

Abstract
Activation of Ca2+-activated K+-channels (KCa) has been suggested to play a key role in endothelium-derived hyperpolarizing factor (EDHF)-mediated vasodilation. However, due to the low selectivity of commonly used KCa-channel blockers it is still elusive which endothelial KCa-subtypes mediate hyperpolarization and thus initiate EDHF-mediated vasodilation.
Using the non-cytochrome P450 blocking clotrimazole-derivatives, 1-[(2-chlorophenyl) diphenylmethyl]-1H-pyrazole (TRAM-34) and 2-(2-chlorophenyl)-2,2-diphenylacetonitrile (TRAM-39) as highly selective IK1-inhibitors, we investigated the role of the intermediate-conductance KCa (rIK1) in endothelial hyperpolarization and EDHF-mediated vasodilation.
Expression and function of rIK1 and small-conductance KCa (rSK3) were demonstrated in situ in single endothelial cells of rat carotid arteries (CA). rIK1-currents were blocked by TRAM-34 or TRAM-39, while rSK3 was blocked by apamin. In current-clamp experiments, endothelial hyperpolarization in response to acetylcholine was abolished by the combination of apamin and TRAM-34.
In phenylephrine-preconstricted CA, acetylcholine-induced NO and prostacyclin-independent vasodilation was almost completely blocked by ChTX, CLT, TRAM-34, or TRAM-39 in combination with the SK3-blocker apamin. Apamin, TRAM-34, and CLT alone or sulphaphenzole, a blocker of the cytochrome P450 isoform 2C9, were ineffective in blocking the EDHF-response.
In experiments without blocking NO and prostacyclin synthesis, the combined blockade of SK3 and IK1 reduced endothelium-dependent vasodilation.
In conclusion, the use of selective IK1-inhibitors together with the SK3-blocker apamin revealed that activation of both KCa, rIK1 and rSK3 is crucial in mediating endothelial hyperpolarization and generation of the EDHF-signal while the cytochrome P450 pathway seems to play a minor or no role in rat CA.
Keywords: Endothelium, Ca2+-activated K+-channels, EDHF, TRAM-34, clotrimazole
Introduction
The endothelium controls vascular tone by the release of vasoactive factors such as nitric oxide, prostacyclin, and the endothelium-derived hyperpolarizing factor (EDHF; Pohl et al., 1986; Feletou & Vanhoutte, 2000). In the control of endothelial-mediated vasodilation, the activation of ion channels plays a substantial role by regulating endothelial membrane potential and Ca2+-homeostasis in response to stimulation by humoral factors or haemodynamic forces (Adams et al., 1989; Nilius & Droogmans, 2001). Particularly, Ca2+-activated K+-channels (KCa) mediate endothelial hyperpolarization in response to humoral stimulation (Köhler et al., 2000; Nilius & Droogmans, 2001). This hyperpolarization provides the electrochemical driving force for Ca2+ entry which is important for the Ca2+-dependent synthesis of vasodilating factors (Lückhoff & Busse, 1990). Moreover, endothelial hyperpolarization has been shown to directly induce vasorelaxation due to an electrotonic spread of hyperpolarization to vascular smooth muscle cells via myoendothelial gap junctions (von der Weid & Beny, 1993; Edwards et al., 1999; Chaytor et al., 2001). It was concluded that this electrotonic spread of hyperpolarization contributes to EDHF-signal transduction in addition to other presumed EDHFs such as products of the cytochrome P450 (CYP) pathway (Fisslthaler et al., 1999). However, the contribution of these CYP-generated metabolites of arachidonic acid to EDHF signalling seems to vary considerably in different regions of the vascular tree and the species studied (Mombouli & Vanhoutte, 1997).
Thus far, pharmacological studies investigating the role of endothelial hyperpolarization in EDHF-signalling have commonly used KCa-channel blockers such as the bee venom apamin (APA) to block small-conductance KCa (SK) and the scorpion venom charybdotoxin (ChTX) to block large-conductance KCa (BK) and intermediate-conductance KCa (IK1) (Quignard et al., 2000; Dong et al., 2000; Coleman et al., 2001). However, conclusions drawn from these studies are limited as ChTX is poorly selective and may inhibit voltage-gated K+-channels (Kv) and BK in the endothelium (Fan & Walsh, 1999; Papassotiriou et al., 2000) as well as in the vascular smooth muscle (Waldron & Cole, 1999). Also the use of other KCa-blockers such as clotrimazole (CLT) which inhibits IK1 in endothelium (Köhler et al., 2001) and in activated T-lymphocytes (Logsdon et al., 1997; Wulff et al., 2000) can not be considered an alternative pharmacological tool. CLT has been reported to block BK-, ATP-sensitive K+-channels (KATP), and Ca2+-signalling in vascular smooth muscle (Rittenhouse et al., 1997; Vanheel & Van de Voorde, 1997; Tofukuji et al., 1998) and effectively inhibits CYP enzymes, thus making it difficult to distinguish whether EDHF-type vasodilation is mediated by eicosanoids or endothelial hyperpolarization (Fisslthaler et al., 1999). Therefore, more selective KCa-channel inhibitors of IK1 are needed to ascertain the contribution of different endothelial KCa-subtypes in EDHF-mediated dilation. Recently, CLT-derivatives, TRAM-34 and TRAM-39, were developed (Wulff et al., 2000) which specifically block IK1 at nanomolar concentrations without inhibiting CYP activity. In these compounds the imidazole ring of CLT which is essential for cytochrome P450 inhibition has been replaced by a pyrazole (TRAM-34) or an acetonitrile group (TRAM-39).
To assess the functional importance of especially the endothelial IK1 in EDHF-mediated vasodilations we performed patch-clamp experiments in endothelial cells of rat carotid arteries in situ and measurements of EDHF-mediated vasodilation using the new CLT-derivatives, TRAM-34 and TRAM-39. In addition, we demonstrate that TRAM-34 did not interfere with the function of K+-channels in vascular smooth muscle cells.
The present study revealed that endothelial hyperpolarizing KCa-currents and EDHF-mediated vasodilations were almost completely blocked by APA in combination with CLT, TRAM-34, or TRAM-39. Moreover, CLT alone or the CYP inhibitor sulphaphenazole were ineffective in blocking EDHF-mediated vasodilations. Thus the present study indicates that activation of both the endothelial KCa, rIK1 and rSK3 channels, is essential in generating the EDHF-signal whereas CYP-generated arachidonic acid metabolites apparently do not contribute to EDHF-mediated vasodilations in rat carotid arteries.
Methods
Carotid artery endothelial cells
Freshly isolated carotid arteries (CA) from male Sprague–Dawley rats (350–400 g) were cut open longitudinally and fixed on a holding capillary to give direct access to the luminal surface. For in situ harvesting of endothelial cells, vessel slices were pre-incubated with 0.05% trypsin and 0.02% ethylenediaminetetraacetic acid (EDTA) in phosphate buffered saline (PBS) without Ca2+/Mg2+ for up to 15 min (Köhler et al., 2001). For wash out, CA were superfused with PBS for 5 min. Under microscopic control, a single EC was selectively fixed with the patch pipette and mechanically detached from the vessel wall.
Carotid artery vascular smooth muscle cells
Vascular smooth muscle cells of rat carotid (CASMC) were isolated as previously described (Gollasch et al., 1992) with some modifications. Briefly, the vessels were cleaned of adventitial tissue and placed in Dulbecco's modified Eagle's medium (Biochrom KG, Berlin, Germany) containing 1.5 mg ml−1 collagenase typ II, 0.5 mg ml−1 elastase typ IIA, and 0.5 mg trypsin inhibitor (all Sigma, Deisenhofen, Germany) and were incubated for 30 min in a humidified atmosphere of 5% CO2. After cleaning of remaining advential tissue, vessels were cut into small segments of 1–2 mm in length and segments were further digested for 1 h under gentle agitation until single cells were dispersed. Cells were washed twice in PBS and immediately used for patch-clamp experiments.
Patch-clamp experiments
Whole-cell currents in electrically isolated CAEC and isolated CASMC were recorded with an EPC-9 (HEKA) patch-clamp amplifier using voltage ramps (duration: 1000 ms) from −100 mV to +50 mV as described previously (Köhler et al., 2000). Membrane potential changes in response to acetycholine (200 nM) were recorded in the electrically coupled CAEC in situ by using the current-clamp mode of the EPC-9 amplifier. Patch pipettes had a tip resistance of 2–4 MΩ in symmetrical KCl solution. For activation of KCa-currents, CAEC and CASMC were dialysed with a KCl-pipette solution containing 3 μM [Ca2+]free (mM): 135 KCl, 4 MgCl2, 2 ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 1.91 CaCl2, and 5 N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid] (HEPES), pH 7.2. For measurements of currents through voltage-gated K+-channels (KV) in CASMC and current-clamp experiments in CAEC, the pipette solution was prepared with 0.86 mM CaCl2 ([Ca2+]free=100 nM). The NaCl bath solution contained (mM): NaCl 137, Na2HPO4 4.5, KCl 3, KH2PO4 1.5, MgCl2 0.4 and CaCl2 0.7, pH 7.4. Whole-cell currents through ATP-sensitive K+-channels (KATP) in CASMC were measured, as previously described (Petkov et al., 2001). The bathing solution contained (in mM) NaCl 90, KCl 50, CaCl2 1, MgCl2 1 and HEPES 10, pH 7.4. The pipette solution contained (in mM) KCl 140, CaCl2 1 (free Ca2+ 20 nM), MgCl2 1, EGTA 10, HEPES 10, GTP 0.2, ADP 0.1 and ATP 0.1 (free ATP=0.0053), pH 7.2. Under these conditions and at holding potentials more negative than −26 mV, KATP-currents were inwardly directed.
Experiments were performed at room temperature. Data analysis was performed as previously described (Köhler et al., 2001).
Reverse transcription and single-cell-RT–PCR
Subsequent to patch-clamp experiments EC were transferred to a tube containing 1 μl ‘first-strand' buffer, 0.5 μl dNTPs (10 mM each), 1 μl ‘random'-hexamer primer (100 μM), 1 μl DTT (0.1 M), and 0.5 μl RNase-inhibitor (40 U μl−1). After one freeze-thaw cycle to induce breakdown of the cell membrane, 0.5 μl SuperScript®RT (200 U μl−1; Life Technologies) was added and the final volume (∼10 μl) was incubated for 1 h at 37°C. Single-cell RT–PCR was performed as described previously (Köhler et al., 2001). First and ‘nested' primer pairs (TIB MOL BIOL, Berlin) for rat endothelial nitric oxide synthase (eNOS) as endothelial cell marker, small KCa (rSK3), and intermediate KCa (rIK1) were selected to span intronic sequences and identities of PCR products were verified by sequencing using an ABI 377 automatic sequencer (ABI Prism). Forward and reverse primer: rIK1: first: 5′-GAGAGGCAGGCTGTCAATG-3′; 5′-GGGAGTCCTTCCTTCGAGTG-3′; nested: 5′-CATCACGTTCCTGACCATTG-3′; 5′-GTGTTTCTCCGCCTTGTTGA-3′; rSK3: first: 5′-AACCCCTCCAGCTCTTCAGT-3′; 5′-TGTGGTAGGCGATGATCAAA-3′; nested: 5′-GATAACCATGCCCACCAGAC-3′; 5′-ATTTCAGGGCCAACGAAAAC-3′; eNOS: first: 5′-GCACTATGGGGTCTGTTCCA-3′; 5′-CTTCATCCAGCTCCATGCT-3′; nested: 5′-CCAGCTCTGTCCTCAGAAGG-3′; 5′-ATGGATGAGCCAACTCAAGG-3′. In a single-cell sample, cDNA of rIK1 and rSK3 were co-amplified along with eNOS cDNA. A first ‘multiplex' PCR was performed in a final volume of 50 μl containing 5 μl PCR-buffer (10×), 2 μl dNTPs (10 mM each), 3 μl MgCl2 (50 mM), 1 μl of each sense- and antisense-primer (10 pmol), ∼10 μl RT-product, and 0.5 μl Taq DNA polymerase (5 U μl−1; all Life Technologies) using a Biozym Maxicycler PTC 9600. After initial denaturation for 5 min at 94°C, PCR amplification was carried out for 30 s at 94°C, 1 min at 55°C, 1 min at 72°C, and 10 min at 72°C for 50 cycles. In a second ‘multiplex' PCR round with ‘nested' primers, 5 μl of the first PCR product were used for reamplification (45 cycles, annealing temperature: 60°C). PCR products were analysed on a 2% agarose gel containing ethidium bromide. A 50 bp DNA ladder served as molecular weight markers.
Pressure myograph experiments
CA segments of 3–4 mm in length were cannulated with micropipettes in an experimental chamber mounted on the stage of a Zeiss Axiovert 100. Vessel diameter was continuously monitored with a video camera. The bath and perfusion solution contained (in mM): NaCl 145.0, NaH2PO4 1.2, KCl 4.7, MgSO4 1.2, CaCl2 2.0, glucose 5.0, pyruvate 2.0 and MOPS buffer 3.0 along with 1 g 100 ml−1 bovine serum albumin (pH 7.4 at 37°C). To block NO and prostacyclin synthesis the NO-synthase inhibitor NG-nitro-L-arginine (L-NNA, 100 μM) and the cyclo-oxygenase inhibitor indomethacin (10 μM) were added to both the bath and perfusion solution. CA were pressurized to 80 mmHg with a pressure myograph system (J.P. Trading P100) and continuously perfused at a flow rate of 0.6 ml min−1 and at constant intraluminal pressure. After an initial equilibration period for 40 min, CA were preconstricted with 1 μM phenylephrine in the bath solution. After development of stable tone, NO- and prostacyclin-independent vasodilatory responses were determined by perfusion with increasing concentrations of acetylcholine (ACh, 1 nM to 1 μM) alone or with 200 nM APA in combination with 100 nM charybdotoxin (ChTX), TRAM-34, TRAM-39, or CLT (all 1 μM). In different subsets of experiments, vessels were preincubated with 10 μM CLT or 10 μM sulphaphenazole (SPZ), a selective CYP 2C9 inhibitor (Fleming et al., 2001) for 30 min. In an additional subset of experiments, the bath and perfusion medium was prepared without NO-synthase- and cyclo-oxygenase inhibitor. Diameter changes were expressed as a percentage of the maximal dilation measured in response to 1 μM sodium nitroprusside (SNP). TRAM-34 and TRAM-39 were a kind gift from Heike Wulff (University of California Irvine, CA, U.S.A.). Levcromakalim, the active enantiomer of cromakalim (BRL 38227), was obtained from TOCRIS (Ballwin, MO, U.S.A.). All other standard chemicals and toxins were obtained from Sigma.
Statistical analysis
Data are given as mean±s.e.mean. The Mann–Whitney U-test was used to assess differences between groups. P-values <0.05 were considered significant.
Results
Inhibition of endothelial KCa by new clotrimazole-derivatives
In in situ whole-cell patch-clamp experiments, KCa-currents in EC of rat carotid arteries (CAEC) were activated by cell dialysis with 3 μM [Ca2+]free via the patch pipette. They exhibited characteristics of both rIK1 and rSK3 regarding K+-selectivity, slight inward-rectification, and Ca2+-dependence (Köhler et al., 2001). In the presence of TRAM-34 (100 nM), the selective IK1-blocker (Wulff et al., 2000), the KCa-current was reduced to 60–70% of the maximal current amplitude under control conditions (Figure 1A). The remaining TRAM-34-insensitive KCa-current was almost completely inhibited by the rSK3-blocker APA (200 nM) thus showing a substantial contribution of rSK3 to KCa-currents in the cells. In the presence of apamin (200 nM) to eliminate SK3-currents, TRAM-34 blocked the rIK1-current with a potency (KD 11±2 nM) similar to the cloned channel (Wulff et al., 2000) (Figure 1C). The other IK1-blocker TRAM-39 had similar blocking effects on rIK1-currents although with a slightly higher KD of 20±3 nM (Figure 1B,C).
Figure 1.
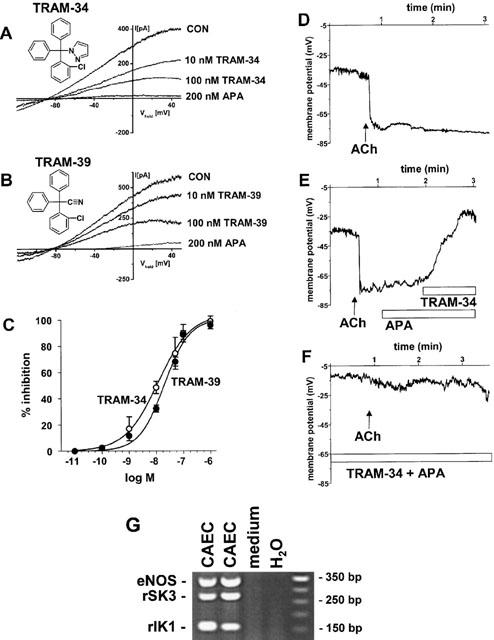
Representative whole-cell recordings of CAEC from rat carotid artery (CA) and inhibition by selective IK1 and SK3 blockers. For KCa-activation CAEC were dialyzed with a 140 mM K+ pipette solution containing 3 μM [Ca2+]free. Bathing solution contained 4.5 mM K+. Inhibition of Ca2+-activated K+-currents by TRAM-34 (A) and TRAM-39 (B) at indicated concentrations. TRAM-34- and TRAM-39-insensitive Ca2+-activated K+-currents were inhibited by APA. (C) Concentration-dependent blockade of apamin-resistant KCa-currents by TRAM-34 (n=4–6) and TRAM-39 (n=4–6) determined at membrane potential of 0 mV and in the presence of 200 nM apamin. Data points were fitted to the Boltzmann equation. Values are given as mean±s.e.mean. (D-F) Representative current-clamp recordings of endothelial membrane potentials and ACh-induced hyperpolarization and its inhibition by APA (200 nM) and TRAM-34 (1 μM). (G) Representative ‘multiplex' RT–PCR analysis of single EC from rat CA in situ. Ethidium bromide-stained gels of RT–PCR products of eNOS and the KCa-genes, rSK3 and rIK1 from 2 EC, one medium sample as negative control and one non-template control (H20); right lane, molecular weight marker.
In a separate set of experiments, we conducted current-clamp experiments in the electrically coupled CAEC in situ (Köhler et al., 2001) and measured ACh (200 nM) induced hyperpolarizations (Figure 1D). Resting membrane potentials were −38±2 mV (n=10). After addition of ACh (200 nM) the membrane potential shifted by −29±3 mV (n=7) to a more negative value of −61±2 mV (n=7). When APA (200 nM) was added thereafter, we observed a small depolarization to −51±4 mV (n=6). An additional strong depolarization to −17±6 mV (n=6) was achieved by adding TRAM-34 (1 μM, Figure 1E). When APA and TRAM-34 were applied first followed by ACh, endothelial hyperpolarization was almost completely suppressed (n=3; Figure 1F).
Subsequent to patch-clamp experiments, expression of the KCa-genes rIK1 and rSK3 in the same CAEC was verified by ‘multiplex'-single-cell RT–PCR analysis (Figure 1G). Other small KCa-subtypes such as rSK1 and rSK2 or the rBK were not considerably expressed in CAEC (Köhler et al., 2001).
To test whether TRAM-34 affects the function of K+-channels in vascular smooth muscle of rat carotid artery, we measured K+-currents through BK-, KV- and KATP-channels in the presence or absence of TRAM-34. TRAM-34 (10 μM) had apparently no effect on the function of BK-channels in CASMC while BK-currents were almost completely blocked by the selective BK-blocker iberiotoxin (100 nM, Figure 2A). Similarly, TRAM-34 (10 μM) exerted no apparent blocking effects on KV or levcromakalim-induced KATP-currents in CASMC whereas the KV-blocker 4-aminopyridine (2 mM) or the KATP-blocker glibenclamide (10 μM) were effective, respectively (Figure 2B,C).
Figure 2.
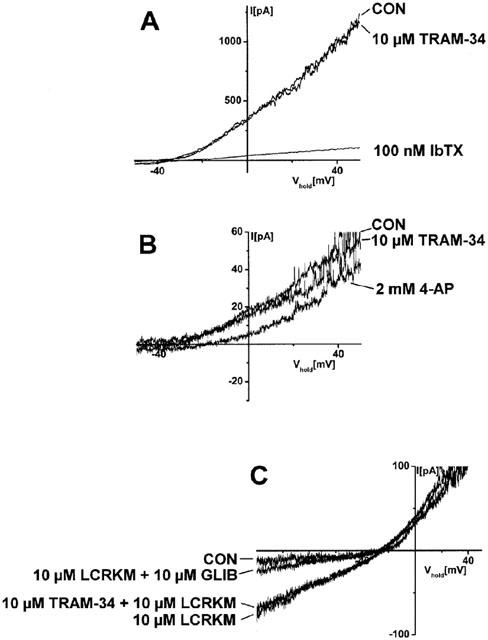
TRAM-34 did not affect the function of BK-, KV-, and KATP-channels in CASMC. (A) Whole-cell recordings of BK-currents in the presence or absence of TRAM-34 (10 μM) and blockade of BK-currents by selective BK-blocker iberiotoxin (IbTX, 100 nM). For KCa-activation CASMC were dialysed with a 140 mM K+ pipette solution containing 3 μM [Ca2+]free. Bathing solution contained 4.5 mM K+. The experiment shown is representative of seven experiments with similar results. (B) Whole-cell recordings of KV-currents in the presence or absence of TRAM-34 (10 μM) and blockade of KV-currents by the KV-blocker 4-aminopyridine (4-AP, 2 mM). CASMC were dialysed with a 140 mM K+-pipette solution containing 0.1 μM [Ca2+]free. The experiment shown is representative of ten experiments with similar results. Bathing solution used for measurements of BK- and KV-currents contained 4.5 mM K+. (C) Levcromakalim (LCRKM, 10 μM)-induced whole-cell currents through KATP-channels in CASMC in absence and presence of TRAM-34 (10 μM) and blockade of KATP-currents by the KATP-blocker glibenclamide (GLIB, 10 μM). CASMC were dialysed with a 140 mM K+-pipette solution containing 0.1 μM [Ca2+]free and 5.3 μM [ATP]free. Bathing solution contained 50 mM K+. Note that under these conditions and at holding potentials more negative than −26 mV, KATP currents were inwardly directed. The experiment shown is representative of eight experiments with similar results.
Inhibition of EDHF-mediated vasodilations by blockade of endothelial KCa
After preincubation with 100 μM L-NNA and 10 μM indomethacin, intraluminally applied ACh (n=31) induced a dose-dependent and EDHF-type vasodilation, with a maximal reduction of 13% of PE-induced tone at the highest concentration of ACh (1 μM) tested (Figure 3A). ACh-induced EDHF-mediated vasodilations at all ACh concentrations up to 1 μM ACh were almost eliminated by the combination of APA with CLT (n=5), TRAM-34 (n=5), or TRAM-39 (n=9, Figure 3A,B). Similar to the blocking effects of APA in combination with TRAM-34, TRAM-39, or CLT, EDHF-type vasodilations were almost abolished by the combination of ChTX (100 nM) and APA (200 nM) (n=5; Figure 3C). When ACh was applied in combination with only one KCa-blocker, APA (2 μM; n=7) or TRAM-34 (1 μM; n=7), EDHF-type vasodilations were not significantly different from those induced in the absence of KCa-blockers (Figure 3C).
Figure 3.
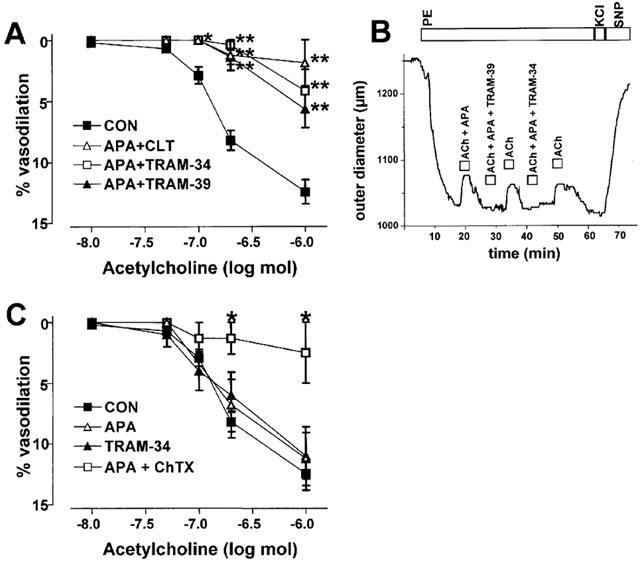
Inhibition of EDHF-type vasodilations in rat carotid arteries by KCa-blockers. (A) Inhibition of EDHF-type vasodilator responses by combinations of KCa-blockers as indicated. KCa-blocker concentrations: 200 nM APA, 1 μM CLT, 1 μM TRAM-34, 1 μM TRAM-39. Values are given as mean±s.e.mean. *P<0.05, **P<0.01, Mann–Whitney U-test. (B) Original trace illustrating EDHF-type vasodilator responses to intraluminal application of 0.2 μM ACh and in combination with APA (200 nM) or with APA and TRAM-34 (1 μM) or with APA and TRAM-39 (1 μM). Vessels were preconstricted with 1 μM phenylephrine (PE). Endothelium-independent vasodilation and maximal vasoconstriction were induced by extraluminal application of 1 μM sodium nitroprusside (SNP) and 60 mM KCl, respectively. Boxes indicate exposure intervals followed by wash-out (right upper lane). (C) Dose-dependent EDHF-type vasodilator response in response to ACh and inhibition by the combination of APA (200 nM) and ChTX (100 nM) but not by APA (2 μM) or TRAM-34 (1 μM) alone.
To test whether EDHF-type vasodilations could be reduced by inhibiting CYP-generated EDHF, vessels were preincubated with CLT (10 μM, n=5), which blocks CYP in addition to its blocking effects on the IK1, or with the CYP 2C9 specific inhibitor sulphaphenazole (SPZ, 10 μM, n=3) for 30 min. We found that under these conditions ACh-induced vasodilations were not substantially altered by CLT or SPZ when compared to controls (Figure 4A). However, when ACh was then applied in combination with APA (200 nM, n=4) in the presence of CLT, EDHF-type vasodilations were almost abolished (Figure 4B).
Figure 4.
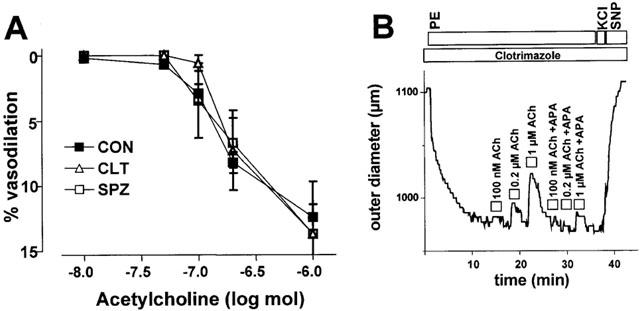
(A) Dose-dependent EDHF-type vasodilations in rat carotid arteries in the presence of CLT (10 μM) or SPZ (10 μM). (B) Original tracings illustrating dose-dependent EDHF-type vasodilator responses in the presence of CLT (10 μM) alone and in combination with APA (200 nM). Values are given as mean±s.e.mean.
In the absence of inhibitors of NO and prostacyclin synthesis we found that the combination of APA and TRAM-34 reduced maximal ACh-induced dilation at all ACh concentrations up to 1 μM ACh (Figure 5). TRAM-34 alone was ineffective in blocking ACh-induced dilation (Figure 5).
Figure 5.
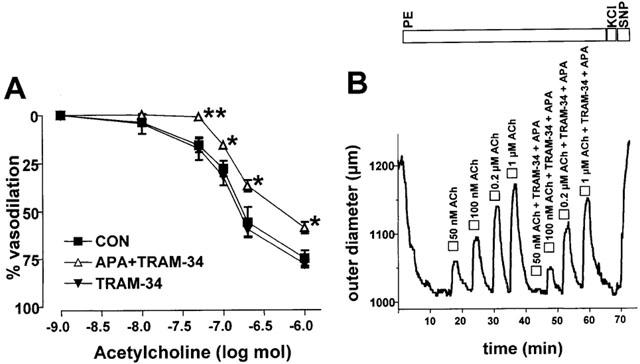
Inhibition of ACh-induced vasodilations by the KCa-blockers APA (200 nM) and TRAM-34 (1 μM) in the absence of inhibitors of NO-synthase and cyclo-oxygenase. (A) Dose-response curves of ACh-induced vasodilations in the presence (n=8) or absence (n=6) of APA (200 nM) and TRAM-34 (1 μM) or in the presence of TRAM-34 (1 μM) alone. Values are given as mean±s.e.mean. *P<0.05, **P<0.01, Mann–Whitney U-test. (B) Original trace illustrating dose-dependent vasodilations to intraluminal application of ACh alone and of ACh in combination with APA (200 nM) and TRAM-34 (1 μM).
Discussion
In the present study, the new IK1 inhibitors TRAM-34 and TRAM-39 have been employed to investigate the functional role of the endothelial rIK1 in NO- and prostacyclin-independent EDHF-type vasodilation of rat carotid arteries. The findings provide new evidence that activation of the endothelial rIK1 in synergy with the rSK3 is essential in generating the EDHF-response.
In in situ whole-cell patch-clamp experiments in native EC from rat CA, rIK1-currents were selectively and dose-dependently blocked by TRAM-34 and TRAM-39 as well as by CLT. TRAM-34 was found to be the most potent inhibitor followed by TRAM-39 and CLT. The KD values for rIK1-blockade in rat CAEC and the rank order of potency were similar to those reported for cloned human IK1 and the IK1 in activated human T-lymphocytes (Wulff et al., 2000). In contrast to inhibition of the endothelial rIK1, these CLT-derivatives had no blocking effects on endothelial rSK3 in rat CAEC. Moreover, TRAM-34 had also been shown to have no blocking effects on the cloned BK, several types of cloned voltage-gated K+- and, inward rectifying K+-channels, and Ca2+-release activated Ca2+-currents (Wulff et al., 2000) and did not interfere with the function of BK-, KV-, and KATP-channels in CASMC as shown in the present study. Thus in our hands, the apparent high selectivity of these compounds to block exclusively IK1 makes them an important pharmacological tool to define the functional role of the endothelial IK1 in endothelium-dependent control of vascular tone.
In a variety of small and large arteries such as guinea-pig cerebral and carotid arteries, rat mesenteric arteries, and pig coronary arteries the functional role of endothelial KCa in EDHF-mediated vasodilations was tested by the combination of the toxins APA and ChTX (Petersson et al., 1997; Doughty et al., 1999; Quignard et al., 1999; 2000). The synergistic inhibitory action of these two toxins on EDHF-type vasodilations led to the conclusion that opening of both endothelial KCa and the subsequent hyperpolarization is required to generate EDHF-type vasodilations in response to endothelial stimulation. In the present study we confirmed that the combination of APA and ChTX, when selectively applied to the endothelium of rat CA, abolished the EDHF-type dilatory response to ACh. Moreover, we showed here that a similar inhibition of EDHF-type dilation was achieved by combining APA with the other commonly used IK1 blocker CLT. These observations indicate that both endothelial KCa are involved in endothelial hyperpolarization and the subsequent EDHF-type vasodilatory response. However, experimental evidence at this point is still limited. In particular ChTX could have exerted its blocking action also by inhibiting BK in vascular smooth muscle and voltage-gated K+-channels in endothelial and vascular smooth muscle cells. This might result in changes in resting potentials of both cell types and thus in a decreased ability to hyperpolarize. Regarding CLT, the inhibition of EDHF-type vasodilations could have been exerted by interfering with BK- or ATP-sensitive K+-channels (KATP) in vascular smooth muscle (Rittenhouse et al., 1997; Vanheel & Van de Voorde, 1997) or with the CYP pathway and thus the synthesis of metabolites of arachidonic acid which also have been shown to serve as EDHFs in various vessel types (Fisslthaler et al., 1999). Thus, in our hands, the poor selectivity of ChTX or CLT limits their usefulness to define the role of a specific K+-channel or the CYP-pathway in EDHF-type vasodilations.
The major finding of the present study is that inhibition of the endothelial rIK1 by the highly selective TRAM-34 or TRAM-39 combined with rSK3 blockade by APA effectively abolished EDHF-type vasodilations in rat CA. This clearly supports the conclusion that in rat CA activation of both rIK1 and rSK3 together is essential in generating the EDHF-signal in response to endothelial stimulation. Moreover, rIK1- and SK3-mediated endothelial hyperpolarization seems to be a relevant and additive component of ACh-induced vasodilation in addition to the vasodilators NO and prostacyclin, since KCa-channel blockade considerably diminished the endothelium-dependent dilations even when NO and protacyclin generation was not blocked.
CYP-generated metabolites of arachidonic acid such as epoxyeicosatrienoic acids have been proposed to serve as EDHFs in the porcine coronary artery and human umbilical vein endothelium (Fisslthaler et al., 1999; Welsh & Segal, 2000; Bolz et al., 2000). Moreover, a CYP isozyme homologous to CYP 2C8/9 has been identified as an EDHF synthase. However, in the present study inhibition of CYP by CLT or SPZ alone was without effect on EDHF-type vasodilations. Therefore, in rat CA CYP-generated arachidonic acid metabolites presumably do not contribute to EDHF-mediated vasodilations and thus apparently do not serve as EDHFs. This is in agreement with observations of other investigators who also found strongly varying or even no contribution of CYP-generated arachidonic metabolites depending on the vascular bed studied (Drummond et al., 2000; Andersson et al., 2000; Ungvari & Koller, 2001; Coleman et al., 2001).
In conclusion, inhibition of endothelial hyperpolarization through blockade of both IK1 and SK3 channels prevents EDHF-mediated vasodilation. This indicates that activation of endothelial KCa is essential for NO- and prostacyclin-independent vasodilation whereas arachidonic acid metabolites of the CYP pathway seem to play a minor or no role in rat carotid arteries.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (FOR 341/1 and 341/7). The authors wish to thank Heike Wulff for the gift of TRAM-34 and TRAM-39 and for helpful discussions on the manuscript.
Abbreviations
- APA
apamin
- CA
carotid artery
- CAEC
carotid artery endothelial cells
- ChTX
charybdotoxin
- CLT
clotrimazole
- CYP
cytochrome P450
- eNOS
endothelial nitric oxide synthase
- KCa
Ca2+-activated K+-channel
- rIK1
rat intermediate-conductance Ca2+-activated K+-channel
- rSK3
rat small-conductance Ca2+-activated K+-channel
- SPZ
sulphaphenazole
- TRAM-34
1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole, TRAM-39, 2-(2-chlorophenyl)-2,2-diphenylacetonitrile
References
- ADAMS D.J., BARAKEH J., LASKEY R., VAN BREEMEN C. Ion channels and regulation of intracellular calcium in vascular endothelial cells. FASEB J. 1989;3:2389–2400. doi: 10.1096/fasebj.3.12.2477294. [DOI] [PubMed] [Google Scholar]
- ANDERSSON D.A., ZYGMUNT P.M., MOVAHED P., ANDERSSON T.L., HOGESTATT E.D. Effects of inhibitors of small- and intermediate-conductance calcium-activated potassium channels, inwardly-rectifying potassium channels and Na(+)/K(+) ATPase on EDHF relaxations in the rat hepatic artery. Br. J. Pharmacol. 2000;129:1490–1496. doi: 10.1038/sj.bjp.0703226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLZ S.S., FISSLTHALER B., PIEPERHOFF S., DE WIT C., FLEMING I., BUSSE R., POHL U. Antisense oligonucleotides against cytochrome P450 2C8 attenuate EDHF-mediated Ca(2+) changes and dilation in isolated resistance arteries. FASEB J. 2000;14:255–260. doi: 10.1096/fasebj.14.2.255. [DOI] [PubMed] [Google Scholar]
- COLEMAN H.A., TARE M., PARKINGTON H.C. K+ currents underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig, rat and human blood vessels. J. Physiol. 2001;531:359–373. doi: 10.1111/j.1469-7793.2001.0359i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAYTOR A,T., MARTIN P.E., EDWARDS D.H., GRIFFITH T.M. Gap junctional communication underpins EDHF-type relaxations evoked by ACh in the rat hepatic artery. Am. J. Physiol. Heart. Circ. Physiol. 2001;280:H2441–H2450. doi: 10.1152/ajpheart.2001.280.6.H2441. [DOI] [PubMed] [Google Scholar]
- DONG H., JIANG Y., COLE W.C., TRIGGLE C.R. Comparison of the pharmacological properties of EDHF-mediated vasorelaxation in guinea-pig cerebral and mesenteric resistance vessels. Br. J. Pharmacol. 2000;130:1983–1991. doi: 10.1038/sj.bjp.0703474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOUGHTY J.M., PLANE F., LANGTON P.D. Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. Am. J. Physiol. 1999;276:H1107–H1112. doi: 10.1152/ajpheart.1999.276.3.H1107. [DOI] [PubMed] [Google Scholar]
- DRUMMOND G.R., SELEMIDIS S., COCKS T.M. Apamin-sensitive, non-nitric oxide (NO) endothelium-dependent relaxations to bradykinin in the bovine isolated coronary artery: no role for cytochrome P450 and K+ Br. J. Pharmacol. 2000;129:811–819. doi: 10.1038/sj.bjp.0703107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., FELETOU M., GARDENER M.J., THOLLON C., VANHOUTTE P.M., WESTON A.H. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br. J. Pharmacol. 1999;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAN J., WALSH K.B. Mechanical stimulation regulates voltage-gated potassium currents in cardiac microvascular endothelial cells. Circ. Res. 1999;84:451–457. doi: 10.1161/01.res.84.4.451. [DOI] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. Endothelium-dependent hyperpolarization of vascular smooth muscle cells. Acta. Pharmacol. Sin. 2000;21:1–18. [PubMed] [Google Scholar]
- FISSLTHALER B., POPP R., KISS L., POTENTE M., HARDER D.R., FLEMING I., BUSSE R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- FLEMING I., MICHAELIS U.R., BREDENKOTTER D., FISSLTHALER B., DEHGHANI F., BRANDES R.P., BUSSE R. Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ. Res. 2001;88:44–51. doi: 10.1161/01.res.88.1.44. [DOI] [PubMed] [Google Scholar]
- GOLLASCH M., HESCHELER J., QUAYLE J., PATLAK J., NELSON M.T. Single Ca-channel currents from arterial smooth muscle at physiological calcium concentrations. Am. J. Physiol. 1992;263:C948–C952. doi: 10.1152/ajpcell.1992.263.5.C948. [DOI] [PubMed] [Google Scholar]
- KÖHLER R., BRAKEMEIER S., KÜHN M., BEHRENS C., REAL R., DEGENHARDT C., ORZECHOWSKI H.D., PRIES A.R., PAUL M., HOYER J. Impaired hyperpolarization in regenerated endothelium after balloon catheter injury. Circ. Res. 2001;89:174–179. doi: 10.1161/hh1401.093460. [DOI] [PubMed] [Google Scholar]
- KÖHLER R., DEGENHARDT C., KÜHN M., RUNKEL N., PAUL M., HOYER J. Expression and function of endothelial Ca2+-activated K+ channels in human mesenteric artery. A single-cell reverse transcriptase-polymerase chain reaction in situ. Circ. Res. 2000;87:496–503. doi: 10.1161/01.res.87.6.496. [DOI] [PubMed] [Google Scholar]
- LOGSDON N.J., KANG J., TOGO J.A., CHRISTIAN E.P., AIYAR J. A novel gene hKCa4, encodes the calcium-activated potassium channel in human T lymphocytes. J. Biol. Chem. 1997;272:32723–32726. doi: 10.1074/jbc.272.52.32723. [DOI] [PubMed] [Google Scholar]
- LÜCKHOFF A., BUSSE R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflugers Arch. 1990;416:305–311. doi: 10.1007/BF00392067. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends Pharmacol. Sci. 1997;18:252–256. [PubMed] [Google Scholar]
- NILIUS B., DROOGMANS G. Ion channels and their functional role in vascular endothelium. Physiological Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- PAPASSOTIRIOU J., KOHLER R., PRENEN J., KRAUSE H., AKBAR M., EGGERMONT J., PAUL M., DISTLER A., NILIUS B., HOYER J. Endothelial K(+) channel lacks the Ca(2+) sensitivity-regulating beta subunit. FASEB J. 2000;14:885–894. [PubMed] [Google Scholar]
- PETERSSON J., ZYGMUNT P.M., HOGESTATT E.D. Characterisation of potassium channels involved in EDHF-mediated relaxation in cerebral arteries. Br. J. Pharmacol. 1997;120:1344–1350. doi: 10.1038/sj.bjp.0701032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETKOV G.V., HEPPNER T.J., BONEV A.D., HERRERA G.M., NELSON M.T. Low levels of K(ATP) channel activation decrease excitability and contractility of urinary bladder. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001;280:R1427–R1433. doi: 10.1152/ajpregu.2001.280.5.R1427. [DOI] [PubMed] [Google Scholar]
- POHL U., HOLTZ J., BUSSE R., BASSENGE E. Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension. 1986;8:37–44. doi: 10.1161/01.hyp.8.1.37. [DOI] [PubMed] [Google Scholar]
- QUIGNARD J.F., FELETOU M., EDWARDS G., DUHAULT J., WESTON A.H., VANHOUTTE P.M. Role of endothelial cell hyperpolarization in EDHF-mediated responses in the guinea-pig carotid artery. Br. J. Pharmacol. 2000;129:1103–1112. doi: 10.1038/sj.bjp.0703175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUIGNARD J.F., FELETOU M., THOLLON C., VILAINE J.P., DUHAULT J., VANHOUTTE P.M. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RITTENHOUSE A.R., PARKER C., BRUGNARA C., MORGAN K.G., ALPER S.L. Inhibition of maxi-K currents in ferret portal vein smooth muscle cells by the antifungal clotrimazole. Am. J. Physiol. 1997;273:C45–C56. doi: 10.1152/ajpcell.1997.273.1.C45. [DOI] [PubMed] [Google Scholar]
- TOFUKUJI M., METAIS C., WANG S.Y., ALPER S.L., SELLKE F.W. Clotrimazole is a potent vasodilator of the rat coronary microcirculation. J. Surg. Res. 1998;77:6–10. doi: 10.1006/jsre.1998.5320. [DOI] [PubMed] [Google Scholar]
- UNGVARI Z., KOLLER A. Mediation of EDHF-induced reduction of smooth muscle [Ca(2+)](i) and arteriolar dilation by K(+) channels, 5,6-EET, and gap junctions. Microcirculation. 2001;8:265–274. doi: 10.1038/sj/mn/7800080. [DOI] [PubMed] [Google Scholar]
- VANHEEL B., VAN DE VOORDE J. Evidence against the involvement of cytochrome P450 metabolites in endothelium-dependent hyperpolarization of the rat main mesenteric artery. J. Physiol. 1997;501:331–341. doi: 10.1111/j.1469-7793.1997.331bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VON DER WEID P.Y., BENY J.L. Simultaneous oscillations in the membrane potential of pig coronary artery endothelial and smooth muscle cells. J. Physiol. 1993;471:13–24. doi: 10.1113/jphysiol.1993.sp019888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALDRON G.J., COLE W.C. Activation of vascular smooth muscle K+ channels by endothelium-derived relaxing factors. Clin. Exp. Pharmacol. Physiol. 1999;26:180–184. doi: 10.1046/j.1440-1681.1999.03006.x. [DOI] [PubMed] [Google Scholar]
- WELSH D.G., SEGAL S.S. Role of EDHF in conduction of vasodilation along hamster cheek pouch arterioles in vivo. Am. J. Physiol. Heart Circ. Physiol. 2000;278:H1832–H1839. doi: 10.1152/ajpheart.2000.278.6.H1832. [DOI] [PubMed] [Google Scholar]
- WULFF H., MILLER M.J., HANSEL W., GRISSMER S., CAHALAN M.D., CHANDY K.G. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc. Natl. Acad. Sci. U.S.A. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]