

Abstract
The mechanism of action by which methotrexate (MTX) exerts its anti-inflammatory and immunosuppressive effects remains unclear. The aim of this study is to investigate the hypothesis that MTX exerts these effects via the production of reactive oxygen species (ROS).
Addition of MTX (100 nM–10 μM) to U937 monocytes induced a time and dose dependent increase in cytosolic peroxide [peroxide]cyt from 6–16 h. MTX also caused corresponding monocyte growth arrest, which was inhibited (P<0.05) by pre-treatment with N-acetylcysteine (NAC; 10 mM) or glutathione (GSH; 10 mM). In contrast, MTX induction of [peroxide]cyt in Jurkat T cells was more rapid (4 h; P<0.05), but was associated with significant apoptosis at 16 h at all doses tested (P<0.05) and was significantly inhibited by NAC or GSH (P<0.05).
MTX treatment of monocytes (10 nM–10 μM) for 16 h significantly reduced total GSH levels (P<0.05) independently of dose (P>0.05). However, in T-cells, GSH levels were significantly elevated following 30 nM MTX treatment (P<0.05) but reduced by doses exceeding 1 μM compared to controls (P<0.05).
MTX treatment significantly reduced monocyte adhesion to 5 h and 24 h LPS (1 μg ml−1) activated human umbilical vein endothelial cells (HUVEC; P<0.05) but not to resting HUVEC. Pre-treatment with GSH prevented MTX-induced reduction in adhesion.
In conclusion, ROS generation by MTX is important for cytostasis in monocytes and cytotoxicity T-cells. Furthermore, MTX caused a reduction in monocyte adhesion to endothelial cells, where the mechanism of MTX action requires the production of ROS. Therefore its clinical efficacy can be attributed to multiple targets.
Keywords: Methotrexate, reactive oxygen species, adhesion, glutathione, growth arrest, apoptosis, inflammation, rheumatoid arthritis, immunosuppression
Introduction
The folate antagonist methotrexate (MTX) is a potent cytotoxic agent, initially developed for the treatment of malignancies (Farber et al., 1956) and is presently used in non-neoplastic diseases as an anti-inflammatory agent and immunosuppressant. MTX is the most widely used drug in the second line treatment of rheumatoid arthritis (RA) as one of the few disease modifying antirheumatic agents available. It has a well-documented efficacy relative to toxicity profile, however, its ability to reduce radiological progression is uncertain (Rau et al., 1997).
The immunosuppressive activities of MTX have been studied in the context of cell proliferation, and recruitment. Paillot et al. (1998) and Genestier et al. (1998a) describe the induction of activation dependent T-cell apoptosis by low dose MTX in vitro. This is supported by da Silva et al. (1996), who examined the cytotoxicity of several chemotherapeutic drugs and observed chromatin condensation, membrane blebbing and nuclear fragmentation, typical of apoptosis in Jurkat T cells. However, MTX inhibits growth and induces terminal differentiation of keratinocytes, indicating a cell-type specific response (Schwartz et al., 1995).
MTX mediated modulation of cytokine secretion, particularly IL-1β, IL-6 and TNFα, and cyclo-oxygenase and lipoxygenase activities have been widely investigated, however there is no consistent effect observed either in vitro or in vivo (Andersson et al., 2000; Bondeson & Sundler, 1995; Hawkes et al., 1993; 1994; Hu et al., 1998; Sperling et al., 1992; Williams et al., 1999).
Recent advances in the molecular studies of inflammation suggest that cell–cell interactions by specific adhesion molecules could be important targets for immunosuppression, where down-regulation of both CD18 on mononuclear cells and ICAM-1 on endothelial cells has been described following treatment with MTX during cardiac allograft transplantation in rats (Ciesielski et al., 1998a, b).
The cytotoxic actions of MTX have been attributed to its inhibition of RNA, DNA and protein synthesis, and release of adenosine. It is thought that the cytotoxicity of MTX is dependent on its property as a powerful antimetabolite for folate, competitively inhibiting dihydrofolate reductase (DHFR), preventing regeneration of tetrahydrafolate (FH4) from dihydrofolate (FH2) and thereby inhibiting de novo purine and pyrimidine synthesis (Budzik et al., 2000; as reviewed in Allison (2000)). However, the anti-inflammatory actions are unlikely to arise from this property, as supplementation with folate in RA patients on MTX to attenuate toxicity does not compromise its clinical efficacy (Morgan et al., 1994). Furthermore, the dosing regimen for RA is in the order of three orders of magnitude lower than for oncological disease (Coombe et al., 1995; Hamilton & Kremer, 1995; Oguey et al., 1992). Fairbanks et al. (1999) described that in activated peripheral blood lymphocytes (PBL), the immunosuppressant properties of MTX resulting cytostasis were due to the inhibition of the enzyme amidophosphoribosyl transferase leading to elevated PP-ribose-P and stimulating UTP synthesis but not via the inhibition of the two folate dependent enzymes.
Conflicting evidence surrounds the effects of MTX on blockade of 5-amino-imadizole carboxamide ribonucleotide (AICAR) transformylase. Intracellular MTX is converted to the polyglutamated forms, potent inhibitors of AICAR transformylase leading to an increase in extracellular adenosine (Baggott et al., 1993; Bannwarth et al., 1994). Adenosine has been reported to be a potent endogenous anti-inflammatory purine nucleotide that inhibits superoxide generation (Cronstein et al., 1985; Roberts et al., 1985), induces apoptosis in activated PBL (Genestier et al., 1998a), neutrophil mediated damage to the endothelium and leukocyte accumulation in the inflamed hamster air pouch model (Cronstein et al., 1993). In contrast, more recent work has shown that AICAR transformylase inhibition by polyglutamated forms of MTX in human T-cells, causes a dose-dependent reduction in adenosine and guanosine pools (Budzik et al., 2000). Further, the contribution of adenosine production in the cytotoxic action of MTX in human or murine activated T-cells has been described as minimal (Genestier et al., 1998a; Paillot et al., 1998).
Few redox-altering properties of MTX have been described. MTX treatment of peripheral blood neutrophils (PBN) induces a dose dependent increase in peroxide levels (Gressier et al., 1994) and is associated with a loss in the cellular and mitochondrial levels of the anti-oxidant glutathione (Babiak et al., 1998; Neuman et al., 1999). Therefore, we propose that generation of ROS is an important mechanism in the immunosuppressive effects of MTX. To address this hypothesis, we have investigated the effects of MTX on monocyte and T cell intracellular redox status, cell cycle distribution and adhesion of monocytes to endothelial cells. We show for the first time, that reactive oxygen species (ROS) are generated by MTX mediating functional changes in leukocytes, where scavengers of ROS are effective inhibitors of MTX induced cell cycle arrest, apoptosis and changes in monocyte-endothelial adhesion.
Methods
Materials
All reagents were obtained from Sigma Chemical Company (Poole, U.K.) and solvents from Fisher (Loughborough, U.K.) unless otherwise stated. RPMI 1640, foetal bovine serum and penicillin (1000 U ml−1)/streptomycin (10,000 μg ml−1) were purchased from GibcoBRL (Paisley, U.K.). Isotype negative controls and monoclonal antibodies (MoAb) conjugated to phycoerythrin (PE) or fluorescein isothiocyanate (FITC) were from Diaclone Research, (Besançon Cedex, France).
2′,7′ dichlorodihydrofluorescein diacetate (DCFH-DA) was dissolved in dimethyl sulphoxide (DMSO) to a stock solution of 75 mM. Subsequent dilutions were made with DMSO and serum free RPMI 1640. In the final ROS assay, the final concentration of DMSO employed did not exceed 0.1%.
Cell culture and stimulation
The acute human T-cell leukaemia cell line Jurkat T-cells and the human monocytic cell line U937 were maintained in RPMI 1640 media, supplemented with 10% heat inactivated foetal calf serum and 1% (of stock) penicillin/streptomycin. The number of viable cells was determined by Trypan blue exclusion using an improved Neubauer haemocytometer (Weber Scientific International Ltd., Teddington, U.K.). Viable cells at a concentration of 2×106 ml−1 were serum starved for 4 h in the presence or absence of 10 mM glutathione (GSH) or N-acetylcysteine (NAC) in the described incubator conditions prior to MTX treatment. Where indicated, cells were treated with MTX for the times and concentrations noted. All incubations were performed at 37°C in a humidified 5% CO2/95% air incubator. Individual additions to cell suspensions did not exceed 1% of the total volume and were dispersed with gentle mixing by pipette. Control experiments were conducted under identical conditions as tests, employing vehicle treatment.
Primary T cell purification
PWB was diluted with PBS (0.1% BSA; 1 : 3) and 30 ml were layered onto 15 ml of Lymphoprep (Nycomed) and centrifuged (157×g 20 min, Heraeus Instruments) at 20°C. Plasma (5 ml) was taken off to remove platelets and tubes were further centrifuged at 381×g for 20 min. Mononuclear cells at the plasma Lymphoprep interface were transferred to fresh 15 ml tubes, diluted 1 : 3 with ice cold PBS (0.1% BSA) and centrifuged for 8 min 235×g at 2°C. Cells were washed and resuspended twice in ice cold PBS (0.1% BSA) at 235×g (8 min, 2°C). Mononuclear cells (MNC) were counted using a haemocytometer (Neubauer). T cells were isolated using the T cell negative isolation kit (Dynal), as previously described. Purity levels assessed as CD3 positivity, using flow cytometry. Only isolated CD3+ve monocytes of >90% purity were used for MTX treatment, analysis of intracellular peroxide and apoptosis determination. Primary T cell activation was achieved through culture in RPMI 1640 media, supplemented with 10% heat inactivated foetal calf serum and 1% (of stock) penicillin/streptomycin in the presence of PHA (2 μg ml−1) for 3 days, where activation was confirmed by CD25 expression.
Flow cytometric DNA cell cycle analysis
Cell stimulation was discontinued by removing 1 ml of treated cell suspension, centrifuging at 1000×g for 5 min and washing twice with 1 ml of ice cold phosphate buffered saline (PBS; 0.137 M NaCl, 0.0027 M KCl, 0.01 M Na2HPO4, 0.002 M KH2PO4). PBS washed cells were centrifuged at 1000×g for 5 min, the supernatant removed and the resulting cell pellet re-suspended in 1 ml hypotonic fluorochrome solution (50 μg ml−1 propidium iodide in 0.1% sodium citrate and 0.1% Triton X-100). Samples were incubated in the dark at 4°C for up to 24 h until flow cytometric analysis (Nicolletti et al., 1991).
Propidium iodide (PI) fluorescence of individual nuclei was measured using an EPICS® XL-MCL flow cytometer (Beckman-Coulter, Miami, U.S.A.) equipped with a 488 nm air-cooled Argon laser. Forward scatter (FS) and side scatter (SS) of the nuclei were simultaneously measured in addition to linear and log red fluorescence (FL3, bandwidth 605 nm–635 nm). Clumps of nuclei were eliminated by appropriate gating. For each analysis, 20,000 events were recorded on the gated FL3 detector. The percentage of apoptotic nuclei was determined by quantifying the number of hypoploid (subdiploid) nuclei. Values were then expressed a percentage of specific apoptosis according to the formula specific apoptosis=(T−C)/(100−c)×100, where T equals the percentage of apoptotic events from treated cells, and C equals the percentage of apoptotic events from control cells (Genestier et al., 1998b). The percentage of nuclei in the G0/G1 phase of the cell cycle was quantified using Multi-Cycle for Windows (Phoenix Flow Systems, San Diego, U.S.A.).
Flow cytometric assay for cytosolic peroxide production
For measurement of cytosolic peroxide ([peroxide]cyt) by flow cytometry, the procedure was adapted from Bass et al. (1983). Briefly, 50 μM of DCFH-DA was added to cell suspensions 40 min before the termination of the MTX treatment period. DCFH-DA rapidly diffuses into the cytosol of cells where it is hydrolysed to the non-fluorescent, oxidation sensitive DCFH. In the presence of [peroxide]cyt, DCFH is rapidly oxidized to the non-diffusible, fluorescent DCF. Immediately following agent/DCFH-DA incubation, the log FL1 fluorescence (green light, band width 505–545 nm) of viable cells, determined by their FS and SS properties were analysed by flow cytometry and the median (MdX) values noted. Ten thousand cells were examined from each sample on the log FL1 detector. ΔDCF represents the difference in MdX fluorescence of MTX treated samples from vehicle treated controls.
Determination of cellular glutathione
This was done according to the recycling assay of Tietze (1969) as modified by Anderson (1985). Briefly, at the end of each treatment period, samples were washed with ice cold PBS by centrifugation at 2300 r.p.m. for 5 min. An aliquot was removed for protein determination and then cells were resuspended in 3.33% 5-sulphosalicyclic acid dihydrate, vortexed vigorously and centrifuged at 13,000 r.p.m. to precipitate interfering proteins. Supernatant (25 μl) was added to 50 μl 5,5′-dithio-bis (2-nitrobenzoic acid; DTNB; 6 mM), and 150 μl NADPH (0.3 mg ml−1) in triplicate, and incubated at 37°C for 3 min, prior to addition of 25 μl glutathione reductase (GSR; 20 U ml−1). The plate was immediately read at 420 nM and then read again at +1 and +5 min to determine DTNB reduction. Test sample values were calibrated against a standard curve of glutathione from 0–60 nmoles, and expressed per milligram of cellular protein.
Protein determination
Protein concentration was determined in quadruplicate using the BCA assay based on the method of Smith et al. (1985), using bovine serum albumin (BSA) as standards to calculate unknowns.
Flow cytometry of cell surface antigens
Monocytes were analysed for CD11a, CD11b, CD18, CD29, CD31 and CD49D expression by flow cytometry following MTX treatment (Beckman Coulter) using appropriate two-way colour compensation with relevant isotype negative controls for each sample. Following treatment, monocytes were washed twice in ice cold PBS, incubated with appropriate combinations MoAb conjugated to PE or FITC at greater than 10 μl/106 cells for 30 min in the dark. Optilyse (Beckman Coulter; 250 μl) was added to fix the samples, mixed and incubated at RT in the dark for 10 min. Each sample was diluted 1 : 1 with Isoton (Beckman Coulter), vortexed and analysed on a single parameter histogram of linear fluorescence against event count.
Adhesion assay
Human Umbilical Endothelial Cells (HUVEC) were obtained from umbilical cords by digestion with collagenase and cultured in Endothelial Growth Medium (EGM; BioWhittaker) as described previously (Jaffe et al., 1973). Ethical approval was granted from the Birmingham Woman's Hospital, Edgbaston, Birmingham, U.K. HUVEC were grown to confluence within 24 well plates up to passage 3 in EGM media and were used 24 h after confluence. HUVEC were washed and lipopolysaccharide (LPS; 1 μg ml−1) added for 0, 5 or 24 h. Each well was then washed twice with 1 ml of M199 medium before addition of monocytes. Monocytes were resuspended to 5×106 cells ml−1 and labelled with 2′-7′-bis - 2 - carboxy-5-(6)-carboxyfluorescein-acetoxymethylester (BCECF-AM; 10 μg ml−1) for 30 min in the dark. To investigate the potential role of chemokine signalling, blockade of G0/G1 was achieved by co-incubation with pertussis toxin (500 ng ml−1). Dye loading was quenched by adding 10 ml of PBS (0.1% BSA) and centrifugation at 235×g for 8 min, followed by washing. For the adhesion assay, MTX treated monocytes (0.25×106) and controls in M199 were added to each well of HUVEC and incubated for 30 min at 37°C. Non-attached monocytes were removed by inverted centrifugation at 300 r.p.m. for 5 min (Weber et al., 1996). Adhered cells were lysed with 1 ml of lysis buffer (0.1% Triton X; 0.1 M Tris, pH 8.8) for 30 min in the dark at RT. Fluorescence was analysed on a dual scanning spectrofluorimeter (Ex 485 nm and Em 535 nm; Spectramax GeminiXS, Molecular Devices, Sunnyvale, U.S.A.) utilizing the cut out filter at 520 nm. The number of monocytes adhered to HUVEC calibrated against a standard curve of vehicle treated U937 monocytes, dye loaded with BCECF-AM and fluorescence analysed as described.
Statistical analysis was performed by one-way ANOVA followed by Tukey's or Dunnett's post hoc test analysis, or Student's t-test. A P<0.05 or P<0.01 was considered to be significant.
Results
Exposure of the human monocytic cell line U937 to MTX from 100 nm–10 μM induced an accumulation of nuclei in the G0/G1 phase of the cell cycle when compared to vehicle treated controls in a time and dose dependent manner. This was first evident following 6 h treatment of U937s with 100 nM MTX (P<0.05; Figure 1), where 60% of nucleoids were in G0/G1 compared with control values of 52%, and was enhanced after 16 h incubation with MTX (P<0.01; Figure 1) from 52% in controls to 67% in 100 nM MTX treated cells. After 16 h MTX treatment, the percentage nucleoids in G0/G1 was not significantly different at any of the doses tested (Figure 1). Pre-incubation with NAC or GSH prior to treatment with MTX caused significant abrogation of nucleoid accumulation in G0/G1 at all concentration of MTX examined 100 nM–10 μM, following 16 h treatment (P<0.01; Figure 2). Flow cytometric evaluations of the uptake of the cell-impermeable dye PI showed no difference between vehicle control treated cells and those treated with MTX at all concentrations analysed (10 nM–100 μM; P>0.05) indicating that U937 monocyte viability was not compromised by MTX (data not shown).
Figure 1.
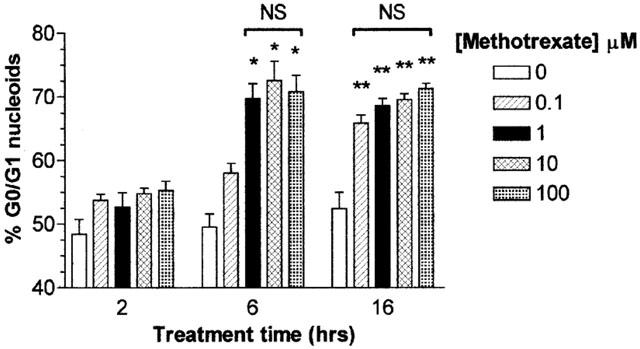
Methotrexate induces growth arrest in U937 monocytes. U937 monocytes (2×106 ml−1) were serum starved for 4 h in RPMI 1640 prior to the addition of 0–100 μM methotrexate for 2, 6 or 16 h. Incubations were terminated by washing the cells twice with ice cold PBS. Cell pellets were resuspended in 1 ml of hypotonic fluorochrome solution and incubated in the dark at 4°C overnight prior to DNA cell cycle analysis by flow cytometry as described in Methods. The percentage G0/G1 DNA content of 20,000 nucleoids from each sample was analysed using MultiCycle™ for Windows (Phoenix Flow Systems, San Diego, U.S.A.). The data is expressed as the mean±s.e.mean of at least four individual experiments, where *(P<0.05) and **(P<0.01) were considered significantly different from control samples by one-way ANOVA followed by Tukey's post hoc test. NS, no significant difference.
Figure 2.
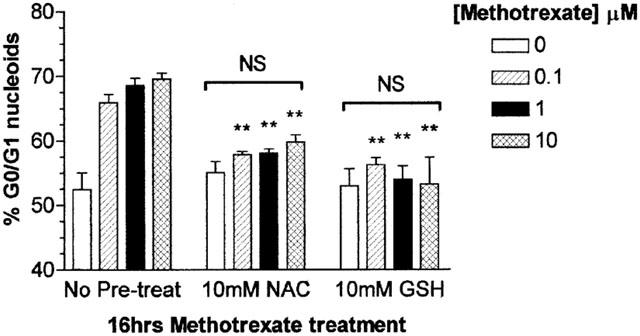
Methotrexate induced growth arrest in U937 monocytes is inhibited by the anti-oxidants glutathione and N-acetylcysteine. U937 monocytes (2×106 ml−1) were serum starved for 4 h in RMPI 1640 in the presence or absence of 10 mM N-acetylcysteine (NAC) or 10 mM glutathione (GSH) prior to the addition of 0–100 μM methotrexate for 16 h. Incubations were terminated by washing the cells twice with ice cold PBS. Cell pellets were resuspended in 1 ml of hypotonic fluorochrome solution and incubated in the dark at 4°C overnight prior to DNA cell cycle analysis by flow cytometry as described in Methods. The percentage G0/G1 DNA content of 20,000 nucleoids from each sample was analysed using MultiCycle™ for Windows (Phoenix Flow Systems, San Diego, U.S.A.). The data is expressed as the mean±s.e.mean of at least four individual experiments where **(P<0.01) were considered significantly different from samples with no pre-treatment by Student's t-test. NS=no significant difference by one-way ANOVA followed by Tukey's post hoc test.
Unlike the effects observed in monocytes, the exposure of Jurkat T-cells to MTX induced a time and dose dependent elevation in the percentage of specific apoptosis (Figure 3). Following 6 h of treatment, no significant apoptosis was induced by MTX at all concentrations examined, whereas at 16 h, all concentrations of MTX greater than 100 nM induced significant T cell apoptosis (P>0.05). A maximum apoptotic response of approximately 35% was observed following 16 h treatment with 1, 10 and 100 μM MTX. Primary T cells, activated for 3 days in vitro with PHA (as demonstrated by a shift in CD25 expression from 2–10% to 30–53%), also exhibited a lower, but significant apoptotic response to MTX (1 μM, 16 h); from a mean cell death in control cells of 5.04±0.25% to 19.7±7.16% apoptotic nucleoids (P<0.05). However, resting T cells were insensitive to MTX, measured as subdiploid nuclei (6.8±1.9% after 16 h). The apoptotic cellular response induced by MTX was significantly inhibited at 16 h by 10 mM NAC or GSH at all MTX doses from 30 nM–10 μM (P<0.05; Figure 3).
Figure 3.
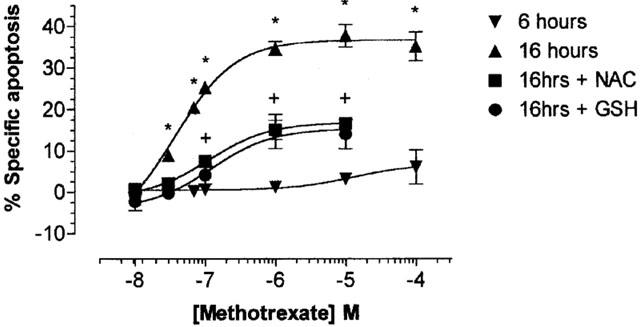
Methotrexate mediated induction of apoptosis in Jurkat T-cells is inhibited by the anti-oxidants glutathione and N-acetylcysteine. Jurkat T-cells (2×106 ml−1) were serum starved for 4 h prior to the addition of methotrexate (0–1×10−4 M) for 6 or 16 h in the presence or absence of 10 mM of the anti-oxidants N-acetylcysteine (NAC) or 10 mM glutathione (GSH). Incubations were performed at 37°C in a 95% air, 5% CO2 humidified atmosphere and terminated by washing the cells twice with ice cold PBS. Cell pellets were resuspended in 1 ml of hypotonic fluorochrome solution and incubated in the dark at 4°C overnight prior to DNA cell cycle analysis by flow cytometry. The sub-diploid DNA content of 20,000 nucleoids from each sample was analysed. The data is expressed as the mean±s.e.mean of at least four individual experiments, expressed as the percentage specific apoptosis where * represents significant difference from control samples (P<0.05) by one-way ANOVA followed by Dunnett's multiple comparison test and + represents significant difference of 16 h methotrexate samples pre-treated with NAC or GSH compared to none pre-treated samples by Student's t-test (P<0.05).
During the first 16 h of monocyte and Jurkat T-cell exposure to MTX, the presence of [peroxide]cyt was monitored by flow cytometry, analysing the fluorescence emission distribution of cells co-exposed to the peroxide sensitive dye DCFH-DA (Figure 4a). The MdX of the DCFH-DA vehicle treated viable T cell population, determined by forward FS and SS properties, increased after 4 h between 100 nM and 10 μM MTX. ΔDCF increased as a function of MTX concentration and incubation period. A maximum increase in ΔDCF of Jurkat T-cells of approximately 150 arbitrary units (a.u.) was observed at 16 h treatment with 1 and 10 μM MTX (P<0.01) and was first significant following 30 nM (P<0.05; Figure 4a). At all treatment periods examined, 1 and 10 μM induced identical changes in DCF fluorescence (P>0.05). The [peroxide]cyt response of primary T cells to MTX (1 μM) was of a lower magnitude than that of Jurkat T cells, indeed a significant loss of [peroxide]cyt was observed after 4 h (P<0.01). As with Jurkat T cells, a significant more than 2 fold elevation in intracellular peroxide was recorded at 16 h post-treatment (P<0.05).
Figure 4.
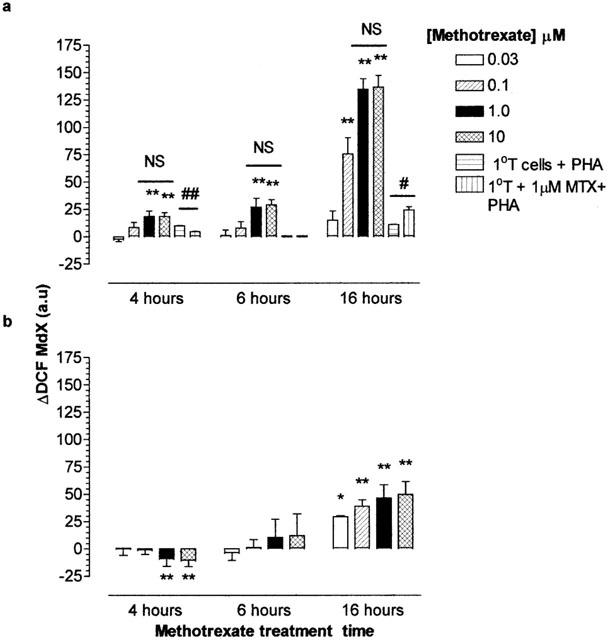
Methotrexate mediated alterations in the cytosolic peroxide levels of Jurkat T-cells, primary T cells and U937 monocytes. Determination of the kinetics for the oxidation of DCFH to DCF. Jurkat T-cells or primary T cells (a) or U937 monocytes (b; 2×106 ml−1) was undertaken in cells which were serum starved in RMPI 1640 for 4 h prior to the addition of 0–100 μM methotrexate for 4, 6 or 16 h. Cells were treated with 50 μM DCFH-DA as described in Methods. At the end of the treatment period, cell samples were analysed immediately for DCF fluorescence by flow cytometry. The median X (MdX) DCF fluorescence of 10,000 cells was analysed per sample. ΔDCF represents the difference in MdX DCF of methotrexate treated cells from that of vehicle treated cells for each time point. All incubations were performed at 37°C in a humidified, 95% air, 5% CO2 atmosphere. The data is expressed as the mean±s.e.mean of four individual experiments where *(P<0.05) and **(P<0.01) were considered significantly different from control samples by one-way ANOVA followed by Tukey's post hoc test analysis. a.u., arbitrary units; NS, no significant difference.
No significant increase in ΔDCF was observed in U937 monocytes until 16 h MTX treatment (30 nM–10 μM; P<0.05; Figure 4b). This deferred increase in ΔDCF corresponds with the appearance of an altered cell cycle profile MTX treated U937 monocytes (Figure 1). Levels of [peroxide]cyt generation were significantly lower for U937 monocytes compared with Jurkat T cells between treatments of 100 nM and 10 μM MTX (P<0.05).
At low doses (10 nM–100 nM, 6 h) MTX treatment of Jurkat T-cells did not affect total GSH levels. At supra 10 nM, total cellular GSH levels decreased although this was not significantly different from controls (P>0.05; Figure 5a). After 16 h of Jurkat T-cell treatment, 30 nM of methotrexate mediated a significant elevation of total GSH levels (P<0.05) to approximately 115% of control treated cells. On increasing the concentration of methotrexate, the total GSH levels of Jurkat T-cells were significant reduced to approximately 60% of control levels (P<0.05; Figure 5b). After 16 h U937 monocyte treatment, MTX at 10 nM and all greater concentrations induced a reduction in total GSH levels compared to controls (P<0.05; Figure 6b). No alteration in the total cellular GSH levels in U937 monocytes was observed following 6 h MTX treatment (P>0.05; Figure 6a). The degree of GSH loss in U937s after 16 h MTX exposure was not statistically different for all concentrations of methotrexate examined (P>0.05; Figure 6b) and was to approximately 65–70% of that of control levels. The total cellular GSH concentration of control U937 monocytes (mean=31.18±1.479 nmoles of GSH mg−1 protein, n=6) was significantly greater than that of Jurkat T-cells (mean=10.95±2.103 nmoles of GSH mg−1 protein, n=6; P<0.01).
Figure 5.
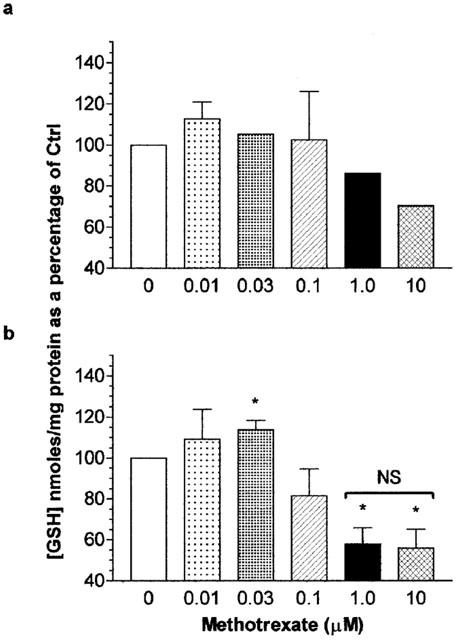
The effect of methotrexate on Jurkat T-cell total cellular glutathione levels. Jurkat T-cells (2×106 ml−1) were serum starved for 4 h prior to the addition of methotrexate (0–10 μM) for 6 h (a) or 16 h (b). At the end of the treatment period, cells were washed twice with ice cold PBS. The total glutathione (GSH) content analysed by spectrophotometric determination of reduced DNTB and quantified against a standard curve of known GSH concentrations as described in Methods. Data represents the mean [GSH] per milligram of cellular protein±s.e.mean of four individual experiments analysed in quadruplicate. Statistical analysis was performed by one-way ANOVA followed by Tukey's post hoc test where *(P<0.05) was considered significant from vehicle control treatments. NS, no significant difference.
Figure 6.
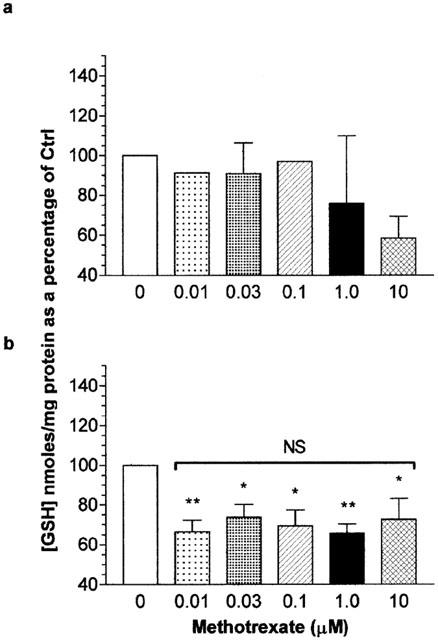
The effect of methotrexate on U937 monocyte total cellular glutathione levels. U937 monocytes (2×106 ml−1) were serum starved for 4 h in RPMI 1640 prior to the addition of methotrexate (0–10 μM) for 6 h (a) or 16 h (b). At the end of the treatment period, cells were washed twice with ice cold PBS. The total glutathione (GSH) content analysed by spectrophotometric determination of reduced DNTB and quantified against a standard curve of known GSH concentrations as described in Methods. Data represents the mean [GSH] per mg of cellular protein±s.e.mean of four individual experiments analysed in quadruplicate. Statistical analysis was performed by one-way ANOVA followed by Tukey's post hoc test where * (P<0.05) and ** (P<0.01) represents significant difference from control samples. NS, no significant difference.
The mechanism by which methotrexate modulates inflammation is widely debated, although the immunosuppressive properties of low dose methotrexate has been postulated to relate to the induction of apoptosis in activated T-cells. However, the effects of methotrexate on the activity of monocytes in the inflammatory responses are unknown. Considering that a characteristic of several inflammatory sites is the recruitment and adhesion of monocytes to sites of inflammation and we have observed that the treatment of U937 monocytes with methotrexate induced no evidence of cell death, the effect of methotrexate on the adhesion of monocytes to endothelial cells was investigated. Methotrexate treatment of U937 monocytes with 30 nM and 1.0 μM did not affect their adhesion to the resting HUVEC (1 μg ml−1 LPS for 0 h; Figure 7a), where the number of control cells bound was 4.2×104±0.2 (approximately 17% of total cells added). However, when the HUVEC monolayers were activated with LPS (1 μg ml−1) for 5 h, the 30 nM and 1.0 μM methotrexate treatment of U937s showed significantly reduced adherence to approximately 60% (2.6×104 P<0.01) and 70% (3.1×104 P<0.05) of control treated U937 monocytes respectively (Figure 7b). Preincubation with pertussis toxin did not affect resting monocyte adhesion nor MTX mediated reduction in adhesion to endothelial cells that had been activated with LPS (1 μg ml−1) for 5 h (data not shown). When the HUVEC were activated with 1 μg ml−1 of LPS for 24 h, the methotrexate treatment of U937 monocytes reduced their adhesion further to approximately 40% of that of control monocytes (1.7×104 P<0.01). There was no significant difference in the anti-adhesive properties of 30 nM and 1.0 μM methotrexate treatment of U937 monocyte to 24 h LPS (1 μg ml−1) activated HUVEC (P>0.05; Figure 7c). MTX did not interfere with fluorescence of BCECF-AM (data not shown).
Figure 7.
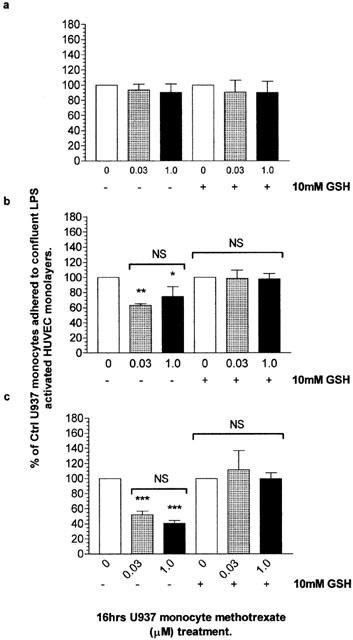
Methotrexate treated monocytes exhibit reduced adhesion to LPS activated HUVEC with a requirement for ROS production. U937 monocytes (2×106 ml−1) were serum starved for 4 h in RPMI 1640 in the presence or absence of 10 mM glutathione (GSH) prior to 0, 0.03 or 1.0 μM methotrexate treatment. Treatments were terminated by centrifugation and the resulting cell pellets washed twice with ice cold PBS. Cells (5×106 ml−1) were loaded with 1 μg ml−1 of BCECF-AM for 30 min in the dark. Cells were then washed and resuspended in M199 to a concentration of 0.5×106 ml−1. Confluent HUVEC monolayers in 24 well plates were treated with 1 μg ml−1 LPS for 0 (a), 5 (b) or 24 h (c). HUVEC were washed twice prior to the addition of treated monocyte suspensions in duplicate for 30 min under the described incubator conditions. Their adherence was quantified against a standard curve of vehicle treated monocytes and expressed as a percentage of controls. The results are presented as the mean±s.d. of four individual experiments where * (P<0.05) and ** (P<0.01) represent significant difference from controls by one-way ANOVA with Dunnett's post-test analysis. NS, no significant difference.
The reduction in adhesion of monocytes treated with methotrexate to activated HUVEC was inhibited by pre-treatment of U937 monocytes with the antioxidant GSH and was not significantly different to control treated U937 monocytes, irrespective of the period of HUVEC activation by LPS (1 μg ml−1; P>0.05; Figure 7a–c).
To further evaluate the anti-adhesive properties of methotrexate treatment of U937 monocytes, we quantified the membrane expression of monocyte integrin and selectin molecules CD11a, CD11b, CD18, CD29, CD31, CD49D and CD62L by flow cytometry. Monocytes treated with 30 nM or 1.0 μM methotrexate induced a significant elevation in the membrane expression of all the adhesion molecules examined of between approximately 120–140% of that of control U937 monocytes (P<0.05) with no significant difference observed between concentrations (P>0.05). Pre-treatment of U937 monocytes with 10 mM GSH inhibited the methotrexate-mediated elevation in adhesion molecule membrane expression (P<0.05; Figure 8).
Figure 8.
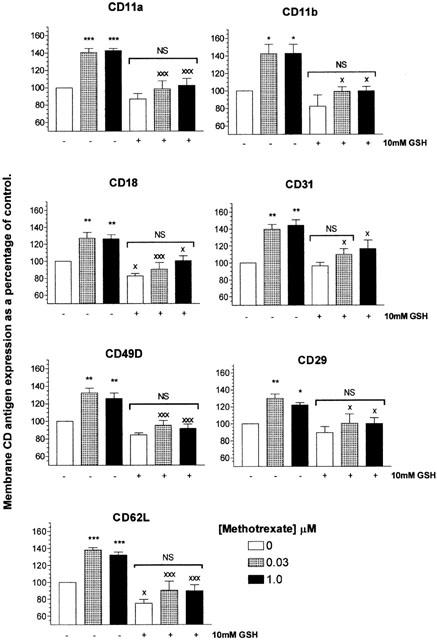
Methotrexate increases the membrane expression of monocytic adhesion molecules with an essential requirement for ROS. U937 monocytes (2×106 ml−1) were serum starved for 4 h in RPMI 1640 in the presence or absence of 10 mM glutathione (GSH) prior to 0, 0.03 or 1.0 μM methotrexate treatment for 16 h. Treatments were terminated by centrifugation and the resulting cell pellets washed twice with ice cold PBS. Cells were treated with >10 μl of fluorescently tagged mouse IgG1 monoclonal antibody (MoAb) or isotype negative control per 106 cells for 30 min on ice, in the dark and fixed as described in Methods. Samples were then analysed by flow cytometry. Background fluorescence of each sample was established utilizing cells stained with isotype negative controls. Positive regions were defined to contain 1% of the negatively stained cells. Samples were then analysed for MoAb membrane expression and the median X of the fluorescent peak recorded. The membrane expression of CD11a, CD11b, CD18, CD31, CD29, CD49D and CD62L were evaluated. At least 10,000 cells were analysed per sample. The results are presented as the mean±s.d. of at least four individual experiments where * (P<0.05) and ** (P<0.01) represent significant difference from controls by one-way ANOVA with Tukey's post hoc test analysis. × (P<0.05) and ××× (P<0.001) signifies statistical difference of samples pre-treated with 10 mM GSH compared to no pre-treatment by Student's t-test. NS, no significant difference.
Discussion
The neutralization of monocyte/macrophage activity is recognized as a useful therapeutic strategy, where macrophages comprise 20% of primary cultured cells from rheumatoid synovium. There is a wealth of data indicating that the two macrophage-derived cytokines IL-1 and TNFα play a pivotal role in RA (Elliott et al., 1994; Feldmann et al., 1996; Plows et al., 1995; Ruschen et al., 1992; Van den Berg, 1995; Zangerle et al., 1992) where this may arise through stimulation of matrix metalloproteinases (Krane et al., 1990). Indeed, more recently developed therapeutics (e.g. etanercept) have been specifically targeted against the effects of TNFα (as reviewed: Alldred, 2001; Richard-Miceli & Dougados, 2001).
Nevertheless, there are several disease modifying antirheumatic drugs which were discovered serendipitously, and whose mechanism of action remains unknown. MTX is one such potent antiinflammatory agent, which is widely used to control the arthritic process. However, a limiting factor in the use of MTX is its associated toxicity at multiple sites including the liver and lungs (as reviewed in Alarcón (2000)). Therefore, further elucidation of the mechanism(s) of action of MTX may allow development of compounds with improved efficacy:toxicity profiles.
In this paper, we have examined MTX effects of DNA cell cycle and have evaluated the capacity of MTX to generate intracellular ROS, using concentrations that encompass the plasma concentration (500 nM) observed in RA subjects receiving a weekly 7.5 mg oral dose of MTX (Coombe et al., 1995) or in leukaemia patients (Synold et al., 1994). In agreement of the effects of MTX treatment of PMN (Gressier et al., 1994), we describe that treatment of U937 monocytes and Jurkat T-cells induces the production of [peroxide]cyt in both dose and time dependent fashion. DNA fragmentation observed in Jurkat T-cells in response to MTX precedes loss of membrane integrity confirming the work of da Silva et al. (1996).
The [peroxide]cyt production in Jurkat T-cells at concentrations which induced apoptosis was approximately two fold greater than the [peroxide]cyt that those which induced growth arrest in U937 monocytes. The greater levels of production of [peroxide]cyt in T-cells may induce the apoptotic response rather than the adaptive response of growth arrest seen in monocytes. Moreover, the amplitude of [peroxide]cyt production in response to MTX at all concentrations may reflect the intrinsic, endogenous levels of antioxidant present within the cell. Indeed monocytes possess three times the concentration of total cellular glutathione than Jurkat T-cells and hence may reduce [peroxide]cyt levels to those which mediate a non-deleterious response rather than the terminal cellular response of apoptosis associated with the massive production of peroxide. To confirm the relevance of these findings, we have further evaluated the sensitivity of primary T cells to MTX and associated [peroxide]cyt production. Whilst primary T cells were insensitive, following PHA activation, T cells showed evidence of an increase in [peroxide]cyt production and induction of apoptosis after MTX treatment, which was inhibited following N-acetyl cysteine treatment. The insensitivity of primary cells to MTX toxicity and requirement for activation, confirms previous observations (Genestier et al., 1998a; Fairbanks et al., 1999). It was not feasible to determine the sensitivity of primary monocytes to MTX, as they differentiate in culture over the period of MTX treatment, altering both their adherence properties and integrin expression. In addition, the yield of primary monocytes from peripheral blood (106 monocytes per 10 ml blood) precludes multiple analyses. However, expression of adhesion molecules and subsequent adhesive properties on U937 cells following cytokine and LPS activation have been shown by us and others to be an adequate model (Kalogeris et al., 1999; Chuluyan & Issekutz, 1993).
Pre-treatment of Jurkat T-cells with either of the anti-oxidants GSH or NAC completely abrogated the apoptosis induced by MTX at low doses (<1 μM). At higher doses (1–10 μM) MTX, GSH/NAC significantly reduced the percentage specific apoptosis by approximately 20%. The incomplete protection at these doses may be explained by incomplete detoxification of peroxide by GSH/NAC or the induction of apoptosis via a peroxide independent process. (Intracellular bulk phase transfer of GSH and increased provision of substrate amino acids is believed to afford its protection, whereas NAC readily crosses the plasma membrane). Conversely, the accumulation of nucleoids in the G0/G1 phase of the cell cycle in U937 monocytes in response to MTX treatment (⩾100 nM), which is indicative of growth arrest, was completely inhibited by NAC or GSH at all concentrations analysed implying that the growth arrest response to MTX is totally dependent on [peroxide]cyt production. Membrane integrity, evaluated by the flow cytometric analysis of PI uptake, was not compromised for all concentrations of MTX analysed after 16 h exposure in both U937s and Jurkat T-cells indicating that MTX effects are independent of necrosis.
Previous work implicates ROS as signalling molecules in apoptotic cell death (Mansat-de-Mas et al., 1999; Yamauchi et al., 1989). A reduction in the cellular GSH levels of HeLa cells in response to MTX has been reported (Babiak et al., 1998). Further, the MTX mediated depletion of total cellular GSH may be necessary for initial increases in [peroxide]cyt (Tan et al., 1998). In U937 monocytes, even at concentrations less than those that conferred growth arrest (<0.1 μM), MTX reduced the total cellular concentration of GSH independently of concentration, to approximately 70% of vehicle, control treated monocytes. Similarly, MTX at a concentration of 1 μM or more, which induce a maximal apoptotic response, decreased total GSH levels in Jurkat T-cells. Loss of GSH can arise from formation of mixed disulphides with proteins or oxidized GSH (GSSG) which would normally be recycled by GSR. In addition, MTX mediates inhibition of GSR (Babiak et al., 1998), resulting in a failure to restore cellular GSH.
At a low dose of MTX (30 nM) the total GSH concentration in Jurkat T-cells was significantly elevated above control cells indicating that the [peroxide]cyt generated at this low MTX concentration may act in a transient signalling capacity to increase GSH in an attempt to prevent MTX-peroxide mediated cell death via redox sensitive transcription of GCS (as reviewed in Haddad et al., 2002; Rahman (2000)). In this instance, the small and early accumulation of [peroxide]cyt may act as a protective mechanism (Tan et al., 1998).
The magnitude of [peroxide]cyt formation in response to MTX in both U937 monocytes and Jurkat T-cells is suggestive of alternative sources of ROS production for the source of [peroxide]cyt other than as a result of GSH depletion. Possible MTX or polyglutamated MTX targets include intracellular organelles, e.g. mitochondria and endoplasmic reticulum or the NADPH oxidase system (Coyle & Puttfarken, 1993; Cross & Jones, 1991).
Treatment of U937 monocytes with MTX at low (30 nM) and high (1 μM) induced a differentiated like phenotype with the enhanced expression of the αM-integrin, CD11b as reported by Seitz et al. (1998). Further, we found that MTX induced an equivalent elevation to similar levels of the β2 integrin CD18 and all other integrin adhesion molecules studied (CD11a, CD31, CD29, CD49D) and the leukocyte associated selectin, CD62L. The MTX induced elevation in adhesion molecule expression was also dependent on the production of ROS since antioxidant pre-treatment of monocytes also inhibited these effects. MTX has been reported to alter membrane expression of a variety of functionally important antigens expressed on Jurkat T-cells (Hall et al., 1997). Seitz et al. (1998) postulated that MTX might inhibit the recruitment of immature and inflammatory monocytes from bone marrow into the inflammatory sites. However, expression alone (as measured by flow cytometry) does not equate function, where changes in conformation of integrins are required for altered behaviour.
Monocytes treated with MTX did not show altered adherence to resting, quiescent endothelial cells, but MTX was effective in inhibiting monocytic adherence to 5 h activated endothelial cells and displayed greater efficacy in reducing monocyte interaction with 24 h LPS activated endothelial cells. These effects were completely abrogated when U937 monocytes were pre-treated with GSH implying an essential requirement for ROS in the inhibition of monocyte adherence to activated endothelial cells. The reduction in monocyte adhesion to endothelial cells was found to be independent of effects on chemokine receptor signalling, as pertussis toxin did not cause a reduction in adhesion alone nor did it affect the MTX-mediated reduction in adhesion.
The observation that folate supplementation of up to 27.5 mg/week administered to MTX-treated RA patients reduces toxicity without compromising clinical efficacy indicates that the anti-inflammatory/immunosuppressive actions of MTX do not directly involve the inhibition of DHFR (Budzik et al., 2000). Others have postulated that the mechanism of MTX action is via the release of anti-inflammatory autocoid, adenosine (Cronstein, 1994; Merril et al., 1997), where adenosine reduces superoxide formation (Cronstein et al., 1985; Roberts et al., 1985). However, MTX treatment in combination with the adenosine antagonist R-P1A did not attenuate the MTX anti-rheumatic effect of MTX alone in rats with antigen induced arthritis (Andersson et al., 2000), implying that the release of adenosine is not important. Further, a concentration of adenosine deaminase (2 U) that completely abrogated adenosine-induced apoptosis in activated PBL in vitro, only decreased MTX mediated apoptosis by 10–20% (Paillot et al., 1998; Genestier et al., 1998a). In contrast, we describe for the first time that the production of peroxide is essential in the mechanism of MTX anti-inflammatory and immunosuppressive action. We have shown that the stress responses of growth arrest and apoptosis in monocytes and T-cells respectively to MTX treatment is dependent on the production of peroxide. We have further clarified the anti-inflammatory action of MTX by investigating the functional consequence of the MTX treatment of monocytes with regards to their recruitment to endothelial cells. We propose that MTX or its polyglutamated metabolites target free radical/ROS producing centres of the cell. The ensuing elevation in peroxide is responsible, at least in part to the cellular responses and functional consequence of MTX treatment. We hypothesize that MTX could affect the progression of inflammatory disease states by inhibiting monocyte interaction with the inflamed endothelium. This is currently being investigated in a clinical study of RA patients pre- and post-MTX treatment.
Acknowledgments
DCP thanks Aston University for studentship, Dr David Poyner for pertussis toxin and Dr Sheila Handley for reading of manuscript.
Abbreviations
- AICAR
5-amino-imidazole carboxamide ribonucleotide
- a.u.
arbitrary units
- BCECF-AM
2′-7′-bis-2-carboxy-5-(6)-carboxyfluorescein-acetoxymethylester
- BSA
bovine serum albumin
- [peroxide]cyt
cytosolic peroxide
- DCFH-DA
2′,7′dichlorodihydrofluorescein diacetate
- DHFR
dihydrofolate reductase
- DMSO
dimethyl sulphoxide
- DNTB
5,5′-dithio-bis (2-nitrobenzoic acid)
- FH2
dihydrofolate
- FH4
tetrahydrofolate
- EGM
endothelial growth medium
- FITC
fluorescein isothiocyanate
- FS
forward scatter
- GSSG
oxidized glutathione
- GSH
glutathione
- GSR
glutathione reductase
- HUVEC
human umbilical vein endothelial cells
- LPS
lipopolysaccharide
- MdX
median
- MTX
Methotrexate, MoAb, monoclonal antibody
- NAC
N-acetyl cysteine
- PBL
peripheral blood lymphocytes
- PBN
peripheral blood neutrophils
- PBS
phosphate buffered saline
- PE
phycoerythrin
- PI
propidium iodide
- PTP
protein tyrosine phosphatases
- RA
rheumatoid arthritis
- ROS
reactive oxygen species
- SS
side scatter
- SOD
superoxide dismutase
References
- ALARCÓN A. Methotrexate in rheumatoid arthritis. A clinician's perspective. Immunopharmacology. 2000;47:259–271. doi: 10.1016/s0162-3109(00)00184-3. [DOI] [PubMed] [Google Scholar]
- ALLISON A. Immunosuppressive drugs: the first 50 years and a glance forward. Immunopharmacology. 2000;47:63–83. doi: 10.1016/s0162-3109(00)00186-7. [DOI] [PubMed] [Google Scholar]
- ALLDRED A. Etanercept in rheumatoid arthritis. Expert Opin. Pharmacother. 2001;2:1137–1148. doi: 10.1517/14656566.2.7.1137. [DOI] [PubMed] [Google Scholar]
- ANDERSON M.Determination of glutathione and glutathione disulfide in biological samples Methods in Enzymology 1985113New York: Academic Press; 548–555.ed. Meister, A. [DOI] [PubMed] [Google Scholar]
- ANDERSSON S., JOHANSSON L.H., LEXMÜLLER K., EKSTRÖM G. Anti-rheumatic effect of methotrexate: is it really mediated by adenosine. Eu. J. Pharm. Sci. 2000;9:333–343. doi: 10.1016/s0928-0987(99)00073-1. [DOI] [PubMed] [Google Scholar]
- BABIAK R., CAMPELLO A., CARNIERI E., OLIVEIRA M. Methotrexate; pentose cycle and oxidative stress. Cell Biochem. Funct. 1998;16:283–293. doi: 10.1002/(SICI)1099-0844(1998120)16:4<283::AID-CBF801>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- BAGGOTT J., MORGAN S., HA T., ALARCÓN G., KOOPMAN W., KRUMDIECK C. Antifolates in rheumatoid arthritis: a hypothetical mechanism of action. Clin. Exp. Rheumatol. 1993;11:S101–S105. [PubMed] [Google Scholar]
- BANNWARTH B., LABAT L., MORIDE Y., SCHAEVERBEKE T. Methotrexate in rheumatoid arthritis: an update. Drugs. 1994;47:25–50. doi: 10.2165/00003495-199447010-00003. [DOI] [PubMed] [Google Scholar]
- BASS D., PARCE J., DECHATELET L., SZEJDA P., SEEDS M., THOMAS M. Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J. Immunol. 1983;130:910–917. [PubMed] [Google Scholar]
- BONDESON J., SUNDLER R. Auranofin inhibits the induction of interleukin 1 and tumour necrosis factor alpha mRNA in macrophages. Biochem. Pharmacol. 1995;52:35–42. doi: 10.1016/0006-2952(95)02030-6. [DOI] [PubMed] [Google Scholar]
- BUDZIK G., COLLETTI L., FALTYNEK C. Effects of methotrexate on nucleotide pools in normal T cells and the CEM T cell line. Life Sciences. 2000;66:2297–2307. doi: 10.1016/s0024-3205(00)00559-2. [DOI] [PubMed] [Google Scholar]
- CHULUYAN H., ISSEKUTZ VLA-4 can mediate CD11/CD18-independent transendothelial migration of human monocytes. J. Clin. Invest. 1993;92:2768–2777. doi: 10.1172/JCI116895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIESIELSKI C., MEI J., PICCININI L. Effects of cyclosporine A and methotrexate on CD18 expression in recipients of rat cardiac allografts. Transpl. Immunol. 1998a;6:122–123. doi: 10.1016/s0966-3274(98)80027-0. [DOI] [PubMed] [Google Scholar]
- CIESIELSKI C., PFLUG J., MEI J., PICCININI L. Methotrexate regulates ICAM-1 expression in recipients of rat cardiac allografts. Transpl. Immunol. 1998b;6:111–121. doi: 10.1016/s0966-3274(98)80026-9. [DOI] [PubMed] [Google Scholar]
- COOMBE B., EDNO L., LAFFORGUE P., BOLOGNA J., ACQUAVIVA P., SANY J., BRESSOLLE F. Total and free methotrexate pharmacokinetics, with and without piroxicam, in rheumatoid arthritis patients. J. Rheumatol. 1995;34:421–428. doi: 10.1093/rheumatology/34.5.421. [DOI] [PubMed] [Google Scholar]
- COYLE J., PUTTFARKEN P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- CRONSTEIN B. Adenosine, an endogenous anti-inflammatory agent. J. Appl. Physiol. 1994;76:5–13. doi: 10.1152/jappl.1994.76.1.5. [DOI] [PubMed] [Google Scholar]
- CRONSTEIN B., ROSENSTEIN E., KRAMER S., WEISSMANN G., HIRSCHHORN R. Adenosine: a physiological modulator of superoxide anion generation by human neutrophils. Adenosine acts via an A2 receptor on neutrophils. J. Immunol. 1985;135:1366–1371. [PubMed] [Google Scholar]
- CRONSTEIN B., NAIME D., OSTAD E. The anti-inflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J. Clin. Invest. 1993;92:2675–2692. doi: 10.1172/JCI116884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROSS A., JONES O. Enzymatic mechanisms of superoxide production. Biochim. Biophys. Acta. 1991;1057:281–298. doi: 10.1016/s0005-2728(05)80140-9. [DOI] [PubMed] [Google Scholar]
- DA SILVA C., DE OLIVEIRA C., DE LIMA M. Apoptosis as a mechanism of cell death induced by different chemotherapeutic drugs in human leukemic T-lymphocytes. Biochem. Pharmacol. 1996;51:1331–1340. doi: 10.1016/0006-2952(96)00041-x. [DOI] [PubMed] [Google Scholar]
- ELLIOTT M., MAINI R., FELDMANN M., KALDEN J., ANTONI C., SMOLEN J., LEEB B., BREEDVELD F., MACFARLANE J., BIJL H., WOODY J. Randomized double-blind comparison of chimeric monoclonal antibody to tumor necrosis factor (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- FAIRBANKS L., RÜCKERMANN K., QUI Y., HAWRYLOWICZ C., RICHARDS D., SWAMINATHAN R., KIRSCHBAUM B., SIMMONDS H. Methotrexate inhibits the first committed step of purine biosynthesis in mitogen-stimulated human T-lymphocytes: a metabolic basis for efficacy in rheumatoid arthritis. Biochemical J. 1999;342:143–152. [PMC free article] [PubMed] [Google Scholar]
- FARBER S., TOCH R., SEARS E., PINKEL D.Advances in chemotherapy of cancer in man Advances in Cancer Research 1956New York: Academic Press; 2–73.ed. Greebstein, J.-P. [DOI] [PubMed] [Google Scholar]
- FELDMANN M., BRENNAN F., MAINI R. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- GENESTIER L., PAILLOT R., FOURNAL S., FERRARO C., MIOSSEC P., REVILLARD J.-P. Immunosuppressive properties of methotrexate: apoptosis and clonal deletion of activated peripheral T cells. J. Clin. Invest. 1998a;102:322–328. doi: 10.1172/JCI2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GENESTIER L., PRIGENT A.-F., PAILLOT R., QUEMENEUR L., DURAND I., BANCHEREAU J., REVILLARD J., BONNEFOY-BÉRARD N. Caspase-dependent ceramide production in Fas- and HLA class I-mediated peripheral T cell apoptosis. J. Biol. Chem. 1998b;273:5060–5066. doi: 10.1074/jbc.273.9.5060. [DOI] [PubMed] [Google Scholar]
- GRESSIER B., LEBAEGUE S., LUYCKX M., DINE T., CAZIN M., CAZIN J. Pro-oxidant properties of methotrexate: evaluation and prevention by an anti-oxidant drug. Pharmazie. 1994;49:679–681. [PubMed] [Google Scholar]
- HADDAD J., SAADE N., SAFIEH-GARABEDIAN B. Redox regulation of TNF-[alpha] biosynthesis: Augmentation by irreversible inhibition of [gamma]-glutamylcysteine synthetase and the involvement of I[kappa]B-[alpha]/NF-[kappa]B-independent pathway in alveolar epithelial cells. Cell Signal. 2002;14:211–218. doi: 10.1016/s0898-6568(01)00233-9. [DOI] [PubMed] [Google Scholar]
- HALL M., LAWRENCE D., LANSIEDEL J., WALSH A., COMSTOCK L., KREMER J. Long-term exposure to methotrexate induces immunophenotypic changes, decreased methotrexate uptake and increased dihydrofolate gene number in Jurkat T cells. Int. J. Immunopharmac. 1997;19:709–720. doi: 10.1016/s0192-0561(97)00075-1. [DOI] [PubMed] [Google Scholar]
- HAMILTON R.A., KREMER J. The effects of food on methotrexate absorption. J. Rheumatol. 1995;22:630–632. [PubMed] [Google Scholar]
- HAWKES J., CLELAND L., JAMES M. The effect of methotrexate in lipoxygenase metabolism in neutrophils from rats: in vitro and ex vivo studies. Agents Actions. 1993;40:181–185. doi: 10.1007/BF01984059. [DOI] [PubMed] [Google Scholar]
- HAWKES J., CLELAND L., PROUDMAN S., JAMES M. The effect of methotrexate on ex vivo lipoxygenase metabolism in neutrophils from patients with rheumatoid arthritis. J. Rheumatol. 1994;21:55–58. [PubMed] [Google Scholar]
- HU S., MITCHKO Y., ORONSKY A., KERWAR S. Studies on the effect of methotrexate on macrophage function. J. Rheumatol. 1998;15:206–209. [PubMed] [Google Scholar]
- JAFFE E., NACHMAN R., BECKER C., MINICK C. Culture of human endothelial cells derived from umbilical veins. J. Clin. Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KALOGERIS T.J., KEVIL C.G., LAROUX F.S., COE L.L., PHIFER T.J., ALEXANDER J.S. Differential monocyte adhesion and adhesion molecule expression in venous and arterial endothelial cells. Am. J. Physiol. 1999;276:L9–L19. doi: 10.1152/ajplung.1999.276.1.L9. [DOI] [PubMed] [Google Scholar]
- KRANE S., CONCA W., STEPHENSON M., AMENTO E., GOLDRING M. Mechanisms of matrix degradation in rheumatoid arthritis. Ann. N.Y. Acad. Sci. 1990;580:340–354. doi: 10.1111/j.1749-6632.1990.tb17943.x. [DOI] [PubMed] [Google Scholar]
- MANSAT-DE MAS V., BEZOMBES C., QUILLET-MARY A., BETTAIEB A., DE THONEL D'ORGEIX A., LAURENT G., JAFFRÉZOU J.-P. Implication of radical oxygen species in ceramide generation, c-Jun terminal kinase activation and apoptosis induced by daunorubicin. Mol. Pharm. 1999;56:867–874. doi: 10.1124/mol.56.5.867. [DOI] [PubMed] [Google Scholar]
- MERRIL J., SHEN C., SCHREIBMAN D., COFFEY D., ZAKJARENKO O., FISHER R., LAHITA R., SALMON J., CRONSTEIN B. Adenosine A1 receptor promotion of multinucleated giant cell formation by human monocytes: a mechanism for methotrexate-induced nodulosis in rheumatoid arthritis. Arthritis Rheum. 1997;40:1308–1315. doi: 10.1002/1529-0131(199707)40:7<1308::AID-ART16>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- MORGAN S., BAGGOTT J., VAUGHN W., AUSTIN J., VEITCH T., LEE J., KOOPMAN W., KRUMDIECK C., ALARCÓN G. Supplementation with folic acid during methotrexate therapy for rheumatoid arthritis. A double-blind, placebo controlled trial. Ann. Int. Med. 1994;121:833–841. doi: 10.7326/0003-4819-121-11-199412010-00002. [DOI] [PubMed] [Google Scholar]
- NEUMAN M., CAMERON R., HABER J., KATZ G., MALKIEWICZ I., SHEAR N. Inducers of cytochrome P450 2E1 enhance methotrexate-induced hepatoxicity. Clin. Biochem. 1999;32:519–536. doi: 10.1016/s0009-9120(99)00052-1. [DOI] [PubMed] [Google Scholar]
- NICOLLETTI I., MIGLIORATI G., PAGLIACCII M., GRIGNANI F., RICCARDI C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- OGUEY D., KOLLIKER F., GERBER N., REICHEN J. Effect of food on the bioavailability of low dose methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 1992;35:611–614. doi: 10.1002/art.1780350603. [DOI] [PubMed] [Google Scholar]
- PAILLOT R., GENESTIER L., FOURNEL S., FERRARO C., MIOSSEC P., REVILLARD J. Activation-dependent lymphocyte apoptosis induced by methotrexate. Transplant Proc. 1998;30:2348–2350. doi: 10.1016/s0041-1345(98)00648-4. [DOI] [PubMed] [Google Scholar]
- PLOWS D., PROBERT L., GEORGOPOULOS S., ALEXOPOULOU L., KOLLIAS G. The role of tumour necrosis factor (TNF) in arthritis: studies in transgenic mice. Rheumatol. Eur. 1995;24:51–54. [Google Scholar]
- RAHMAN I. Regulation of nuclear factor-kappa B, activator protein-1, and glutathione levels by tumour necrosis factor-alpha and dexamethasone in aveolar epithelial cells. Biochem. Pharmacol. 2000;60:1041–1049. doi: 10.1016/s0006-2952(00)00392-0. [DOI] [PubMed] [Google Scholar]
- RAU R., SCHLEUSSER B., HERBORN G., KARGER T. Long term treatment of destructive rheumatoid arthritis with methotrexate. J. Rheumatol. 1997;24:1881–1889. [PubMed] [Google Scholar]
- RICHARD-MICELI C., DOUGADOS M. Tumour necrosis factor-alpha blockers in rheumatoid arthritis: review of the clinical experience. BiodDrugs. 2001;15:251–259. doi: 10.2165/00063030-200115040-00005. [DOI] [PubMed] [Google Scholar]
- ROBERTS P., NEWBY A., HALLET M., CAMPBELL A. Inhibition of adenosine by reactive oxygen metabolite production by human polymorphonuclear leukocytes. Biochemical J. 1985;227:669–674. doi: 10.1042/bj2270669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUSCHEN S., STELLBERG W., WARNATZ H. Kinetics of cytokine secretion by mononuclear cells of the blood from rheumatoid patients are different from those from healthy controls. Clin. Exp. Immunol. 1992;89:32–37. doi: 10.1111/j.1365-2249.1992.tb06873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHWARTZ P., BARNETT S., MILSTONE L. Keratinocytes differentiate in response to inhibitors of deoxyribonucleotide synthesis. J. Dermatol. Sci. 1995;9:129–135. doi: 10.1016/0923-1811(94)00370-t. [DOI] [PubMed] [Google Scholar]
- SEITZ M., ZWICKER M., LOETSCHER P. Effects of methotrexate on differentiation of monocytes and production of cytokine inhibitors by monocytes. Arthritis Rheum. 1998;41:2032–2038. doi: 10.1002/1529-0131(199811)41:11<2032::AID-ART19>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- SMITH P., KROHN R., HERMANSON G., MALLIA A., GARTNER F., PROVENZANO M., FUJIMOTO E., GOEKE N., OLSON B., KLENK D. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- SPERLING R., BENINCASO A., ANDERSON COBLYN J.K.F.A., WEINBLATT M. Acute and chronic suppression of leukotriene B4 synthesis ex vivo in neutrophils from patients with rheumatoid arthritis beginning treatment with methotrexate. Arthritis Rheum. 1992;35:376–384. doi: 10.1002/art.1780350403. [DOI] [PubMed] [Google Scholar]
- SYNOLD T., RELLING M., BOYETT J., RIVERA G., SANDLUND J., MAHMOUD M., CRIST W., PUI C., EVANS W. Blast cell methotrexate-polyglutamate accumulation in vivo differs by lineage, ploidy and methotrexate dose in acute lymphoblastic leukemia. J. Clin. Invest. 1994;94:1996–2001. doi: 10.1172/JCI117552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAN S., SAGARA Y., LIN Y., MAHER P., SCHUBERT D. The regulation of reactive oxygen species production during programmed cell death. J. Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIETZE F. Enzymic methods for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- VAN DEN BERG W. Uncoupling of inflammation and joint destruction in arthritis: pivotal role of interleukin 1 in destruction. Trends Cell Biol. 1995;5:392–399. [Google Scholar]
- WEBER C., ERL W., WEBER K., WEBER P. Increased adhesiveness of isolated monocytes to endothelium is prevented by vitamin C intake in smokers. Circulation. 1996;93:1488–1492. doi: 10.1161/01.cir.93.8.1488. [DOI] [PubMed] [Google Scholar]
- WILLIAMS A., JONES S., GOODFELLOW R., ARMOS N., WILLIAMS B. Interleukin-1beta (IL-1beta) inhibition: a possible mechanism for the anti-inflammatory potency of liposomally conjugated methotrexate formulation in arthritis. B. J. Pharmacol. 1999;128:234–240. doi: 10.1038/sj.bjp.0702776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAUCHI N., KURIYAMA H., WATANABE N., NEDA H., MAEDA M., NIITSU Y. Intracellular hydroxyl radical protection induced by recombinant human tumour necrosis factor and its implication in the killing of tumour cells in vitro. Cancer Res. 1989;49:1671–1675. [PubMed] [Google Scholar]
- ZANGERLE P., DE GROOTE D., LOPEZ M.R.J.M., VRINDTS Y., FAUCHET F., DEHART I., JADOUL M., RADOUX D., FRANCHIMONT P. Direct stimulation of cytokines (IL-1 beta, TNF-alpha, IL-6, IL-2, IFN-gamma and GM-CSF) in whole blood II: application to rheumatoid arthritis and osteoarthritis. Cytokine. 1992;4:568–575. doi: 10.1016/1043-4666(92)90021-i. [DOI] [PubMed] [Google Scholar]