

Abstract
Activation of human platelets by thrombin is mediated by the proteolytic cleavage of two G-protein coupled protease-activated receptors, PAR-1 and PAR-4. However, thrombin also binds specifically to the platelet surface glycoprotein GPIb. It has been claimed that thrombin can induce aggregation of platelets via a novel GPIb-mediated pathway, which is independent of PAR activation and fibrinogen binding to αIIbβ3 integrin, but dependent upon polymerizing fibrin and the generation of intracellular signals.
In the presence of both fibrinogen and the αIIbβ3 receptor antagonist lotrafiban, thrombin induced a biphasic platelet aggregation response. The initial primary response was small but consistent and associated with the release of platelet granules. The delayed secondary response was more substantial and was abolished by the fibrin polymerization blocking peptide GPRP.
Cleavage of the extracellular portion of GPIb by mocarhagin partially inhibited thrombin-induced αIIbβ3-dependent aggregation and release, but had no effect on the secondary fibrin-dependent response.
Fixing of the platelets abolished αIIbβ3-dependent aggregation and release of adenine nucleotides, whereas the fibrin-dependent response remained, indicating that platelet activation and intracellular signalling are not necessary for this secondary ‘aggregation'.
In conclusion, the secondary fibrin-dependent ‘aggregation' response observed in the presence of fibrinogen and lotrafiban is a platelet trapping phenomenon dependent primarily on the conversion of soluble fibrinogen to polymerizing fibrin by thrombin.
Keywords: PAR-1, PAR-4, GPRP, lotrafiban, mocarhagin
Introduction
Thrombin plays a key role in both haemostasis and thrombosis. It is a major end product of the coagulation cascades and is responsible for the conversion of circulating soluble fibrinogen to fibrin monomers which polymerize to form an insoluble mesh which serves to stabilize primary haemostatic plugs formed by aggregating platelets at sites of vascular injury (Colman et al., 2001). Thrombin also directly activates platelets via protease-activated receptors (PARs) of which there are two main subtypes on human platelets, PAR-1 and PAR-4 (Coughlin, 2000). Activation of the G-protein-coupled PARs on platelets by thrombin leads to mobilization of intracellular calcium, inhibition of adenylyl cyclase and activation of small G proteins including Rho. This results in platelet functional responses including shape change, aggregation, generation of thromboxane A2 (TxA2), the release of the contents of alpha and dense granules, and expression of pro-coagulant phospholipids (Coughlin, 2001).
Thrombin also binds with high affinity to the surface glycoprotein GPIb (Harmon & Jamieson, 1986), the second most abundant platelet surface protein (approx. 25,000 copies per platelet (Modderman et al., 1992)). Interference with the interaction of thrombin and GPIb inhibits the functional response of the platelet to thrombin (de candia et al., 1999; De Cristofaro & De Candia, 1999; de cristofaro et al., 2000; de marco et al., 1991; 1994; Mazurov et al., 1991; Nurden et al., 1983). It has been suggested that binding of thrombin to GPIb serves to facilitate the interaction of thrombin with PAR-1 resulting in its more efficient and rapid activation (de candia et al., 2001). In this model GPIb acts as a catalytic co-factor such that together, GPIb, PAR-1 and thrombin form a ternary complex resulting in the efficient activation of PAR-1. No role for GPIb in generating direct intracellular signals was identified in this study.
However, it has also been suggested that thrombin can induce a novel pathway of aggregation via an interaction with GPIb (Soslau et al., 2001). This response was claimed to be mediated by intracellular signals from GPIb, to be dependent upon the presence of polymerizing fibrin, and independent of the integrin αIIbβ3. Such a pathway, it is claimed, may account for the inability of anti-thrombin and anti-platelet treatments to completely abrogate thrombosis in cardiac patients (Soslau et al., 2001). Previous studies have demonstrated platelet aggregation following interactions with polymerizing fibrin (Niewiarowski et al., 1972), although the dependence of this phenomenon on platelet activation is less clear, with some investigators claiming that pre-activation of the platelets is necessary (Hantgan et al., 1985), while others have shown that polymerizing fibrin will cause aggregation of inert particles (Chao et al., 1976). Therefore, the claim that thrombin induces an increase in intracellular calcium via its interaction with GPIb, resulting in a fibrin dependent aggregation (Soslau et al., 2001), represents a novel and exciting development in platelet signalling and functional biology of potentially great significance. It was therefore decided to further investigate this claim.
In the present study, aggregation was measured primarily using the turbidimetric method originally described by Born & Cross (1963), a technique which has been widely used. Briefly, the aggregation of platelets, induced by a platelet agonist, allows more light to pass through a turbid suspension of platelets and less light to be scattered. This reduction in optical density is monitored in real time providing a characteristic aggregation trace. Therefore, turbidimetric aggregometry essentially measures an optical phenomenon which can be influenced by other factors. For example, it is widely recognized that the shape change of platelets results in an increase in the optical density of a suspension of platelets (Born, 1970).
We report here two thrombin-induced phenomena which appear, using a turbidimetric aggregometer, to resemble aggregation responses. The first is a consequence of the degranulation of platelets, while the second is a thrombin-induced, fibrin-dependent response which results in an apparently rapid and complete aggregation of all platelets. It is this second phenomenon which has been claimed as a novel GPIb-mediated pathway of platelet aggregation dependent upon active intracellular signalling (Soslau et al., 2001). We have investigated this phenomenon and have shown that the response to thrombin in the presence of fibrinogen and antagonists of αIIbβ3 is not abrogated by the enzymatic removal of GPIb, and furthermore, can be reproduced using fixed platelets unresponsive to potent platelet agonists. We conclude that this response does not require GPIb or the generation of intracellular signals but represents the trapping of platelets in a fibrin mesh which results following cleavage of fibrinogen to fibrin by thrombin.
Methods
Materials
Formaldehyde was from Polysciences Inc., Warrington, PA, U.S.A. Luciferase assay reagents were from BioThema, Haninge, Sweden. Pyruvate kinase (EC 2.7.1.40) was from Roche Molecular Biochemicals, Lewes, U.K. EDTA, thrombin, fibrinogen, prostacyclin, phosphoenolpyruvate and other salts were obtained from Sigma Chemical Company, Poole, U.K. Lotrafiban (Liu et al., 2000) was a gift from GlaxoSmithKline. The PAR-1 activating peptide TRAP-6 (H-Ser-Phe-Leu-Leu-Arg-Asn-OH) and the fibrin polymerization inhibiting peptide GPRP (H-Gly-Pro-Arg-Pro-NH2) were obtained from Bachem, St Helens, U.K. Mocarhagin (de luca et al., 1995) was kindly provided by Dr Rob Andrews (Baker Medical Research Institute, Australia).
Platelet preparation
Blood was collected from healthy human donors into syringes containing 0.11 M sodium citrate to give a final concentration of 11 mM citrate. The blood was centrifuged at 240×g for 15 min and the PRP removed. Prostacyclin (800 nM) was added to the PRP which was then centrifuged at 125×g to sediment out any residual red blood cells. The red cell free PRP was removed and centrifuged at 640×g for 15 min to pellet the platelets. The PPP was poured off and the pellet re-suspended in 10 ml of the following modified Tyrode's solution (mM): NaCl 137, NaHCO3 11.9, NaH2PO4 0.4, KCl 2.7, MgCl2 1.1, glucose 5.6; pH 7.4, 37°C, containing 800 nM prostacyclin. The re-suspended platelets were centrifuged at 640×g, the supernatant discarded, and the platelets re-suspended in 10 ml of the modified Tyrode's solution. The platelet count was adjusted to 200×106 ml−1 and the platelets allowed to rest at room temperature for at least 1 h. Prior to use of the platelets, 1 mM CaCl2 was added to the suspension.
Fixed platelets were prepared by adding formaldehyde (0.1% final concentration) and EDTA (3 mM final concentration) to washed platelets and allowing them to incubate for at least 3 h. The platelets were then centrifuged at 640×g for 15 min and re-suspended in the modified Tyrode's. The re-suspended platelets were centrifuged again at 640×g and re-suspended at a concentration of 200×106 ml−1 in modified Tyrode's. Prior to use of the platelets, 1 mM CaCl2 was added to the suspension.
Turbidimetric aggregometry
Platelet aggregation was measured using an optical method with a BioData PAP-4 aggregometer (Alpha Laboratories, Eastleigh, U.K.). The assay was performed at 37°C with a sample stir speed of 1000 r.p.m. The aggregometer automatically generated values for the final extent of aggregation, measured as a percentage of a theoretical maximum represented by platelet poor plasma, and the rate of aggregation which is an arbitrary function of the maximum slope of the aggregation trace over the course of the assay.
Platelet counting aggregometry
Platelet aggregation was also assessed using a single platelet counting technique (Bevan & Heptinstall, 1985). This is a more sensitive measure of aggregation, able to detect micro-aggregate formation that is difficult to detect with conventional optical aggregometers. Following activation, the platelets were fixed using formaldehyde (0.1% final concentration) and EDTA (3 mM final concentration). Platelet counting was performed using a Coulter Z2 particle count and size analyser.
Measurement of ATP and ADP
Following activation, platelets were fixed using formaldehyde (0.1% final concentration) and EDTA (3 mM final concentration). The platelets were pelleted using a micro-centrifuge and 200 μl of the supernatant removed and added to 200 μl absolute ethanol. The samples were kept frozen until required. Levels of ATP and ADP were determined using a modified version of a luminometric assay (Jarvis et al., 1996; Lundin et al., 1986). ATP was detected using a luciferin-luciferase assay, and ADP was detected following conversion to ATP using pyruvate kinase and phosphoenolpyruvate. Levels of luminescence were measured with an EG&G Berthold Lumat LB9507 luminometer (PerkinElmer Life Sciences, Cambridge, U.K.).
Data analysis
Concentration-response data was modelled using the following four parameter logistic equation (de lean et al., 1978):
 |
where the variables are: R=response (dependent variable); A=concentration of agonist (independent variable) and the parameters to be estimated are: Min=response when A=0; Max=response when A=∞; pA50=−log10 of concentration of A that gives a response equal to (Max+Min)/2; nH=Hill coefficient. For aggregation data, the parameter Min was constrained to zero in all cases, whereas for the release data, it was estimated along with the other parameters.
For those experiments with fibrinogen and lotrafiban, data from each individual donor for the four different conditions (±fibrinogen, ±lotrafiban) were modelled simultaneously, thereby generating estimates of 12 separate parameters in the case of aggregation and 16 for release. The number of parameters estimated was reduced by constraining values across the four conditions and the best overall model was selected using the Schwarz Bayesian Criterion (Schwarz, 1978) which was calculated as follows:
 |
where: SBC=Schwarz Bayesian Criterion; RSS=the residual sum of squares; n=the number of data points; p=the number of parameters in the model.
The model with the lowest value of the Schwarz criterion was chosen as best representing the data. The parameter values from the best fit models from three separate individuals are expressed as mean±s.e.mean. Subsequent comparison of these values was done using Student's t-test. P values <0.05 were considered significant. Analysis was carried out using SPSS software and Microsoft Excel.
Results
Effect of fibrinogen and lotrafiban on thrombin-induced platelet aggregation and release of adenine nucleotides
Thrombin-induced aggregation of washed human platelets (Figure 1a,c) was measured in a PAP-4 aggregometer and values for the rate and extent of aggregation were automatically obtained. Addition of fibrinogen (0.2 mg ml−1) significantly increased the maximum rate of aggregation from 32.4±1.7 to 64.0±2.6 (mean±s.e.mean; n=3; arbitrary units), whereas in the presence of the αIIbβ3 antagonist lotrafiban (10 μM) the maximum rate was significantly reduced to 10.7±0.4. In the presence of both fibrinogen and lotrafiban, thrombin induced a biphasic response (Figure 1c): the maximum rate under these conditions (83.2±14.8) was similar in magnitude, although more variable, to that in the presence of fibrinogen alone. In control platelets the pA50 for the rate of thrombin-induced aggregation was 1.60±0.25 (A50=0.025 u ml−1) which was unaffected by either fibrinogen or lotrafiban alone. However, in the presence of fibrinogen and lotrafiban together, the pA50 was significantly reduced to 0.65±0.15 (A50=0.22 u ml−1). The Hill coefficient of the response was 3.4±1.0 and was not significantly altered by the presence of lotrafiban or fibrinogen alone or together. A similar pattern was observed for the extent of aggregation (Table 1a,b).
Figure 1.
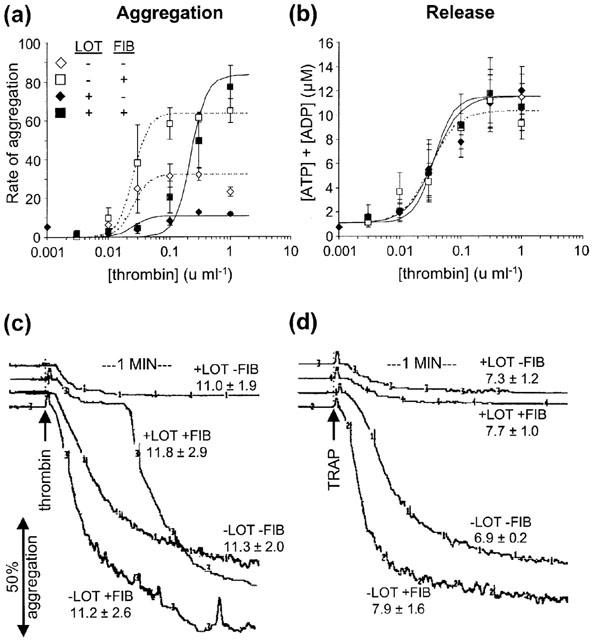
Effect of fibrinogen (FIB, 0.2 mg ml−1) and lotrafiban (LOT, 10 μM) on thrombin and TRAP-induced platelet aggregation and release of adenine nucleotides, ATP and ADP, in washed human platelets. (a) Thrombin concentration-response curves for rate of aggregation. Rate of aggregation is measured in arbitrary units. Fibrinogen increases the rate of aggregation, whereas lotrafiban substantially inhibits the response. The residual response in the presence of lotrafiban is due to release of platelet granules. A combination of fibrinogen and lotrafiban cause a rightward shift in the main response which is caused by the thrombin-induced polymerization of fibrin. (b) Thrombin concentration-response curves for release of ATP and ADP. Fibrinogen and lotrafiban have no effect on the thrombin-induced release of adenine nucleotides. The data points in (a) and (b) are the mean±s.e.mean, and the curves represent the best fit model, for all data from three separate experiments. (c) and (d) Trace recordings showing the effect of fibrinogen and lotrafiban on the aggregation response induced by 0.3 u ml−1 thrombin and by 50 μM TRAP. Figures show the total concentration of released ATP and ADP (μM) (mean±s.e.mean). TRAP fails to induce a secondary response (measured up to 6 min) in the presence of both fibrinogen and lotrafiban, in contrast to thrombin.
Table 1.
Parameter estimates for the thrombin-induced (a) rate and (b) extent of aggregation and (c) release of ATP and ADP

The addition of fibrinogen or lotrafiban, either alone or in combination, had no effect on the thrombin-induced release of adenine nucleotides (Figure 1b,c; Table 1c). Furthermore, the sensitivity to thrombin of the release response was similar to that seen for aggregation as indicated by the pA50 which was 1.50±0.16 (A50=0.032 u ml−1).
For control platelets and also in the presence of either fibrinogen or lotrafiban alone, the thrombin-induced responses were prompt, occurring within seconds of adding the agonist. However, in the presence of both fibrinogen and lotrafiban, although there was a prompt primary response (similar to that seen in the presence of lotrafiban alone) there was a concentration-dependent delay between addition of the agonist and the initiation of the more substantial secondary response (Figures 1c and 2b).
Figure 2.
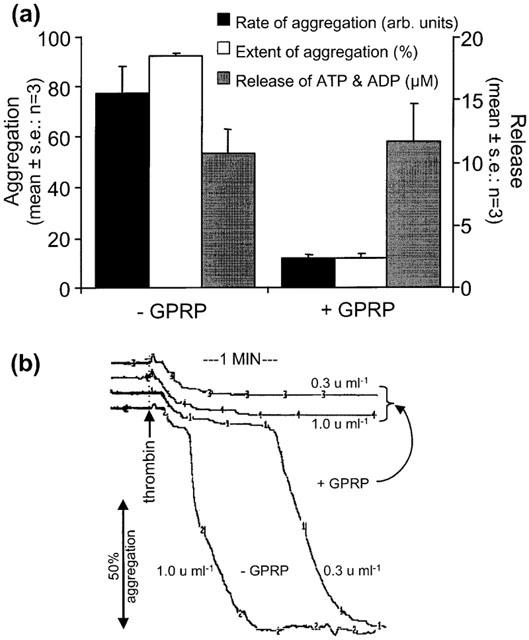
The effect of fibrin polymerization blocking peptide GPRP (1 mM) on thrombin-induced responses in washed human platelets. (a) In the presence of fibrinogen (0.2 mg ml−1) and lotrafiban (10 μM), GPRP inhibited both the rate and extent of the thrombin-induced aggregation response by preventing the polymerization of fibrin monomers, whereas it had no effect on the release of ATP and ADP. The residual response in the presence of GPRP represents the small primary response induced by thrombin which is caused by the release of platelet granules. Data shown are the mean±s.e.mean of the response to 1 u ml−1 thrombin from three different experiments. (b) These traces illustrate the inhibitory effect of GPRP on the secondary thrombin-induced response in the presence of fibrinogen (0.2 mg ml−1) and lotrafiban (10 μM). The smaller primary response is unaffected by GPRP. Concentrations of thrombin are shown and illustrate the concentration dependence of the delay to the onset of the fibrin-dependent secondary response. The traces are representative of three separate experiments.
Effect of fibrinogen and lotrafiban on TRAP-induced platelet aggregation and release of adenine nucleotides
Thrombin-related activatory peptide (TRAP) is a six amino acid synthetic peptide (H-Ser-Phe-Leu-Leu-Arg-Asn-OH) which directly activates PAR-1 but does not cleave fibrinogen (Vassallo et al., 1992). TRAP (50 μM) induced aggregation in control platelets with a rate of 35.8±2.8 (mean±s.e.mean; n=2). In the presence of fibrinogen, this increased to 64.3±1.8. Lotrafiban inhibited TRAP in a similar manner to thrombin: a small response was still observed with a rate of 9.8±0.8. However, in the presence of fibrinogen and lotrafiban together, TRAP only induced the small response (rate=10.8±1.8) observed in the presence of lotrafiban alone, and did not induce the large secondary response observed with thrombin (Figure 1c,d). The addition of fibrinogen and lotrafiban alone or together had no effect on the release of ATP and ADP induced by 50 μM TRAP (Figure 1d).
Effect of GPRP on thrombin-induced aggregation and release of adenine nucleotides
The effect of the fibrin polymerization blocking peptide GPRP (Laudano & Doolittle, 1978) was investigated. In the presence of fibrinogen (0.2 mg ml−1) and lotrafiban (10 μM), GPRP (1 mM) abolished the main secondary response induced by thrombin whilst having no effect on the smaller primary response or the release of adenine nucleotides (Figure 2a). In the presence of GPRP, the small response was indistinguishable from that in the presence of lotrafiban alone (Figure 1c and 2b). It has previously been shown that GPRP at 1 mM has a minimal effect on platelet aggregation (Adelman et al., 1990): in accordance with these observations, GPRP at 1 mM had no effect on thrombin-induced aggregation or release in the absence of lotrafiban, either in the absence or presence of fibrinogen (data not shown).
Effect of mocarhagin on thrombin-induced aggregation and release of adenine nucleotides
We investigated the role played by GPIb in the response of platelets to thrombin using mocarhagin, a cobra venom toxin which cleaves the extracellular portion of GPIb (Ward et al., 1996). Washed platelets were incubated for at least 10 min with mocarhagin (10 μg ml−1) prior to use. The effectiveness of the mocarhagin treatment was demonstrated by its ability to inhibit the agglutination of platelets induced by a combination of von Willebrand Factor (vWF, 10 μg ml−1) and ristocetin (1 mg ml−1) (data not shown), a response which is dependent upon GPIb (Cooper et al., 1976; Howard & Firkin, 1971).
Mocarhagin inhibited the thrombin-induced rate of aggregation in control platelets as well as in the presence of fibrinogen (Figure 3a,b). The small residual response observed in the presence of lotrafiban was also inhibited (Figure 3c). In the presence of lotrafiban and fibrinogen together, mocarhagin inhibited the small primary response but had no effect on the main secondary thrombin-induced fibrin-dependent response (Figures 3d and 4). Similar effects of mocarhagin were also observed for the extent of aggregation (data not shown). Thrombin-induced release of ATP and ADP was also inhibited by mocarhagin. The inhibition was manifest as an approximate 10 fold rightward shift in the concentration-response curve to thrombin, which was similar regardless of the absence or presence of lotrafiban and fibrinogen (Figure 3e–h).
Figure 3.
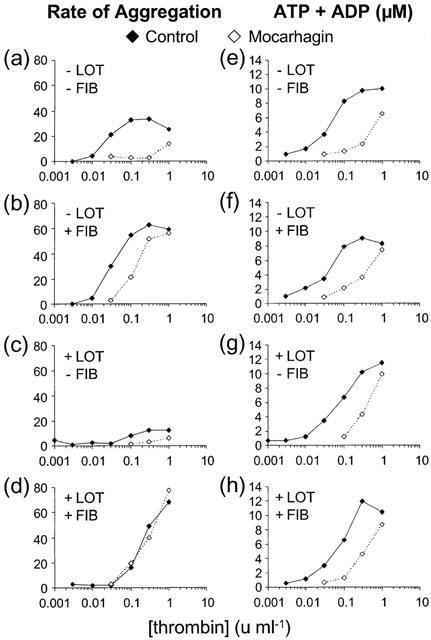
The effect of mocarhagin on the rate of aggregation and release of ATP and ADP induced by thrombin in the absence and presence of fibrinogen (FIB, 0.2 mg ml−1) and lotrafiban (LOT, 10 μM). Mocarhagin (10 μg ml−1) inhibited the response to thrombin in all cases, except the aggregation response in the presence of fibrinogen and lotrafiban which is dependent on the fibrin polymerizing activity of thrombin. Data are the mean of two different experiments.
Figure 4.
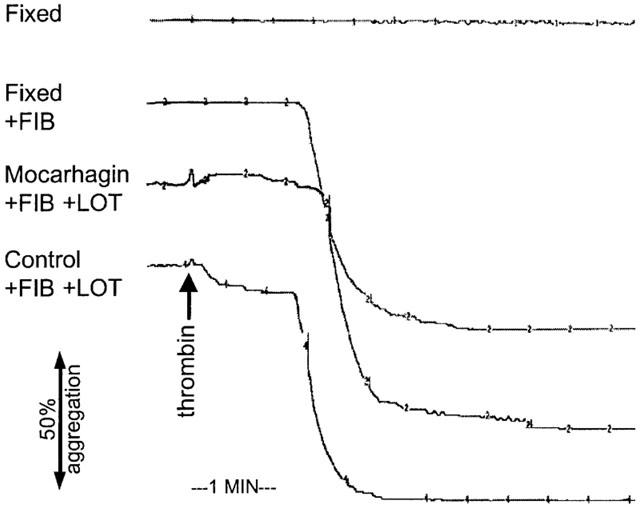
The effect of mocarhagin (10 μg ml−1) and fixation on responses to thrombin. The control trace shows the primary and secondary responses induced by thrombin in the presence of lotrafiban (LOT, 10 μM) and fibrinogen (FIB, 0.2 mg ml−1). Pre-treatment with mocarhagin substantially inhibited the primary response but did not affect the secondary fibrin-dependent response. Fixation of the platelets with formaldehyde (0.1%) and EDTA (3 mM) abolished the response to thrombin in the absence of fibrinogen. However, in the presence of fibrinogen, although there was no primary response, as seen in control platelets, the secondary response was virtually unaffected. The concentration of thrombin used was 0.3 u ml−1 in all cases except for the fixed trace in which it was 1 u ml−1. Traces shown are representative of those obtained from two separate experiments.
Thrombin-induced responses in fixed platelets
We investigated the ability of thrombin (1 u ml−1) to induce responses in platelets fixed with formaldehyde (0.1%) and EDTA (3 mM). In the absence of fibrinogen and lotrafiban, fixation abolished all thrombin-induced aggregation and release (Figures 4 and 5). Fixation also abolished the small residual response observed in control platelets in the presence of lotrafiban. In the presence of fibrinogen, this small residual response was not observed, but thrombin did induce the secondary fibrin-dependent aggregation response (Figure 4) that was only slightly lower than that seen in normal platelets in the presence of fibrinogen and lotrafiban (Figure 5: rate, P=0.054; extent, P=0.019; normal (plus fibrinogen and lotrafiban) vs fixed (plus fibrinogen) platelets).
Figure 5.
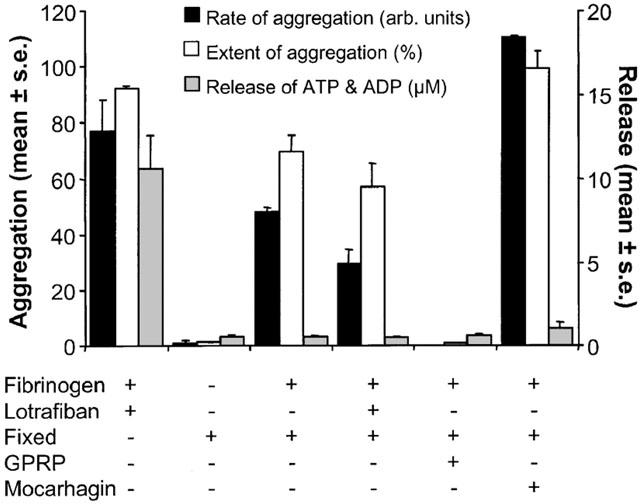
The effect of fixation on the response of platelets to thrombin. The fibrin polymerization-mediated response in normal platelets in the presence of fibrinogen (0.2 mg ml−1) and lotrafiban (10 μM) is shown. Fixation of the platelets abolishes all responsiveness of the platelets. However, in the presence of fibrinogen, the fixed platelets manifest a rapid aggregation response. This was abolished by GPRP (1 mM), slightly attenuated by lotrafiban (10 μM), and augmented following treatment with mocarhagin (10 μg ml−1). The concentration of thrombin used is 1 u ml−1 in all cases. Data shown are mean±s.e.mean of three separate experiments, except for the data with mocarhagin which is from two experiments.
The fibrin polymerization-inhibiting peptide GPRP (1 mM) abolished the response in fixed platelets (Figure 5). In the presence of lotrafiban (10 μM) the fibrinogen-dependent response in fixed platelets remained, although it was slightly attenuated (Figure 5): the rate was significantly lower (P=0.025) although the extent of aggregation was not (P=0.28). vWF/ristocetin induced an agglutination response in fixed platelets in the absence of fibrinogen, which, as expected, was abolished by mocarhagin (data not shown). Interestingly, following treatment of the fixed platelets with mocarhagin, the thrombin-induced response in the presence of fibrinogen was significantly greater (Figure 5: rate (P<0.001); extent (P=0.042)). Under no circumstances did thrombin induce release of ATP and ADP in fixed platelets (Figure 5). The low levels of ATP and ADP present following treatment with thrombin were not significantly different from those following treatment with a vehicle control (data not shown).
Thrombin-induced responses in the presence of lotrafiban
In the presence of the αIIbβ3 antagonist lotrafiban, thrombin (and TRAP) consistently induced an apparent small aggregation response as indicated by the aggregometer (Figures 1c,d, 2b and 4; Table 1a,b). In order to further investigate this phenomenon, we performed single platelet counting aggregometry on these samples as described above.
In the absence of lotrafiban, maximal concentrations of thrombin always induced a substantial fall in the platelet count from approximately 170×106 ml−1 to values of approximately 10×106 ml−1 and less (data not shown). However, as expected, the effect of thrombin on the platelet count in the presence of lotrafiban was less marked. In two out of three experiments there was a small fall in the platelet count observed at the highest concentrations of thrombin (Figure 6). In the third experiment, there was a greater reduction in the platelet count, which was blocked in the presence of GPRP (1 mM). GPRP did not affect the thrombin-induced change in optical density which remained unchanged at 12.5±0.5% (mean±s.e.mean; n=2).
Figure 6.
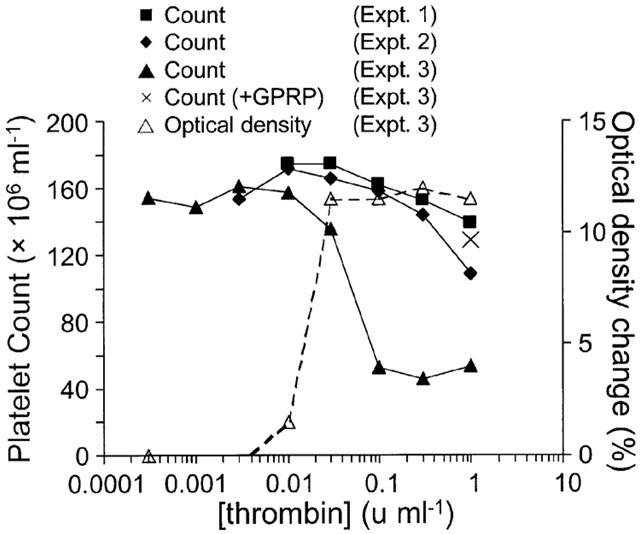
The effect on the platelet count of thrombin treatment in the presence of lotrafiban (10 μM). Thrombin-stimulated platelets from three separate experiments were fixed and the platelet count subsequently determined using a Coulter counter. For experiments 1 and 2 there was only a small reduction in platelet count at the highest concentrations of thrombin. In experiment 3, the platelet count was more substantially reduced, although, as indicated by the response to 0.03 u ml−1 thrombin, there was not a consistent relationship between this effect and the small change in optical density as measured by the aggregometer. In the presence of GPRP (1 mM), the fall in the platelet count observed in experiment 3 was almost completely reversed suggesting that the micro-aggregation was predominantly mediated by the generation of small quantities of polymerizing fibrin.
Discussion
Thrombin is a well characterized and potent platelet agonist which activates the G-protein-coupled receptors PAR-1 (Vu et al., 1991) and PAR-4 (Xu et al., 1998) which are present on human platelets (Coughlin, 2000; Kahn et al., 1998; 1999). Thrombin acts by cleaving the extracellular N-terminal portion of the receptor, revealing a so-called tethered ligand which then interacts with the receptor resulting in activation. Thrombin is also known to bind with high affinity to GPIb (Mazzucato et al., 1998). The role of GPIb in thrombin-induced platelet activation has been less extensively investigated than the role of the PARs; however, it has been demonstrated that inhibition of the interaction of thrombin with GPIb can substantially attenuate the ability of thrombin to activate platelets (De Cristofaro & De Candia, 1999; de marco et al., 1991; 1994). Binding of thrombin to GPIb is thought to facilitate the thrombin-mediated cleavage of PAR-1 (de candia et al., 2001) and in this way to promote platelet activation.
However, it has recently been suggested that thrombin can induce a unique pathway of platelet aggregation via a GPIb-mediated signalling pathway, independently of the PARs, which utilizes polymerizing fibrin instead of fibrinogen binding to activated αIIbβ3 integrin (Soslau et al., 2001). The identification of such a pathway would be of considerable interest and, as indicated by the authors of the study, could be of direct and substantial clinical relevance. In the present study, we have further investigated this phenomenon but have, by contrast, concluded that it is not associated with a functionally significant thrombin-induced activation of GPIb. In fact, the response observed is primarily due to the thrombin-induced polymerization of fibrin, resulting in the trapping of platelets in suspension, giving the appearance of a rapid aggregation response.
Platelet aggregation typically proceeds following the conversion of the integrin αIIbβ3 into a high affinity fibrinogen-binding receptor resulting in the cross linking of activated platelets by bi-polar fibrinogen molecules (Leung & Nachman, 1986; Zucker & Nachmias, 1985). We initially investigated the effect of fibrinogen and the αIIbβ3 fibrinogen antagonist lotrafiban (Liu et al., 2000) on thrombin-induced platelet responses. Aggregation of control platelets occurred since platelet alpha granules release sufficient fibrinogen to enable aggregation to proceed. Addition of exogenous fibrinogen, however, resulted in a substantial increase in the rate of aggregation, presumably by facilitating a more rapid cross-linking of the integrin. Addition of lotrafiban inhibited the aggregation response (Figure 1a,c), although there remained a small reduction in the optical density of approximately 10% which we believe is associated with the release of platelet granules rather than actual aggregation (see below).
In the presence of both fibrinogen and lotrafiban the small initial response was still observed, but followed subsequently by a rapid secondary response (Figure 1c). The time to its onset was clearly concentration-dependent (Figure 2b), and the sensitivity of this response was approximately 10 fold less than that of αIIbβ3-dependent aggregation and release (Table 1), suggesting that it may be mediated via a different mechanism. This is consistent with the hypothesis that αIIbβ3-dependent aggregation and release are caused by platelet activation whilst the less sensitive secondary aggregation response could be mediated by the conversion of fibrinogen to fibrin by thrombin and the subsequent trapping of the platelets by the polymerizing fibrin. This view was supported by the observation that the response only occurred in the presence of added fibrinogen, so we further investigated this possibility by using the fibrin polymerization blocking peptide, GPRP (Achyuthan et al., 1986; Laudano & Doolittle, 1978). The inhibition by GPRP of the secondary response (Figure 2) without any effect on the other parameters of platelet activation indicates that the secondary response was dependent upon fibrin polymerization. This is consistent with the results previously reported (Soslau et al., 2001).
Since it has been suggested that this secondary fibrin-dependent response is mediated by an interaction of thrombin with GPIb, resulting in the generation of intracellular signals in platelets (Soslau et al., 2001), and given that GPIb is a known thrombin-binding protein (Harmon & Jamieson, 1986), we investigated the role of GPIb in this phenomenon using the cobra venom mocarhagin (Ward et al., 1996). Consistent with previous studies (de candia et al., 2001; Ward et al., 1996), mocarhagin partially inhibited thrombin-induced platelet activation as indicated by the inhibition of the aggregation responses (Figure 3a–c) and release (Figure 3e–h). By contrast, in the presence of fibrinogen and lotrafiban, mocarhagin did not inhibit the secondary thrombin-induced fibrin-dependent response (Figure 3d). Although mocarhagin abolished the vWF/ristocetin agglutination response (data not shown) it is possible that a low level of GPIb remained uncleaved. However, it is inconceivable that the concentration-response relationship of a GPIb-dependent thrombin-induced aggregation should have remained unchanged (Figure 3d), particularly when in the same preparations, the release response was inhibited (Figure 3h). These data indicate therefore, that the thrombin-induced fibrin-dependent aggregation response is not dependent on GPIb, whereas the thrombin-induced platelet activation resulting in αIIbβ3-dependent aggregation and release is partially dependent on GPIb, a conclusion entirely in accord with previous observations (de candia et al., 2001; Ward et al., 1996).
Indeed, the ability of mocarhagin to partially inhibit all indices of thrombin-induced platelet activation except for the fibrin-dependent response suggests that this latter secondary response may be due solely to the conversion of fibrinogen to fibrin and not dependent upon metabolically active platelets at all. This view was confirmed by the observation that all thrombin-induced responses were abolished in fixed platelets, except the fibrin-dependent response which still occurred and was still abolished by GPRP (Figure 5). Interestingly, the addition of lotrafiban to the fixed platelets caused a small inhibition of the rate and extent of the fibrin-dependent response (Figure 5), suggesting that the polymerized fibrin may trap the fixed platelets more effectively while the αIIbβ3 integrin remains unblocked. Mocarhagin abolished vWF/ristocetin-induced agglutination of fixed platelets (data not shown), but did not cause any inhibition of the fibrin-dependent response observed in fixed platelets. These data confirm that the thrombin-induced fibrin-dependent response of platelets is not dependent on platelet activation or binding of thrombin to GPIb, but is essentially a platelet trapping phenomenon caused by the polymerization of fibrin in close proximity to the platelets in suspension. This is consistent with previous observations of the ability of thrombin to induce ‘aggregation' of inert particles in the presence of fibrinogen (Chao et al., 1976).
The question arises as to why the conclusion of our study is so significantly divergent from that of Soslau et al. (2001). The conclusions drawn by Soslau et al. (2001) are especially dependent upon two assumptions: namely, that thrombin-induced PAR-1 activation was abolished by the PAR-1 antagonist SCH203099 (Ahn et al., 1999; 2000), and that the concentrations of thrombin used were insufficient to activate the PAR-4 receptor. However, their data show that TRAP induced platelet shape change in the presence of SCH203099, suggesting that blockade of PAR-1 was incomplete, and they also show that desensitization of PAR-4 attenuated responses induced by 0.05 u ml−1 thrombin, indicating that this concentration of thrombin was able to activate PAR-4. Hence, their claim that the calcium signal induced by 0.1 u ml−1 thrombin was mediated by GPIb is difficult to sustain, given that they neither inactivated PAR-4 nor demonstrated sensitivity to mocarhagin. Furthermore, the authors did not report the effect of mocarhagin on the fibrin-dependent response observed in the presence of the αIIbβ3 peptide antagonist RGDS, a response identical in form to those of our study reported here. By contrast, we have shown that mocarhagin has no effect whatsoever. We therefore believe that the phenomena reported by Soslau et al. (2001) can be explained as responses mediated by PAR-1 and PAR-4, and by the passive trapping of platelets by polymerizing fibrin. Indeed the pattern of the calcium responses observed by Soslau et al. (2001) is indicative of a PAR-1-mediated rapid and transient calcium signal, and a slower, more sustained signal mediated by PAR-4 as previously reported (Covic et al., 2000).
During the course of this study, we also observed a response which was apparently independent of αIIbβ3 integrin. Although lotrafiban substantially inhibited thrombin-induced aggregation, there was a consistent residual response which appeared as approximately 10% aggregation (Figures 1, 2 and 4). The pA50 of this residual response was similar to that of thrombin-induced release under all conditions. This similarity suggested that the reduction in the optical density of the platelet suspension was not caused by aggregation, but was associated with the loss of intracellular platelet granules, in particular the dense granules.
To further investigate this possibility, we measured aggregation using a particle counting technique (Bevan & Heptinstall, 1985) which is able to detect micro-aggregate formation which conventional optical aggregometers cannot (Milton & Frojmovic, 1983; Thompson et al., 1986). Using a Coulter counter, we observed a small and variable reduction in the platelet count despite the presence of the lotrafiban. However, in two out of three experiments, this reduction was minimal (Figure 6) and did not correlate at all with the reduction in optical density in the same samples as measured by the aggregometer. In the third experiment, the reduction in the platelet count was more obvious (Figure 6); however, even in this case it was clear that there was no direct relationship between this response and the change in optical density, since the response to 0.03 u ml−1 thrombin had little effect on the platelet count but induced a maximal optical density response in the aggregometer in two independent samples. Furthermore, addition of GPRP (1 mM) to these platelets substantially attenuated the reduction in the platelet count without affecting the change in optical density. Therefore, thrombin-induced polymerization of trace quantities of fibrin is the most likely explanation for the micro-aggregate formation, particularly in the third experiment.
A 50% reduction in platelet count could be achieved if all platelets formed small micro-aggregates of approximately two platelets each or if a sub-population produced larger micro-aggregates outside the range of detection of the Coulter counter. However, it is known that micro-aggregates comprising approximately two to eight platelets are not detected by turbidimetric aggregometers (Frojmovic & Panjwani, 1975), and therefore it seems unlikely that the consistent residual 10% response observed in the presence of lotrafiban was due to such a small and variable degree of micro-aggregation. By contrast, it has been recognized that the release of platelet granules can influence the optical density of a suspension of platelets (Hugues, 1971; Kitek & Breddin, 1980; Milton & Frojmovic, 1983). Therefore, we conclude that the 10% reduction in optical density is associated primarily with degranulation of platelets.
In conclusion, we have investigated two apparent ‘aggregation' phenomena as measured by a turbidimetric aggregometer. The first is a substantial and rapid response caused by the thrombin-induced conversion of fibrinogen to fibrin and its subsequent polymerization to form a fibrin mesh which traps the platelets, thereby generating an appearance of aggregation. This phenomenon is not dependent on any activation of platelets or on interactions of thrombin with GPIb, contrary to the conclusions of a previous study (Soslau et al., 2001). We have also shown that a small residual response observed in the presence of an αIIbβ3 antagonist is associated primarily with the degranulation of the platelets and not their aggregation.
Acknowledgments
This work was supported by the British Heart Foundation. S.P. Watson is a British Heart Foundation Senior Research Fellow. B.T. Atkinson is a holder of a British Heart Foundation Research Studentship. J. Frampton is a Wellcome Trust Senior Basic Biomedical Research Fellow. We would like to thank Dr Rob Andrews (Baker Medical Research Institute, Melbourne, Australia) for supplying the mocarhagin.
Abbreviations
- FIB
fibrinogen
- LOT
lotrafiban
- PAR
protease-activated receptor
- TRAP
thrombin-related activatory peptide
- TxA2
thromboxane A2
- vWF
von Willebrand Factor
References
- ACHYUTHAN K.E., DOBSON J.V., GREENBERG C.S. Gly-Pro-Arg-Pro modifies the glutamine residues in the alpha- and gamma-chains of fibrinogen: inhibition of transglutaminase cross-linking. Biochim. Biophys. Acta. 1986;872:261–268. doi: 10.1016/0167-4838(86)90279-7. [DOI] [PubMed] [Google Scholar]
- ADELMAN B., GENNINGS C., STRONY J., HANNERS E. Synergistic inhibition of platelet aggregation by fibrinogen-related peptides. Circ. Res. 1990;67:941–947. doi: 10.1161/01.res.67.4.941. [DOI] [PubMed] [Google Scholar]
- AHN H.S., ARIK L., BOYKOW G., BURNETT D.A., CAPLEN M.A., CZARNIECKI M., DOMALSKI M.S., FOSTER C., MANNA M., STAMFORD A.W., WU Y. Structure-activity relationships of pyrroloquinazolines as thrombin receptor antagonists. Bioorg. Med. Chem. Lett. 1999;9:2073–2078. doi: 10.1016/s0960-894x(99)00339-x. [DOI] [PubMed] [Google Scholar]
- AHN H.S., FOSTER C., BOYKOW G., STAMFORD A., MANNA M., GRAZIANO M. Inhibition of cellular action of thrombin by N3-cyclopropyl-7-[[4-(1-methylethyl)phenyl]methyl]-7H-pyrrolo[3,2-f]quinazoline-1,3-diamine (SCH 79797), a nonpeptide thrombin receptor antagonist. Biochem. Pharmacol. 2000;60:1425–1434. doi: 10.1016/s0006-2952(00)00460-3. [DOI] [PubMed] [Google Scholar]
- BEVAN J., HEPTINSTALL S. Serotonin-induced platelet aggregation in whole blood and the effects of ketanserin and mepyramine. Thromb. Res. 1985;38:189–194. doi: 10.1016/0049-3848(85)90060-x. [DOI] [PubMed] [Google Scholar]
- BORN G.V. Observations on the change in shape of blood platelets brought about by adenosine diphosphate. J. Physiol. 1970;209:487–511. doi: 10.1113/jphysiol.1970.sp009176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORN G.V.R., CROSS M.J. The aggregation of blood platelets. J. Physiol. 1963;168:178–195. doi: 10.1113/jphysiol.1963.sp007185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAO F.C., TULLIS J.L., CONNEELY G.S., LAWLER J.W. Aggregation of platelets and inert particles induced by thrombin. Thromb. Haemost. 1976;35:717–736. [PubMed] [Google Scholar]
- COLMAN R.W., HIRSH J., MARDER V.J., CLOWES A.W., GEORGE J.N. Hemostasis and thrombosis: basic principles and clinical practice. Philadelphia: Lippincott Williams & Wilkins; 2001. [Google Scholar]
- COOPER H.A., MASON R.G., BRINKHOUS K.M. The platelet: membrane and surface reactions. Annu. Rev. Physiol. 1976;38:501–535. doi: 10.1146/annurev.ph.38.030176.002441. [DOI] [PubMed] [Google Scholar]
- COUGHLIN S.R. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- COUGHLIN S.R. Protease-activated receptors in vascular biology. Thromb. Haemost. 2001;86:298–307. [PubMed] [Google Scholar]
- COVIC L., GRESSER A.L., KULIOPULOS A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry. 2000;39:5458–5467. doi: 10.1021/bi9927078. [DOI] [PubMed] [Google Scholar]
- DE CANDIA E., DE CRISTOFARO R., LANDOLFI R. Thrombin-induced platelet activation is inhibited by high-and low-molecular-weight heparin. Circulation. 1999;99:3308–3314. doi: 10.1161/01.cir.99.25.3308. [DOI] [PubMed] [Google Scholar]
- DE CANDIA E., HALL S.W., RUTELLA S., LANDOLFI R., ANDREWS R.K., DE CRISTOFARO R. Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J. Biol. Chem. 2001;276:4692–4698. doi: 10.1074/jbc.M008160200. [DOI] [PubMed] [Google Scholar]
- DE CRISTOFARO R., DE CANDIA E. Thrombin interaction with platelet GpIb: structural mapping and effects on platelet activation (review) Int. J. Mol. Med. 1999;3:363–371. doi: 10.3892/ijmm.3.4.363. [DOI] [PubMed] [Google Scholar]
- DE CRISTOFARO R., DE CANDIA R., RUTELLA S., WEITZ J.I. The Asp(272)-Glu(282) region of platelet glycoprotein Ibalpha interacts with the heparin-binding site of alpha-thrombin and protects the enzyme from the heparin-catalyzed inhibition by antithrombin III. J. Biol. Chem. 2000;275:3887–3895. doi: 10.1074/jbc.275.6.3887. [DOI] [PubMed] [Google Scholar]
- DE LEAN A., MUNSON P.J., RODBARD D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- DE LUCA M., DUNLOP L.C., ANDREWS R.K., FLANNERY J.V., JR, ETTLING R., CUMMING D.A., VELDMAN G.M., BERNDT M.C. A novel cobra venom metalloproteinase, mocarhagin, cleaves a 10-amino acid peptide from the mature N terminus of P-selectin glycoprotein ligand receptor, PSGL-1, and abolishes P-selectin binding. J. Biol. Chem. 1995;270:26734–26737. doi: 10.1074/jbc.270.45.26734. [DOI] [PubMed] [Google Scholar]
- DE MARCO L., MAZZUCATO M., MASOTTI A., FENTON J.W., 2ND, RUGGERI Z.M. Function of glycoprotein Ib alpha in platelet activation induced by alpha-thrombin. J. Biol. Chem. 1991;266:23776–23783. [PubMed] [Google Scholar]
- DE MARCO L., MAZZUCATO M., MASOTTI A., RUGGERI Z.M. Localization and characterization of an alpha-thrombin-binding site on platelet glycoprotein Ib alpha. J. Biol. Chem. 1994;269:6478–6484. [PubMed] [Google Scholar]
- FROJMOVIC M.M., PANJWANI R. Blood cell structure-function studies: light transmission and attenuation coefficients of suspensions of blood cells and model particles at rest and with stirring. J. Lab. Clin. Med. 1975;86:326–343. [PubMed] [Google Scholar]
- HANTGAN R.R., TAYLOR R.G., LEWIS J.C. Platelets interact with fibrin only after activation. Blood. 1985;65:1299–1311. [PubMed] [Google Scholar]
- HARMON J.T., JAMIESON G.A. The glycocalicin portion of platelet glycoprotein Ib expresses both high and moderate affinity receptor sites for thrombin. A soluble radioreceptor assay for the interaction of thrombin with platelets. J. Biol. Chem. 1986;261:13224–13229. [PubMed] [Google Scholar]
- HOWARD M.A., FIRKIN B.G. Ristocetin–a new tool in the investigation of platelet aggregation. Thromb. Diath. Haemorrh. 1971;26:362–369. [PubMed] [Google Scholar]
- HUGUES J. What does the optical platelet aggregation test actually measure? Introductory remarks. Acta Med. Scand. Suppl. 1971;525:39–40. doi: 10.1111/j.0954-6820.1972.tb05787.x. [DOI] [PubMed] [Google Scholar]
- JARVIS G.E., EVANS R.J., HEATH M.F. The role of ADP in endotoxin-induced equine platelet activation. Eur. J. Pharmacol. 1996;315:203–212. doi: 10.1016/s0014-2999(96)00637-1. [DOI] [PubMed] [Google Scholar]
- KAHN M.L., NAKANISHI-MATSUI M., SHAPIRO M.J., ISHIHARA H., COUGHLIN S.R. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Invest. 1999;103:879–887. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAHN M.L., ZHENG Y.W., HUANG W., BIGORNIA V., ZENG D., MOFF S., FARESE R.V., JR, TAM C., COUGHLIN S.R. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- KITEK A., BREDDIN K. Optical density variations and microscopic observations in the evaluation of platelet shape change and microaggregate formation. Thromb. Haemost. 1980;44:154–158. [PubMed] [Google Scholar]
- LAUDANO A.P., DOOLITTLE R.F. Synthetic peptide derivatives that bind to fibrinogen and prevent the polymerization of fibrin monomers. Proc. Natl. Acad. Sci. U.S.A. 1978;75:3085–3089. doi: 10.1073/pnas.75.7.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEUNG L., NACHMAN R. Molecular mechanisms of platelet aggregation. Annu. Rev. Med. 1986;37:179–186. doi: 10.1146/annurev.me.37.020186.001143. [DOI] [PubMed] [Google Scholar]
- LIU F., CRAFT R.M., MORRIS S.A., CARROLL R.C. Lotrafiban: an oral platelet glycoprotein IIb/IIIa blocker. Expert Opin. Investig. Drugs. 2000;9:2673–2687. doi: 10.1517/13543784.9.11.2673. [DOI] [PubMed] [Google Scholar]
- LUNDIN A., HASENSON M., PERSSON J., POUSETTE A. Estimation of biomass in growing cell lines by adenosine triphosphate assay. Methods Enzymol. 1986;133:27–42. doi: 10.1016/0076-6879(86)33053-2. [DOI] [PubMed] [Google Scholar]
- MAZUROV A.V., VINOGRADOV D.V., VLASIK T.N., REPIN V.S., BOOTH W.J., BERNDT M.C. Characterization of an antiglycoprotein Ib monoclonal antibody that specifically inhibits platelet-thrombin interaction. Thromb. Res. 1991;62:673–684. doi: 10.1016/0049-3848(91)90371-3. [DOI] [PubMed] [Google Scholar]
- MAZZUCATO M., MARCO L.D., MASOTTI A., PRADELLA P., BAHOU W.F., RUGGERI Z.M. Characterization of the initial alpha-thrombin interaction with glycoprotein Ib alpha in relation to platelet activation. J. Biol. Chem. 1998;273:1880–1887. doi: 10.1074/jbc.273.4.1880. [DOI] [PubMed] [Google Scholar]
- MILTON J.G., FROJMOVIC M.M. Turbidometric evaluations of platelet activation: relative contributions of measured shape change, volume, and early aggregation. J. Pharmacol. Methods. 1983;9:101–115. doi: 10.1016/0160-5402(83)90002-5. [DOI] [PubMed] [Google Scholar]
- MODDERMAN P.W., ADMIRAAL L.G., SONNENBERG A., VON DEM BORNE A.E. Glycoproteins V and Ib-IX form a noncovalent complex in the platelet membrane. J. Biol. Chem. 1992;267:364–369. [PubMed] [Google Scholar]
- NIEWIAROWSKI S., REGOECZI E., STEWART G.J., SENYL A.F., MUSTARD J.F. Platelet interaction with polymerizing fibrin. J. Clin. Invest. 1972;51:685–699. doi: 10.1172/JCI106857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NURDEN A.T., DIDRY D., ROSA J.P. Molecular defects of platelets in Bernard-Soulier syndrome. Blood Cells. 1983;9:333–358. [PubMed] [Google Scholar]
- SCHWARZ G. Estimating the dimension of a model. Ann. Stat. 1978;6:461–464. [Google Scholar]
- SOSLAU G., CLASS R., MORGAN D.A., FOSTER C., LORD S.T., MARCHESE P., RUGGERI Z.M. Unique pathway of thrombin-induced platelet aggregation mediated by glycoprotein Ib. J. Biol. Chem. 2001;276:21173–21183. doi: 10.1074/jbc.M008249200. [DOI] [PubMed] [Google Scholar]
- THOMPSON N.T., SCRUTTON M.C., WALLIS R.B. Particle volume changes associated with light transmittance changes in the platelet aggregometer: dependence upon aggregating agent and effectiveness of stimulus. Thromb. Res. 1986;41:615–626. doi: 10.1016/0049-3848(86)90358-0. [DOI] [PubMed] [Google Scholar]
- VASSALLO R.R., JR, KIEBER-EMMONS T., CICHOWSKI K., BRASS L.F. Structure-function relationships in the activation of platelet thrombin receptors by receptor-derived peptides. J. Biol. Chem. 1992;267:6081–6085. [PubMed] [Google Scholar]
- VU T.K., HUNG D.T., WHEATON V.I., COUGHLIN S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- WARD C.M., ANDREWS R.K., SMITH A.I., BERNDT M.C. Mocarhagin, a novel cobra venom metalloproteinase, cleaves the platelet von Willebrand factor receptor glycoprotein Ibalpha. Identification of the sulfated tyrosine/anionic sequence Tyr-276-Glu-282 of glycoprotein Ibalpha as a binding site for von Willebrand factor and alpha-thrombin. Biochemistry. 1996;35:4929–4938. doi: 10.1021/bi952456c. [DOI] [PubMed] [Google Scholar]
- XU W.F., ANDERSEN H., WHITMORE T.E., PRESNELL S.R., YEE D.P., CHING A., GILBERT T., DAVIE E.W., FOSTER D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZUCKER M.B., NACHMIAS V.T. Platelet activation. Arteriosclerosis. 1985;5:2–18. doi: 10.1161/01.atv.5.1.2. [DOI] [PubMed] [Google Scholar]