

Abstract
Acetaminophen, an analgesic and antipyretic drug with weak antiinflammatory properties, has been suggested to act as a tissue-selective inhibitor of prostaglandin H synthases (PGHSs) (e.g. COX-1 and COX-2) through its reducing activity, that is influenced by the different cellular levels of peroxides.
We have studied the effects of acetaminophen on inducible and constitutive prostanoid biosynthesis in monocytes and platelets in vitro. To discriminate between the inhibitory effect of the drug on PGHS-isozymes vs PGE-synthases (PGESs), parallel measurements of PGE2 and thromboxane (TX) B2 were carried out. Since antioxidant enzymes and cofactors, present in plasma, may affect acetaminophen-dependent inhibition of prostanoids, comparative experiments in whole blood vs isolated monocytes were performed.
Acetaminophen inhibited LPS-induced whole blood PGE2 and TXB2 production, in a concentration-dependent fashion [IC50 μM (95% confidence intervals): 44 (27–70) and 94 (79–112), respectively]. Therapeutic plasma concentrations (100 and 300 μM) of the drug more profoundly reduced PGE2 than TXB2 (71±3 vs 54±4 and 95±0.8 vs 78±2%, respectively, mean±s.e.mean, n=6, P<0.01).
Differently, in isolated monocytes stimulated with LPS, both PGE2 and TXB2 production was maximally reduced by only 60%.
At 100 and 300 μM, the drug caused a similar and incomplete inhibition of platelet PGE2 and TXB2 production during whole blood clotting (45±4 vs 54±4 and 75±2 vs 75±1%, respectively, mean±s.e.mean, n=4).
In conclusion, therapeutic concentrations of acetaminophen caused an incomplete inhibition of platelet COX-1 and monocyte COX-2 but in the presence of plasma, the drug almost completely suppressed inducible PGE2 biosynthesis through its inhibitory effects on both COX-2 and inducible PGES.
Keywords: Acetaminophen, PGE-synthase, platelet COX-1, monocyte COX-2, whole blood
Introduction
Acetaminophen is an effective analgesic and antipyretic agent that, unlike nonsteroidal antiinflammatory drugs (NSAIDs), has been reported to have little antiinflammatory effects (Temple, 1983). It has been suggested to act as a tissue-selective inhibitor of prostaglandin H synthases (PGHSs), the key enzymatic step in the biosynthesis of prostanoids, through its reducing activity (Ouellet & Percival, 2001), that is influenced by the different cellular levels of peroxides (Boutaud et al., 2002).
Prostanoids are formed by the coordinate activity of three consecutive enzymatic reactions: (1) release of arachidonic acid (AA) from membrane phospholipids carried out by phospholipase (PL)s, primarily PLA2s (Dennis, 2000; Fitzpatrick & Soberman, 2001); (2) transformation of AA to the unstable endoperoxide PGH2 by PGHS, which exists in two isoforms, COX-1 and COX-2 (Smith & Langenbach, 2001); and (3) metabolism of PGH2 to the different prostanoids by terminal synthases, which have different structures and exhibit a cell- and tissue-specific distribution (Jakobsson et al., 1999; Tanioka et al., 2000; Mancini et al., 2001; Ueno et al., 2001).
COX-1 displays the characteristics of a ‘housekeeping gene' and is constitutively expressed in virtually all tissues. Differently, the induction of COX-2 is an absolute requirement for delayed prostanoid biosynthesis, which lasts for several hours in response to proinflammatory stimuli (Smith & Langenbach, 2001).
COX-1 and COX-2 are haemoproteins that catalyze the same reactions, i.e. the conversion of AA to PGG2 by their cyclooxygenase activity and, then, the reduction of PGG2 to PGH2 by their peroxidase activity. The reactions catalyzed by PGHS-isozymes are controlled by a complex balance between hydroperoxide generation and removal at the level of the cell. The initiation of PGHS activity requires the generation of a hydroperoxide tone (higher for COX-1 than for COX-2) (Kulmacz & Wang, 1995) while high levels of hydroperoxides cause an irreversible loss of PGHS activity (Wu et al., 1999). PGHS-cyclooxygenase activation depends on the reduction of a peroxide by PGHS-peroxidase activity that yields the generation of a higher oxidative state of the haeme (Marnett et al., 1999). This is an essential requirement for the generation of the tyrosyl radical (Tyr-385 in COX-1 and Tyr-371 in COX-2) that catalyzes the first step of cyclooxygenase reaction, i.e. removal of the 13-pro-S-hydrogen with the generation of an AA-radical.
At least two glutathione (GSH)-dependent forms of terminal synthases, involved in the production of PGE2, have been recently identified (Jakobsson et al., 1999; Forsberg et al., 2000; Tanioka et al., 2000; Mancini et al., 2001; Uematsu et al., 2002). One isoform, present in the cytosol (cPGES), constitutively expressed and unresponsive to proinflammatory stimuli, is identical to the heat shock protein 90-associated protein p23 and promotes COX-1-mediated rapid PGE2 biosynthesis (Tanioka et al., 2000). The other isoform (mPGES), localized to the microsomal compartment, is coordinately induced with COX-2 in response to proinflammatory stimuli (Murakami et al., 2000; Stichtenoth et al., 2001).
Acetaminophen has been reported to inhibit PGHS-isozymes by reducing the higher oxidative state of PGHS to the resting state, a process in which acetaminophen serves as a cosubstrate for the peroxidase activity (Ouellet & Percival, 2001). Recently, it has been reported that cellular levels of peroxides may antagonize the reducing activity of acetaminophen towards PGHSs (Boutaud et al., 2002). This may explain the cellular selectivity of the drug. Thus, acetaminophen has been reported to be an efficient inhibitor of PGHS in the brain, containing low peroxide levels (Flower & Vane, 1972). On the contrary, in an inflammatory milieu, presumably containing high levels of peroxides, acetaminophen has been shown to be a weak inhibitor of prostanoid biosynthesis (Mitchell et al., 1993). This is supported by the finding that the potency of acetaminophen against both purified ovine COX-1 and human COX-2 is increased approximately by 30 fold in the presence of GSH peroxidase, that causes the GSH-dependent reduction of alkyl hydroperoxides and H2O2 (Ouellet & Percival, 2001).
We have studied the effects of acetaminophen on inducible and constitutive prostanoid biosynthesis in monocytes and platelets in vitro. To discriminate between the inhibitory effect of the drug on PGHS-isozymes vs PGESs, parallel measurements of PGE2 and TXB2 were carried out. Since antioxidant enzymes and cofactors, present in plasma, may affect acetaminophen-dependent inhibition of prostanoids, comparative experiments in whole blood vs isolated monocytes were performed.
Methods
Subjects
Seven healthy volunteers (five females and two males: aged 29±9 years) were enrolled to participate in the study after its approval by the Ethical Committee of ‘G. D'Annunzio' University of Chieti. Informed consent was obtained from each subject. The same healthy volunteers were studied on different occasions.
Time-course of COX-2, COX-1 and mPGES expression and prostanoid biosynthesis in LPS-stimulated monocytes
Mononuclear cells were separated from human whole blood containing heparin (10 IU ml−1) by Ficoll-Paque, as previously described (Patrignani et al., 1994). The cell suspension was constituted by >90% of monocytes. The cells(2–3×106 ml−1) were incubated in RPMI-1640 [0.5% foetal calf serum (FCS)] with LPS (10 μg ml−1) at 37°C for 0, 4 and 24 h. PGE2 and thromboxane (TX) B2 levels were measured in the medium by radioimmunoassay (RIA), while COX-1, COX-2 and mPGES levels were evaluated in monocyte lysates by Western blot.
Effects of acetaminophen on prostanoid biosynthesis and COX-isozyme expression in LPS-stimulated monocytes
Acetaminophen (0.05–150 mM) was dissolved in dimethyl sulphoxide (DMSO) and 2 μl aliquots of the solutions were pipetted into test tubes containing 2–3×106 monocytes per millilitre to give a final concentration of 0.1 to 300 μM. The same amount of DMSO was added in the control tubes (i.e. without the drug). The cells were incubated at 37°C for 24 h in the presence of LPS (10 μg ml−1). PGE2 and TXB2 released into the medium were assayed by RIA, while COX-1 and COX-2 levels were evaluated in monocyte lysates by Western blot. Moreover, we studied the effects of exogenous GSH (5 μM, dissolved in 0.9% NaCl) on the inhibition of LPS-induced monocyte PGE2 and TXB2 production by acetaminophen (100 and 300 μM).
Effects of acetaminophen, indomethacin and MK-886 on prostanoid biosynthesis in LPS-stimulated whole blood
Acetaminophen (0.05–150 mM), indomethacin (0.015–15 mM) and MK-886, a 5-lipoxygenase activating protein (FLAP) inhibitor that also affects mPGES activity (Mancini et al., 2001), (50–150 mM) were dissolved in DMSO and 2 μl aliquots of the solutions were pipetted into test tubes to give a final concentration of 0.03 to 300 μM. The same amount of DMSO was added in the tubes not containing the compounds. 1-ml aliquots of heparinized whole blood samples, drawn from healthy volunteers, pretreated with aspirin 300 mg 48 h before sampling to suppress platelet COX-1 activity (Patrignani et al., 1982), were added into the test tubes and incubated at 37°C for 24 h in the presence of LPS (10 μg ml−1). Then, plasma was separated by centrifugation (10 min at 2,000 r.p.m.) and kept at −80°C until assayed for immunoreactive TXB2 and PGE2 by RIA.
Effects of acetaminophen on platelet COX-1 activity
Two-μl of increasing concentrations (0.05–150 mM) of acetaminophen were added to 1-ml aliquots of whole blood samples, drawn from the same healthy volunteers when they had not taken any NSAID during the 2 weeks preceding the study, to give a final concentration of 0.1 to 300 μM. The same amount of DMSO was added in the control tubes (i.e. without the drug). Blood samples were allowed to clot at 37°C for 1 h and serum was separated by centrifugation (10 min at 3000 r.p.m.) and kept at −80°C until assayed for TXB2 and PGE2 by RIA. Whole blood TXB2 and PGE2 production was measured as a reflection of maximally stimulated cyclooxygenase activity of platelet COX-1 by endogenously formed thrombin (Patrono et al., 1980).
Radioimmunoassay of TXB2 and PGE2
Unextracted serum and plasma samples as well as cell culture media were diluted in the standard diluent of the assay (0.02 M phosphate buffer, pH 7.4) and assayed for TXB2 and PGE2 in a volume of 1.5 ml, at a final dilution of 1 : 30–1 : 20,000, as previously described (Ciabattoni et al., 1979; Patrono et al., 1980; Patrignani et al., 1994). We used 4000 d.p.m. of [3H]-PGE2 and [3H]-TXB2 and anti-PGE2, anti-TXB2 sera diluted 1 : 100,000, 1 : 120,000, respectively. The least detectable concentration was 1–2 pg ml−1 for both the assays.
Western blot analysis
Isolated monocytes (2–3×106 cells) were lysed and 10 μg aliquots of proteins were analysed by SDS-polyacrylamide gel electrophoresis and immunoblotting techniques using specific rabbit polyclonal antisera (1 : 1,000 dilution) directed against the carboxyl-terminal portion of human COX-2, or against COX-1, as previously described (Patrignani et al., 1994). For PGES detection, 100 μg of the same monocyte lysates were resolved by SDS–PAGE, using a 16% separating gel and transferred electrophoretically to nitrocellulose membranes as described by Jakobsson et al. (1999). After washing the membranes with phosphate-buffered saline-1% Tween-20 (PBS-Tween-20) they were probed with a 1 : 1,000 dilution of PGES antiserum for 1 h at room temperature. Then, the membranes were washed four times with PBS-Tween-20 containing 5% fat-free dried milk, twice with PBS-Tween-20, and incubated with biotynilated anti-rabbit-IgG diluted 1 : 2,000 for 1 h at room temperature. All the blots were developed with streptavidin-peroxidase.
Materials
Acetaminophen, indomethacin DMSO, biotynilated anti-rabbit IgG, streptavidin peroxidase, Tween 20, GSH and LPS derived from Escherichia Coli 026:B6 were purchased from Sigma Chemical Company (St. Louis, MO, U.S.A.). Ficoll-Paque was purchased from Pharmacia Biotech (Milan, Italy). RPMI-1640 medium, FCS were obtained from Mascia Brunelli (Milan, Italy). [3H]-PGE2 and [3H]-TXB2 (specific activity: 140–185 Ci mmol−1) were purchased from Perkin Elmer Life Science Products (Brussels, Belgium). TXB2 and PGE2 were from Cayman Chemical Company (Ann Arbor, MI, U.S.A.). Anti-TXB2 and PGE2 sera were obtained in our laboratory and their characteristics have been described previously (Ciabattoni et al., 1979; Patrignani et al., 1982). Electrophoresis reagents were from Bio-Rad Laboratories (Richmond, CA, U.S.A.). Rabbit polyclonal antibodies prepared against the COX-2 peptide (C)-NASSSRSGLDDINPTVLLK, which is only present in the carboxyl-terminal (amino acid sequence 580–598) of human COX-2, were obtained from Drs J. Maclouf and A. Habib (INSERM, Paris, France). Specific rabbit polyclonal antibodies directed against COX-1 were a gift from Dr W.L. Smith (Department of Biochemistry, Michigan State University, East Lansing, MI, U.S.A.). Specific rabbit polyclonal antibodies directed against mPGES and MK-886 were a gift from Dr J.A. Mancini (Department of Biochemistry and Molecular Biology, Merck Frosst, Pointe-Claire-Dorval, Canada).
Statistical analysis
The data are expressed as mean±s.e.mean. For each experiment, the production of PGE2 and TXB2 in LPS-stimulated whole blood and isolated monocytes was subtracted from the levels of the prostanoids measured in the presence of DMSO, without LPS and the test compounds. The effects of the compounds were reported as per cent inhibition of prostanoid production assessed in the absence of the test compounds (control). Concentration-response curves were fitted, and IC50 values were derived using PRISM (GraphPad, San Diego, CA, U.S.A.). Statistical comparisons were made by Student's t-test.
Results
In isolated monocytes, LPS stimulated the production of PGE2 and TXB2 with a similar time-course. At 24 h, PGE2 and TXB2 levels detected in the medium were 12±1 and 50±4 ng/106 cells (mean±s.e.mean, n=3), respectively. As shown in Figure 1, LPS caused a time-dependent induction of COX-2 and mPGES. In contrast, COX-1 levels, detected in unstimulated monocytes, were not affected by the incubation with LPS up to 24 h.
Figure 1.

Time-course of PGHS-isozyme and mPGES expression and prostanoid biosynthesis in LPS-stimulated monocytes. Isolated human monocytes (2–3×106 ml−1) were incubated for 0, 4 and 24 h at 37°C with LPS (10 μg ml−1) and PGE2 and TXB2 levels were measured in the medium by RIA while COX-1, COX-2 and mPGES levels were evaluated in monocyte lysates by Western blot.
At 24 h of incubation with LPS, acetaminophen caused a concentration-dependent inhibition of monocyte PGE2 and TXB2 production with a ceiling inhibitory effect of only 60% (Figure 2). Therapeutic plasma concentrations (100 and 300 μM) of the drug caused a similar inhibitory effect of PGE2 and TXB2 production (38±9 vs 44±6% and 62±9 vs 57±6%, respectively, mean±s.e.mean, n=3). The similar extent of suppression of the two prostanoids suggests an inhibitory effect of the drug on COX-2 and excludes an effect on mPGES. The finding that acetaminophen did not affect LPS-induced COX-2 levels support an inhibitory effect of the drug on the catalytic activity of COX-2 (Figure 2).
Figure 2.

Effects of acetaminophen on prostanoid biosynthesis and PGHS-isozyme expression in LPS-stimulated monocytes. Increasing concentrations (0.1 to 300 μM) of acetaminophen were incubated with 2–3×106 monocytes per millilitre at 37°C for 24 h in the presence of LPS (10 μg ml−1). PGE2 and TXB2 released into the medium were assayed by RIA while COX-1 and COX-2 levels were evaluated in monocyte lysates by Western blot.
Since it has been postulated that acetaminophen inhibits the cyclooxygenase activity of PGHS-isozymes through its reducing properties, that may be influenced by the cellular levels of peroxides, we studied the influence of plasma, because of its enzymatic and non-enzymatic antioxidant components (Halliwell & Gutteridge, 1990; Ghiselli et al., 2000). Thus, we performed comparative experiments in LPS-stimulated whole blood. These experiments can also allow to unmask the effect of the drug on mPGES, in the light of the requirement of GSH, a plasma constituent, in the activity of the enzyme (Jakobsson et al., 1999; Mancini et al., 2001). Heparinized whole blood incubated for 24 h at 37°C without LPS produced 0.4±0.1 and 0.7±0.2 ng ml−1 (mean±s.e.mean, n=6) of PGE2 and TXB2, respectively. In the presence of LPS, PGE2 and TXB2 levels increased and averaged 26±5 and 18±2 ng ml−1 (mean±s.e.mean, n=6), respectively. As shown in Figure 3, acetaminophen caused a concentration-dependent inhibition of LPS-induced prostanoid biosynthesis. However, the drug was 2 fold more potent in suppressing LPS-induced whole blood PGE2 than TXB2 production [IC50 μM (95% confidence intervals, CI): 44 (27–70) and 94 (79–112) μM, respectively]. Therapeutic plasma concentrations (100 and 300 μM) of the drug more profoundly reduced PGE2 than TXB2 production (71±7 vs 54±9%, P<0.01, and 95±0.8 vs 78±2%, P<0.01, respectively, mean±s.e.mean, n=6) (Figure 4). These results suggest that in the presence of plasma, the reduction of inducible PGE2 biosynthesis by acetaminophen is the sum of a major inhibitory effect on COX-2 and a marginal effect on mPGES. To verify whether this effect on mPGES is shared by other nonselective COX inhibitors, we compared the inhibitory action of indomethacin on inducible PGE2 and TXB2 production. Indomethacin reduced LPS-induced PGE2 and TXB2 production with superimposable concentration-response curves (not shown) that had similar IC50 values [0.34 (CI 95%: 0.2 to 0.56) μM and 0.37 (CI 95%: 0.18 to 0.74 μM), respectively].
Figure 3.
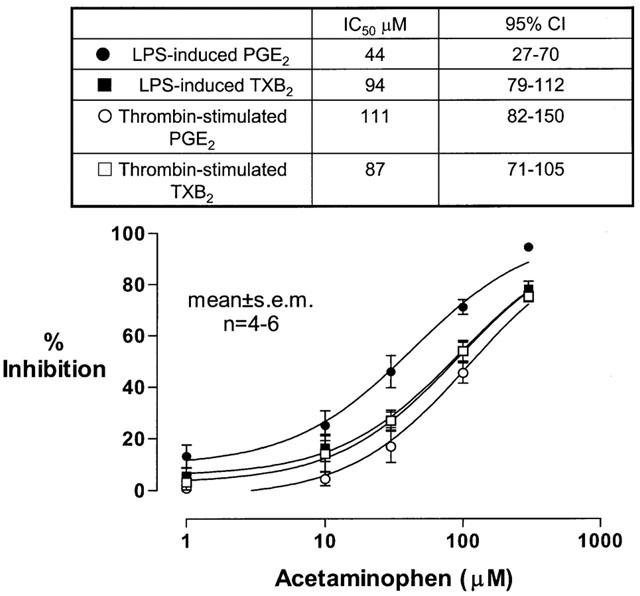
Effects of acetaminophen on prostanoid biosynthesis in whole blood stimulated with LPS or with endogenous thrombin. Increasing concentrations of acetaminophen (0.1–300 μM) were incubated with 1-ml heparinized whole blood samples, drawn from healthy volunteers pretreated with 300 mg of aspirin 48 h before sampling, in the presence of LPS (10 μg ml−1) for 24 h, and plasma PGE2 and TXB2 levels were assayed by RIA as an index of inducible monocyte prostanoid biosynthesis. The same concentrations of the drug were also incubated with 1-ml whole blood samples (drawn from healthy subjects when they had not taken any NSAID during the 2 weeks preceding the study), allowed to clot for 1 h, and serum TXB2 and PGE2 levels were measured as a reflection of constitutive platelet prostanoid biosynthesis in response to endogenously formed thrombin. Results are depicted as percentage inhibition (mean±s.e.mean) from 4–6 separate experiments.
Figure 4.

Effects of therapeutic concentrations of acetaminophen on inducible and constitutive PGE2 and TXB2 production in whole blood. One hundred and 300 μM of acetaminophen were incubated with 1-ml heparinized whole blood samples, drawn from healthy volunteers pretreated with 300 mg of aspirin 48 h before sampling, in the presence of LPS (10 μg ml−1) for 24 h, and plasma PGE2 and TXB2 levels were assayed by RIA as an index of inducible monocyte prostanoid biosynthesis. The same concentrations of the drug were also incubated with 1-ml whole blood samples (drawn from healthy subjects when they had not taken any NSAID during the 2 weeks preceding the study), allowed to clot for 1 h, and serum TXB2 and PGE2 levels were measured as a reflection of constitutive platelet prostanoid biosynthesis. Results are depicted as percentage inhibition (mean±s.e.mean) from 4–6 separate experiments.
To verify whether the limited inhibitory effect of acetaminophen on mPGES activity may be due to a marginal contribution of the enzyme to LPS-induced whole blood PGE2 production, we studied the effects of MK-886, an inhibitor of mPGES activity (Mancini et al., 2001). One hundred μM of MK-886, a concentration that has been shown to completely suppress recombinant rat mPGES (Mancini et al., 2001), caused only 20±5% (mean±s.e.mean, n=3, P<0.05) reduction of LPS-induced PGE2 without affecting TXB2 production (2±2%). No further inhibitory effect was demonstrated at higher concentrations of this compound (data not shown).
In order to ascertain whether the increased inhibition of PGE2 production in whole blood vs isolated monocytes may be due to the availability of GSH, we compared the inhibitory effects of acetaminophen (100 and 300 μM) on PGE2 and TXB2 biosynthesis in isolated monocytes incubated for 24 h with LPS both in the absence and in the presence of GSH (5 μM, a concentration comparable with that detected in plasma of healthy subjects, Adams et al., 1987). In the presence of GSH, the inhibition of PGE2 by acetaminophen was significantly increased (at 100 μM: 63±4.5 vs 49±6%, P=0.029; at 300 μM: 85±5 vs 68±5, P=0.043; mean±s.e.mean, n=5) while that of TXB2 was not affected (at 100 μM: 47±6 vs 46±9%; at 300 μM: 72±10 vs 68±9, P=0.774). TXB2 and PGE2 production in whole blood samples, obtained in aspirin-free periods, allowed to clot at 37°C for 1 h, averaged 366±66 and 17±2 ng ml−1, respectively (mean±s.e.mean, n=4). Acetaminophen inhibited platelet TXB2 and PGE2 production, in response to endogenously formed thrombin, with similar IC50 values of 87 (95% CI: 71–105) and 111 μM (95% CI: 82–150), respectively (Figure 3). At 100 and 300 μM, the drug caused a similar reduction of PGE2 and TXB2 production (45±4 vs 54±4% and 75±2 vs 75±1%, respectively, mean±s.e.mean, n=4, P>0.05) (Figure 4). The similar extent of suppression of the two prostanoids suggests an inhibitory effect of the drug on platelet COX-1 and excludes an effect on PGES.
Discussion
Acetaminophen is the first-line therapy recommended by the American College of Rheumatology for the treatment of pain and stiffness associated with osteoarthritis (ACR Subcommittee on Osteoarthritis Guidelines, 2000) mainly because of its assumed lower toxicity on gastrointestinal (GI) tract and kidney as compared with NSAIDs (Schnitzer, 1998). The drug is considered an analgesic and antipyretic agent with weak antiinflammatory effects (Robak et al., 1980). It has been suggested to act as a tissue-selective inhibitor of PGHSs through its reducing activity, that is presumably influenced by the different cellular levels of peroxides (Ouellet & Percival, 2001; Boutaud et al., 2002). Thus, acetaminophen affects prostanoid biosynthesis in brain microsomes (Flower & Vane, 1972) and in spinal cord (Muth-Selbach et al., 1999) but it is a weak inhibitor in spleen homogenates (Flower et al., 1972). In the present study, we have shown that therapeutic concentrations of acetaminophen (100–300 μM) caused a partial reduction (50–70%) of inducible PGE2 biosynthesis in isolated monocytes stimulated with LPS, through an inhibitory effect on COX-2 activity. In fact, the drug did not affect LPS-induced COX-2 levels analysed by Western blot. The finding that the drug caused a similar suppression of inducible PGE2 and TXB2 excludes an effect on mPGES activity. However, in the presence of plasma, the drug inhibited PGE2 more profoundly than TXB2 (at 100 μM, 71 vs 54%, at 300 μM, 95 vs 78%). This result may suggest that in whole blood, the reduction of inducible PGE2 biosynthesis by acetaminophen is due to the sum of a major inhibitory effect on COX-2 and a limited effect on mPGES. This inhibitory effect of acetaminophen on mPGES is not shared by other nonselective COX inhibitors, such as indomethacin.
Acetaminophen can be metabolized into the reactive intermediate N-acetyl-P-benzoquinoneimine (NAPQI) by liver cytochromes P450 (Thomas, 1993) and the respiratory burst of blood cells, particularly neutrophils (Corbett et al., 1992). NAPQI has been reported to react extensively with GSH (Rosen et al., 1984) and cause its depletion (Rosen et al., 1984; Mohandas et al., 1981). We propose that in whole blood, the oxidation of acetaminophen to its reactive metabolite may affect mPGES by reducing the availability of plasma GSH that is an essential cofactor of its activity (Jakobsson et al., 1999; Mancini et al., 2001). Failure of acetaminophen to affect mPGES, in isolated monocytes cultured in the presence of 0.5% FCS, may be due to a rapid cellular GSH depletion after LPS stimulation and, in consequence, the levels of GSH at the time of mPGES expression (24 h, as shown in Figure 1) could be insufficient to activate the enzyme. This hypothesis has been confirmed by the finding that in the presence of exogenous GSH [5 μM, a concentration comparable to that detected in plasma of healthy subjects (Adams et al., 1987)], the inhibitory effect of acetaminophen on PGE2, but not on TXB2, was significantly increased.
The results of the present study evidence, for the first time, a possible modulation of inducible PGES by acetaminophen, even if limited. This is because mPGES contributes marginally (by approximately 20%) to PGE2 biosynthesis in LPS-stimulated whole blood, as demonstrated by the use of MK-886, a mPGES inhibitor. However, it should be pointed out that this compound is not a selective inhibitor of mPGES. In fact, it was originally identified as a potent inhibitor of leukotriene biosynthesis (IC50 of approximately 3 nM) for its ability to inhibit FLAP (Vickers, 1995) and recently it has been shown to act also as a non-competitive inhibitor of peroxisome-proliferator-activated receptor (PPAR)α, even if at higher concentrations (10–20 μM) (Kehrer et al., 2001). Only the availability of specific and potent inhibitors of mPGES will allow to characterize the contribution of mPGES to PGE2 biosynsthesis in different human tissues. Recently, a critical role of mPGES expression in PGE2 production by mouse peritoneal macrophages in response to LPS has been demonstrated (Uematsu et al., 2002). Anyhow, it should be underlined that the fraction of PGE2 sensitive to MK-886 corresponds to that inhibited by acetaminophen in LPS-stimulated whole blood. In clotting whole blood, acetaminophen caused a similar reduction of PGE2 and TXB2 that suggests its inhibitory effect on COX and excludes that on PGES. The production of the two prostanoids in serum of healthy subjects is mainly of platelet origin, presumably through the activity of constitutive COX-1. In fact, COX-2 is undetectable in platelets of healthy subjects using Western blot techniques (Patrignani et al., 1999) and it has been observed coexpressed with mPGES in <10% of circulating platelets using immunocytochemistry (Rocca et al., 2002). Thus, a different PGES isoform may be involved in the production of PGE2 by activated platelets. Recently, a cPGES expressed ubiquitously in a wide variety of cells that is capable of converting COX-1-, but not COX-2-derived PGH2 to PGE2 has been identified (Tanioka et al., 2000).
Our results demonstrate that therapeutic concentrations of acetaminophen cause a similar inhibition of inducible PGE2 biosynthesis to that obtained by clinical effective doses of coxibs and NSAIDs (e.g.⩾70%) (van hecken et al., 2000). However, acetaminophen is only a modulator of PGHS (Ouellet & Percival, 2001; Boutaud et al., 2002) and mPGES activities and its inhibitory effects may be influenced by the presence of different components of cellular and extracellular milieu (e.g. reduced inhibition by hydroperoxides and enhanced inhibition by GSH). This may lead to a wide intersubject variability in the clinical efficacy of the drug that is presumably involved in the preference of NSAIDs over acetaminophen by rheumatic patients (Pincus et al., 2000; Wolfe et al., 2000). This is further confirmed by the lower clinical efficacy shown by acetaminophen (4 g per day) vs the selective COX-2 inhibitor rofecoxib (25 mg per day) during a 6-week controlled trial in osteoarthritis patients (Geba et al., 2002).
In addition to an inhibitory effect on COX-isozymes and mPGES, it has been claimed that the potent analgesic/antipyretic effects of acetaminophen may involve the inhibition of a COX-1 variant, referred to as COX-3 (Chandrasekharan et al., 2002). The improved renal and GI safety profile of acetaminophen over NSAIDs is not confirmed by population-based observational studies. In fact, Garcia Rodriguez & Hernadez-Diaz (2001) have shown that similarly to oral steroids and aspirin, acetaminophen use was associated with a 2 fold increased risk of upper GI complications when taken at daily doses ⩾2 g. This effect might be due to an inhibitory action of acetaminophen on gastric prostanoid biosynthesis (Konturek et al., 1981). However, this finding has been contradicted by other studies that have shown no effect or even an increase of gastric PGE2 biosynthesis by acetaminophen (Peskar, 1977; van kolfschoten et al., 1981).
As shown in the present study, therapeutic concentrations of acetaminophen may inhibit platelet COX-1 activity in vitro by 50–80%. However, the administration of 1 g of the drug to healthy subjects caused only 44% inhibition of platelet COX-1 activity ex vivo that was not associated with any significant inhibitory effect on AA-induced platelet aggregation (Catella-Lawson et al., 2001). These results may suggest that the toxicity of the drug in vivo is only marginally dependent on the inhibition of prostanoid biosynthesis but may involve its capacity to induce drug-protein adducts and oxidative stress (Hinson et al., 1995; Delanty et al., 1996). Thus, acetaminophen use was associated with increased risk of chronic renal failure (Fored et al., 2001) despite its inability to reduce renal prostanoid biosynthesis (Bippi & Frolich, 1990).
In summary, therapeutic concentrations of acetaminophen caused an incomplete inhibition of monocyte COX-2 but in the presence of plasma, the drug almost completely suppressed inducible PGE2 biosynthesis through its inhibitory effects on both COX-2 and mPGES. However, acetaminophen is only a modulator of PGHS and mPGES activities and its inhibitory effects may be influenced by the levels of tissue-specific constituents (i.e. antioxidant enzymes, GSH and hydroperoxides).
Acknowledgments
Supported by a grant from the Italian Ministry of University and Research (MURST) to the Centre of Excellence on Aging, ‘G.D'Annunzio' University of Chieti
Abbreviations
- AA
arachidonic acid
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- cPGES
cytosolic PGE-synthase
- DMSO
dimethyl sulphoxide
- FLAP
5-lipoxygenase activating protein
- GI
gastrointestinal
- GSH
glutathione
- LPS
lipopolysaccharide
- mPGES
membrane bound PGE-synthase
- NSAIDs
nonsteroidal antiinflammatory drugs
- PGE2
prostaglandin E2
- PLA2
phospholipase A2
- PPAR
peroxisome-proliferator-activated receptor
- TXB2
thromboxane B2
- RIA
radioimmunoassay
References
- ACR SUBCOMMITTEE ON OSTEOARTHRITIS GUIDELINES Recommendations for the medical management of osteoarthritis of the hip and knee. Arthritis Rheum. 2000;43:1905–1915. doi: 10.1002/1529-0131(200009)43:9<1905::AID-ANR1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- ADAMS D., JOHANNESSEN J.N., BACON J.P. Quantification of glutathione disulfide in human plasma. Clinical Chem. 1987;33:1675–1676. [PubMed] [Google Scholar]
- BIPPI H., FROLICH J.C. Effects of acetylsalicylic acid and paracetamol alone and in combination on prostanoid synthesis in man. Br. J. Clin. Pharmacol. 1990;29:305–310. doi: 10.1111/j.1365-2125.1990.tb03640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOUTAUD O., ARONOFF D.M., RICHARDSON J.H., MARNETT L.J., OATES J.A. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H2 synthases. Proc. Natl. Acad. Sci. U.S.A. 2002;99:7130–7135. doi: 10.1073/pnas.102588199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CATELLA-LAWSON F., REILLY M.P., KAPOOR S.C., CUCCHIARA A.J., DEMARCO S., TOURNIER B., VYAS S.N., FITZGERALD G.A. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N. Engl. J. Med. 2001;345:1809–1817. doi: 10.1056/NEJMoa003199. [DOI] [PubMed] [Google Scholar]
- CHANDRASEKHARAN N.V., DAI H., LAMAR TUREPU ROOS K., EVANSON N.K., TOMSIK J., ELTON T.S., SIMMONS D.L.COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression Proc. Natl. Acad. Sci. U.S.A. 2002. epub ahead of print, 1-6 [DOI] [PMC free article] [PubMed]
- CIABATTONI G., PUGLIESE F., SPALDI M., CINOTTI G.A., PATRONO C. Radioimmunoassay measurement of prostaglandin E2 and F2α in human urine. J. Endocrinol. Invest. 1979;2:173–182. doi: 10.1007/BF03349310. [DOI] [PubMed] [Google Scholar]
- CORBETT M.D., CORBETT B.R., HANNOTHIAUX M.H., QUINTANA S.J. The covalent binding of acetaminophen to cellular nucleic acids as the result of the respiratory burst of neutrophils derived from the HL-60 cell line. Toxicol. Appl. Pharmacol. 1992;113:80–86. doi: 10.1016/0041-008x(92)90011-g. [DOI] [PubMed] [Google Scholar]
- DELANTY N., REILLY M., PRATICO D., FITZGERALD D.J., LAWSON J.A., FITZGERALD G.A. 8-Epi PGF2α: specific analysis of an isoeicosanoid as an index of oxidant stress in vivo. Br. J. Clin. Pharmacol. 1996;42:15–19. doi: 10.1046/j.1365-2125.1996.03804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DENNIS E.A. Phospholipase A2 in eicosanoid generation. Am. J. Respir. Crit. Care Med. 2000;161:S32–S35. doi: 10.1164/ajrccm.161.supplement_1.ltta-7. [DOI] [PubMed] [Google Scholar]
- FITZPATRICK F.A., SOBERMAN R. Regulated formation of eicosanoids. J. Clin. Invest. 2001;107:1347–1351. doi: 10.1172/JCI13241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLOWER R., GRYGLEWSKI R., HERBACZYNSKA-CEDRO K., VANE J.R. Effects of anti-inflammatory drugs on prostaglandin biosynthesis. Nature New Biol. 1972;238:104–106. doi: 10.1038/newbio238104a0. [DOI] [PubMed] [Google Scholar]
- FLOWER R.J., VANE J.R. Inhibition of prostaglandin synthetase in brain explains the antipyretic activity of paracetamol (4-acetamidophenol) Nature. 1972;240:410–411. doi: 10.1038/240410a0. [DOI] [PubMed] [Google Scholar]
- FORED C.M., EJERBLAD E., LINDBLAD P., FRYZEK J.P., DICKMAN P.W., SIGNORELLO L.B., LIPWORTH L., ELINDER C.G., BLOT W.J., MCLAUGHLIN J.K., ZACK M.M., NYREN O. Acetaminophen, aspirin, and chronic renal failure. N. Engl. J. Med. 2001;345:1801–1808. doi: 10.1056/NEJMoa010323. [DOI] [PubMed] [Google Scholar]
- FORSBERG L., LEEB L., THOREN S., MORGENSTERN R., JAKOBSSON P.J. Human glutathione dependent prostaglandin E synthase: gene structure and regulation. FEBS Lett. 2000;471:78–82. doi: 10.1016/s0014-5793(00)01367-3. [DOI] [PubMed] [Google Scholar]
- GARCIA RODRIGUEZ L.A., HERNANDEZ-DIAZ S. The risk of upper gastrointestinal complications associated with nonsteroidal anti-inflammatory drugs, glucocorticoids, acetaminophen, and combinations of these agents. Arthritis Res. 2001;3:98–101. doi: 10.1186/ar146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEBA G.P., WEAVER A.L., POLIS A.B., DIXON M.E., SCHNITZER T.J. Vioxx, Acetaminophen, Celecoxib Trial (VACT) Group. JAMA. 2002;287:64–71. doi: 10.1001/jama.287.1.64. [DOI] [PubMed] [Google Scholar]
- GHISELLI A., SERAFINI M., NATELLA F., SCACCINI C. Total antioxidant capacity as a tool to assess redox status: critical view and experimental data. Free Radic. Biol. Med. 2000;29:1106–1114. doi: 10.1016/s0891-5849(00)00394-4. [DOI] [PubMed] [Google Scholar]
- HALLIWELL B., GUTTERIDGE J.M. The antioxidants of human extracellular fluids. Arch. Biochem. Biophys. 1990;280:1–8. doi: 10.1016/0003-9861(90)90510-6. [DOI] [PubMed] [Google Scholar]
- HINSON J.A., PUMFORD N.R., ROBERTS D.W. Mechanisms of acetaminophen toxicity: immunochemical detection of drug-protein adducts. Drug Metab. Rev. 1995;27:73–92. doi: 10.3109/03602539509029816. [DOI] [PubMed] [Google Scholar]
- JAKOBSSON P.J., THOREN S., MORGENSTERN R., SAMUELSSON B. Identification of human prostaglandin E synthase: A microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEHRER J.P., BISWAL S.S., LA E., THUILLIER P., DATTA K., FISCHER S.M., VANDEN HEUVEL J.P. Inhibition of peroxisome-proliferator-activated receptor (PPAR)α by MK886. Biochem. J. 2001;356:899–906. doi: 10.1042/0264-6021:3560899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONTUREK S.J., OBTULOWICZ W., SITO E., OLEKSY J., WILKON S., KIEC-DEMBINSKA A. Distribution of prostaglandins in gastric and duodenal mucosa of healthy subjects and duodenal ulcer patients: effects of aspirin and paracetamol. Gut. 1981;22:283–289. doi: 10.1136/gut.22.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KULMACZ R.J., WANG L.H. Comparison of hydroperoxide initiator requirements for the cyclooxygenase activities of prostaglandin H synthase-1 and -2. J. Biol. Chem. 1995;270:24019–24023. doi: 10.1074/jbc.270.41.24019. [DOI] [PubMed] [Google Scholar]
- MANCINI J.A., BLOOD K., GUAY J., GORDON R., CLAVEAU D., CHAN C.C., RIENDEAU D. Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J. Biol. Chem. 2001;276:4469–4475. doi: 10.1074/jbc.M006865200. [DOI] [PubMed] [Google Scholar]
- MARNETT L.J., ROWLINSON S.W., GOODWIN D.C., KALGUTKAR A.S., LANZO C.A. Arachidonic acid oxygenation by COX-1 and COX-2. Mechanisms of catalysis and inhibition. J. Biol. Chem. 1999;13:22903–22906. doi: 10.1074/jbc.274.33.22903. [DOI] [PubMed] [Google Scholar]
- MITCHELL J.A., AKASEREENONT P., THIEMERMANN C., FLOWER R.J., VANE J.R. Selectivity of nonsteroidal anti-inflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. U.S.A. 1993;90:11693–11697. doi: 10.1073/pnas.90.24.11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOHANDAS J., DUGGIN G.G., HORVATH J.S., TILLER D.J. Metabolic oxidation of acetaminophen (paracetamol) mediated by cytochrome P-450 mixed-function oxidase and prostaglandin endoperoxide synthetase in rabbit kidney. Toxicol. Appl. Pharmacol. 1981;61:252–259. doi: 10.1016/0041-008x(81)90415-4. [DOI] [PubMed] [Google Scholar]
- MURAKAMI M., NARABA H., TANIOKA T., SEMMYO N., NAKATANI Y., KOJIMA F., IKEDA T., FUEKI M., UENO A., OH S., KUDO I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- MUTH-SELBACH U.S., TEGEDER I., BRUNE K., GEISSLINGER G. Acetaminophen inhibits spinal prostaglandin E2 after peripheral noxious stimulation. Anesthesiology. 1999;91:231–239. doi: 10.1097/00000542-199907000-00032. [DOI] [PubMed] [Google Scholar]
- OUELLET M., PERCIVAL M.D. Mechanism of acetaminophen inhibition of cyclooxygenase isoforms. Arch. Biochem. Bioph. 2001;387:273–280. doi: 10.1006/abbi.2000.2232. [DOI] [PubMed] [Google Scholar]
- PATRIGNANI P., FILABOZZI P., PATRONO C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J. Clin. Invest. 1982;69:1366–1372. doi: 10.1172/JCI110576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATRIGNANI P., PANARA M.R., GRECO A., FUSCO O., NATOLI C., IACOBELLI S., CIPOLLONE F., GANCI A., CRÈMINON C., MACLOUF J., PATRONO C. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthases. J. Pharmacol. Exp. Ther. 1994;271:1705–1712. [PubMed] [Google Scholar]
- PATRIGNANI P., SCIULLI M.G., MANARINI S., SANTINI G., CERLETTI C., EVANGELISTA V. COX-2 is not involved in thromboxane biosynthesis by activated human platelets. J. Physiol. Pharmacol. 1999;50:661–667. [PubMed] [Google Scholar]
- PATRONO C., CIABATTONI G., PINCA E., PUGLIESE F., CASTRUCCI G., DE SALVO A., SATTA M.A., PESKAR B.A. Low dose aspirin and inhibition of thromboxane B2 production in healthy subjects. Thromb. Res. 1980;17:317–327. doi: 10.1016/0049-3848(80)90066-3. [DOI] [PubMed] [Google Scholar]
- PESKAR B.M. On the synthesis of prostaglandins by human gastric mucosa and its modification by drugs. Biochim. Biophys. Acta. 1977;487:307–314. doi: 10.1016/0005-2760(77)90007-8. [DOI] [PubMed] [Google Scholar]
- PINCUS T., SWEARINGEN C., CUMMINS P., CALLAHAN L.F. Preference for nonsteroidal antiinflammatory drugs versus acetaminophen and concomitant use of both types of drugs in patients with osteoarthritis. J. Rheumatol. 2000;27:1020–1027. [PubMed] [Google Scholar]
- ROBAK J., KOSTKA-TRABKA E., DUNIEC Z. The influence of three prostaglandin biosynthesis stimulators on carrageenin-induced edema of rat paw. Biochem. Pharmacol. 1980;29:1863–1865. doi: 10.1016/0006-2952(80)90155-0. [DOI] [PubMed] [Google Scholar]
- ROCCA B., SECCHIERO P., CIABATTONI G., RANELLETTI F.O., CATANI L., GUIDOTTI L., MELLONI E., MAGGIANO N., ZAULI G., PATRONO C. Cyclooxygenase-2 expression is induced during human megakaryopoiesis and characterizes newly formed platelets. Proc. Natl. Acad. Sci. U.S.A. 2002;99:7634–7639. doi: 10.1073/pnas.112202999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSEN G.M., RAUCKMAN E.J., ELLINGTON S.P., DAHLIN D.C., CHRISTIE J.L., NELSON S.D. Reduction and glutathione conjugation reactions of N-acetyl-p-benzoquinone imine and two dimethylated analogues. Mol. Pharmacol. 1984;25:151–157. [PubMed] [Google Scholar]
- SCHNITZER T.J. Non-NSAID pharmacologic treatment options for the management of chronic pain. Am. J. Med. 1998;105:45S–52S. doi: 10.1016/s0002-9343(98)00073-4. [DOI] [PubMed] [Google Scholar]
- SMITH W.L., LANGENBACH R. Why there are two cyclooxygenase isozymes. J. Clin. Invest. 2001;12:1491–1495. doi: 10.1172/JCI13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STICHTENOTH D.O., THOREN S., BIAN H., PETERS-GOLDEN M., JAKOBSSON P.J., CROFFORD L.J. Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J. Immunol. 2001;167:469–474. doi: 10.4049/jimmunol.167.1.469. [DOI] [PubMed] [Google Scholar]
- TANIOKA T., NAKATANI Y., SEMMYO N., MURAKAMI M., KUDO I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J. Biol. Chem. 2000;275:32775–32782. doi: 10.1074/jbc.M003504200. [DOI] [PubMed] [Google Scholar]
- TEMPLE A.R. Review of comparative antipyretic activity in children. Am. J. Med. 1983;75:38–46. doi: 10.1016/0002-9343(83)90231-0. [DOI] [PubMed] [Google Scholar]
- THOMAS S. Paracetamol (acetaminophen) poisoning. Pharmacol. Ther. 1993;60:91–120. doi: 10.1016/0163-7258(93)90023-7. [DOI] [PubMed] [Google Scholar]
- UEMATSU S., MATSUMOTO M., TAKEDA K., AKIRA S. Lipopolysaccharide-dependent prostaglandin E2 production is regulated by the glutathione-dependent prostaglandin E2 synthase gene induced by the toll-like receptor 4/MyD88/NF-IL-6 pathway. J. Immunol. 2002;168:5811–5816. doi: 10.4049/jimmunol.168.11.5811. [DOI] [PubMed] [Google Scholar]
- UENO N., MURAKAMI M., TANIOKA T., FUJIMORI K., URADE Y., KUDO I. Coupling between cyclooxygenases, terminal prostanoid synthases and phospholipase A2s. J. Biol. Chem. 2001;276:34918–34927. doi: 10.1074/jbc.M100429200. [DOI] [PubMed] [Google Scholar]
- VAN HECKEN A., SCHWARTZ J.I., DEPRE M., DE LEPELEIRE I., DALLOB A., TANAKA W., WYNANTS K., BUNTINX A., ARNOUT J., WONG P.H., EBEL D.L., GERTZ B.J., DE SCHEPPER P.J. Comparative inhibitory activity of rofecoxib, meloxicam, diclofenac, ibuprofen, and naproxen on COX-2 versus COX-1 in healthy volunteers. J. Clin. Pharmacol. 2000;40:1109–1120. [PubMed] [Google Scholar]
- VAN KOLFSCHOTEN A.A., DEMBINSKA-KIEC A., BASISTA M. Interaction between aspirin and paracetamol on the production of prostaglandins in the rat gastric mucosa. J. Pharm. Pharmacol. 1981;33:462–463. doi: 10.1111/j.2042-7158.1981.tb13834.x. [DOI] [PubMed] [Google Scholar]
- VICKERS P.J. 5-Lipoxygenase activating protein (FLAP) J. Lipid Mediators Cell Signalling. 1995;12:185–194. doi: 10.1016/0929-7855(95)00018-l. [DOI] [PubMed] [Google Scholar]
- WOLFE F., ZHAO S., LANE N. Preference for nonsteroidal antiinflammatory drugs over acetaminophen by rheumatic disease patients: a survey of 1,799 patients with osteoarthritis, rheumatoid arthritis, and fibromyalgia. Arthritis Rheum. 2000;43:378–385. doi: 10.1002/1529-0131(200002)43:2<378::AID-ANR18>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- WU G., WEI C., KULMACZ R.J., OSAWA Y., TSAI A.L. A mechanistic study of self-inactivation of the peroxidase activity in prostaglandin H synthase-1. J. Biol. Chem. 1999;274:9231–9237. doi: 10.1074/jbc.274.14.9231. [DOI] [PubMed] [Google Scholar]