- OLMEZ E., OGUZ GUC M., ILHAN M. Inhibitory muscarinic cholinoceptors on postganglionic sympathetic nerves in the guinea pig isolated atrium are of the M3 subtype. Pharmacology. 1995;51:112–117. doi: 10.1159/000139323. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., GÖTHERT M. Interactions between the presynaptic α2-autoreceptor and presynaptic inhibitory heteroreceptors on noradrenergic neurones. Brain Res. Bull. 1998;47:129–132. doi: 10.1016/s0361-9230(98)00068-9. [DOI] [PubMed] [Google Scholar]
- SOMOGYI G.T., DE GROAT W.C. Modulation of the release of [3H]norepinephrine from the base and body of the rat urinary bladder by endogenous adrenergic and cholinergic mechanisms. J. Pharmacol. Exp. Ther. 1990;255:204–210. [PubMed] [Google Scholar]
- SOMOGYI G.T., TANOWITZ M., ZERNOVA G., DE GROAT W.C. M1 muscarinic receptor-induced facilitation of ACh and noradrenaline release in the rat bladder is mediated by protein kinase C. J. Physiol. 1996;496:245–254. doi: 10.1113/jphysiol.1996.sp021681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STARKE K. Presynaptic α-autoreceptors. Rev. Physiol. Biochem. Pharmacol. 1987;107:73–146. [PubMed] [Google Scholar]
- TRENDELENBURG A.U., COX S.L., SCHELB V., KLEBROFF W., KHAIRALLAH L., STARKE K. Modulation of 3H-noradrenaline release by presynaptic opioid, cannabinoid and bradykinin receptors and β-adrenoceptors in mouse tissues. Br. J. Pharmacol. 2000;130:321–330. doi: 10.1038/sj.bjp.0703305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRENDELENBURG A.U., HEIN L., GAISER E.G., STARKE K. Occurrence, pharmacology and function of presynaptic α2-autoreceptors in α2A/D-adrenoceptor-deficient mice. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;360:540–551. doi: 10.1007/s002109900093. [DOI] [PubMed] [Google Scholar]
- TRENDELENBURG A.U., KLEBROFF W., HEIN L., STARKE K. A study of presynaptic α2-autoreceptors in α2A/D-, α2B-, α2C-adrenoceptor-deficient mice. Naunyn-Schmiedeberg's Arch. Pharmacol. 2001;364:117–130. doi: 10.1007/s002100100423. [DOI] [PubMed] [Google Scholar]
- TRENDELENBURG A.U., SUTEJ I., WAHL C.A., MOLDERINGS G.J., RUMP L.C., STARKE K. A re-investigation of questionable subclassifications of presynaptic α2-autoreceptors: rat vena cava, rat atria, human kidney and guinea-pig urethra. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:721–737. doi: 10.1007/pl00005111. [DOI] [PubMed] [Google Scholar]
- VIZI E.S., KOBAYASHI O., TÖRÖCSIK A., KINJO M., NAGASHIMA H., MANABE N., GOLDINER P.L., POTTER P.E., FOLDES F.F. Heterogeneity of presynaptic muscarinic receptors involved in modulation of transmitter release. Neuroscience. 1989;31:259–267. doi: 10.1016/0306-4522(89)90048-1. [DOI] [PubMed] [Google Scholar]
- WAHL C.A., TRENDELENBURG A.U., STARKE K. Presynaptic α2-autoreceptors in mouse heart atria: evidence for the α2D subtype. Naunyn-Schmiedeberg's Arch. Pharmacol. 1996;354:253–261. doi: 10.1007/BF00171055. [DOI] [PubMed] [Google Scholar]
- WAUD D.R.Analysis of dose-response relationships Advances in General and Cellular Pharmacology 1976Vol 1New York, London: Plenum; 145–178.eds. Narahshi, T. & Bianchi, C.P. [Google Scholar]
- ZHOU H., MEYER A., STARKE K., GOMEZA J., WESS J., TRENDELENBURG A.U. Heterogeneity of release-inhibiting muscarinic autoreceptors in heart atria and urinary bladder: a study with M2- and M4-receptor-deficient mice. Naunyn-Schmiedeberg's Arch. Pharmacol. 2002;365:112–122. doi: 10.1007/s00210-001-0517-7. [DOI] [PubMed] [Google Scholar]