

Abstract
Accumulated evidence indicates that the adenylyl cyclase (AC)/cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA)/cAMP-responsive element binding protein (CREB) signal transduction system may be linked to learning and memory function.
The effects of nefiracetam, which has been developed as a cognition enhancer, on spatial memory function and the AC/cAMP/PKA/CREB signal transduction system in rats with sustained cerebral ischaemia were examined.
Microsphere embolism (ME)-induced sustained cerebral ischaemia was produced by injection of 700 microspheres (48 μm in diameter) into the right hemisphere of rats. Daily oral administration of nefiracetam (10 mg kg−1 day−1) was started from 15 h after the operation.
The delayed treatment with nefiracetam attenuated the ME-induced prolongation of the escape latency in the water maze task that was examined on day 7 to 9 after ME, but it did not reduce the infarct size.
ME decreased Ca2+/calmodulin (CaM)-stimulated AC (AC-I) activity, cAMP content, cytosolic PKA Cβ level, nuclear PKA Cα and Cβ levels, and reduced the phosphorylation and DNA-binding activity of CREB in the nucleus in the right parietal cortex and hippocampus on day 3 after ME. The ME-induced changes in these variables did not occur by the delayed treatment with nefiracetam.
These results suggest that nefiracetam preserved cognitive function, or prevented cognitive dysfunction, after sustained cerebral ischaemia and that the effect is, in part, attributable to the prevention of the ischaemia-induced impairment of the AC/cAMP/PKA/CREB signal transduction pathway.
Keywords: Nefiracetam, microsphere embolism, learning and memory, cyclic AMP/PKA/CREB signal transduction system
Introduction
The biochemical transmission of an external signal, eventually acting on some intracellular second messenger, is absolutely essential for interneuronal signal transduction. cAMP is a representative second messenger, the formation of which is catalyzed by the transmembrane-bound enzyme adenylyl cyclase (AC). Tissue distribution studies based on mRNA levels have shown that type I AC (AC-I) mRNA is present primarily and specifically in neuronal tissues including the neocortex and hippocampus (Xia et al., 1991), that type VIII AC (AC-VIII) mRNA is abundantly expressed in the cerebral cortex and hippocampus (Cali et al., 1994), and that type V/VI AC (AC-V/VI) mRNAs are found ubiquitously at high levels in the brain (Iyengar, 1993). Each of these different AC subtypes has specific patterns of sensitivity to GTP-binding proteins and Ca2+/calmodulin (CaM; Iyengar, 1993). It has been suggested that the multiplicity of AC subtypes contributes to the diversity and specificity of the mechanisms underlying AC signal transduction (Mons & Cooper, 1995). Accumulating evidence indicates that the cAMP signal transduction system is associated with synaptic plasticity and long-term memory formation. For example, (1) AC activity was enhanced by learning and memory in the bar-pressing task (Guillou et al., 1998), and Ca2+-stimulated AC activity increased after completion of a spatial learning test (Guillou et al., 1999). (2) Injection of cAMP into the lateral ventricle improved the experimental amnesia in mice (Chute et al., 1981). (3) The cAMP content increased after performance of an inhibitory avoidance learning task (Bernabeu et al., 1997). (4) PKA inhibitors hindered long-term memory for inhibitory avoidance in the chick brain (Zhao et al., 1995). These findings suggest that cAMP signal transduction pathway may largely contribute to learning and memory function.
The cAMP-responsive element binding protein (CREB) in the nucleus is phosphorylated and activated by the cAMP-dependent protein kinase (PKA), and consequently binds to the cAMP response element (CRE) of target genes (Habener, 1990), the genes of which are considered to be involved in memory, especially long-term memory, formation following new protein synthesis (Guzowski & McGaugh, 1997). In fact, long-term memory of fear conditioning and spatial learning was disrupted in CREB knockout mice (Bourtchuladze et al., 1994). Also, intrahippocampal infusion of CREB antisense oligonucleotides several hours prior to learning reduced the level of CREB protein in the hippocampus and disrupted long-term spatial memory (Guzowski & McGaugh, 1997). These observations suggest that the AC/cAMP/PKA/CREB signal transduction system may play an important role in the regulation of learning and memory function.
Sustained cerebral ischaemia provokes an irreversible neurodegenerative disorder that may lead clinically to a progressive dementia and global cognitive deterioration. Microsphere-induced cerebral embolism results in decreases in cerebral blood flow (Miyake et al., 1993), high-energy phosphates (Takeo et al., 1992), and neurotransmitters (Taguchi et al., 1993) and induces widespread formation of small emboli and multiple infarct areas in the brain. Thus, this model is considered to mimic focal ischaemia-induced human stroke (Lyden et al., 1992) or multi-infarct dementia (Naritomi, 1991). In fact, this model induced learning and memory dysfunction as assessed by the water maze task and impaired the AC-I activity (Takagi et al., 1997; Nagakura et al., 2002a). Agents that prevent such deleterious events have been expected as therapeutic drugs for ischaemia-induced dementia.
Nefiracetam, N-(2,6-dimethylphenyl)-2-(2-oxo-1-pyrrolidinyl) acetamide, is a member of oxopyrrolidine acetic acid derivatives such as piracetam, aniracetam, and oxiracetam. Several studies have shown the pharmacological basis of nefiracetam, including an increase in uptake and release of cholinergic and GABAergic neurotransmitters (Yoshii et al., 1997) and enhancement of neuronal transmission through activation of neuronal Ca2+ channels and nicotinic acetylcholine receptor (Yoshii et al., 2001). However, the exact mechanisms for anti-amnesic effects of this agent remain unclear. Several experiments showed that pretreatment of scopolamine-, benzodiazepine-, and cycloheximide-treated, animals with nefiracetam revealed its anti-amnesic effects in the passive avoidance task (Nabeshima et al., 1990; 1991; Doyle et al., 1993). Furthermore, this agent has been reported to reveal the long-term-potentiation-like facilitation of the synaptic transmission in the hippocampus (Nishizaki et al., 1999), and to improve β-amyloid-induced learning and memory impairments (Yamada et al., 1999). It is suggested that piracetam-like nootropics may lead to a significant improvement of cognitive functions in patients with Alzheimer's disease (Parnetti et al., 1997). Thus, this agent has been developed as a therapeutic drug against cognitive disorders in Alzheimer's disease and/or senile dementia. The therapeutic efficacy of this agent has been under clinical investigation. According to our previous results as above, we hypothesized that nefiracetam may exert a cognition-enhancing effect on ischaemia-induced memory dysfunction and that the effect, if any, may relate to protection of the AC/cAMP/PKA/CREB signal transduction system against ischaemia-induced neuronal damage of the microsphere-embolized animal. This hypothesis was tested in the present study.
Methods
Animals
Male Wistar rats (Charles River Japan Inc., Atsugi, Japan), weighing 180 to 220 g, were maintained in a room with a 12-h light/12-h darkness cycle, at a temperature of 23±1°C, and a humidity of 55±5% throughout the experiment. The animals had free access to food and water according to the National Institute of Health Guide for the Care and Use of Laboratory Animals and the Guideline of Experimental Animal Care issued by the Prime Minister's Office of Japan. All efforts were made to minimize suffering of the animals, to reduce the number of animals used, and to utilize alternatives to in vivo techniques, if available. The study protocol was approved by the Committee of Animal Care and Welfare of Tokyo University of Pharmacy & Life Science.
In the present study, we designed two series of experiments. In the first series of experiments, the operated rats were subjected to a water maze test from day 7 to day 9 after the embolism. The infarct areas of the ME animals were also determined. In the second series, the operated rats were subjected to biochemical and immunochemical experiments on day 3, including determination of adenylyl cyclase activity, G-protein, cAMP content, PKA, and CREB.
Microsphere embolism (ME)
Microsphere-induced cerebral embolism was performed by the method described previously (Miyake et al., 1993) with some modification. In brief, after anaesthetization with 35 mg kg−1 i.p. of sodium pentobarbitone, the right external carotid and pterygopalatine arteries were temporarily occluded with strings. A needle connected to a polyethylene catheter (3 Fr., Atom Co., Tokyo) was inserted into the right common carotid artery. Seven hundred microspheres (47.5±0.5 μm in diameter, NEN-005, New England Nuclear Inc., Boston, U.S.A.), suspended in 20% dextran solution, were injected into the right internal carotid artery through the cannula. The needle was then removed, and the puncture wound was repaired with surgical glue (Aron αA, Sankyo, Co., Tokyo). Finally, the strings occluding the right external carotid and pterygopalatine arteries were released. Following the temporal occlusion of the arteries, it took 2 to 3 min for the blood flow to be re-established in the areas supplied by the right external carotid and pterygopalatine arteries.
Neurological deficits
Fifteen hours after the operation, the neurological deficits of the operated rats were scored on the basis of paucity of movement, truncal curvature, and forced circling during locomotion according to the criteria described previously (Miyake et al., 1993). The score of each item (neurological deficit) was rated from 3 to 0 (3, very severe; 2, severe; 1, moderate; 0, little or none). The rats with a total score of 7–9 points were used in the present study. The neurological deficits were determined at 10 : 00 every morning up to either day 10 or day 3.
Delayed treatment with nefiracetam
After stroke-like symptoms of microsphere-injected rats had been determined, the animals were randomly divided into two groups, nefiracetam-treated and untreated groups. Nefiracetam (10 mg kg−1) was administered orally from 15 h after the operation, once daily to the test group, whereas vehicle (0.5% carboxyl methyl cellulose) was administered to the control group. This treatment was conducted until day 9 in the first series of experiments or until day 3 in the second series. The dose employed in the present study was determined from the data of others (Nabeshima et al., 1990; 1991; Yamada et al., 1999) and those obtained in our preliminary study: we found that nefiracetam at 10 mg kg−1 day−1, p.o., was most effective among these three doses, 3, 10, and 30 mg kg−1 day−1, in the improvement of the spatial memory function of the ME animal.
Water maze test
The water maze test was performed according to the method described by other investigators (Morris, 1981; Takagi et al., 1997). The test was started on day 7 after the operation. ME and sham-operated animals were tested in the water maze by using a three trials/day-regimen. To eliminate rats that could not swim due to injury following ME, we performed a habituation study by placing the surgically treated rats in a pool with a diameter of 100 cm on day 6 after the operation. There were no animals that could not swim in the habituation test of the first series of experiments. The water maze apparatus (model TARGET/2, Neuroscience Co., Tokyo) consisted of a circular pool with a diameter of 170 cm, which was filled with water to a 30-cm depth. The water temperature was maintained at 23±1°C. A hidden platform circle with a diameter of 12 cm was placed 1.5 cm below the surface of the water and kept in a constant position in the centre of one of the four quadrants of the pool. The animals were released from three randomly assigned start locations (excluding the platform-containing quadrant). When a rat mounted the platform, it was kept there for 30 s. If the rat did not reach the platform, it was transferred onto the platform by hand and kept there for 30 s. Data collection was automated by an on-line video-tracking device designed to track the object in the field. Escape latency (the time to climb onto the platform) and swimming speed (the distance that the animals swam divided by escape time) were determined for each trial with a behavioural tracing analyser (BAT-2, Neuroscience Co., Tokyo). The cut-off time for each trial was set at 180 s. Each trial was performed with approximately 1 h of inter-trial interval. On day 10, the visible platform test was performed to examine the ability of spatial navigation of the operated animals.
Triphenyltetrazolium chloride (TTC)-staining
To elucidate the effects of nefiracetam on the infarct size of the ME animal, TTC staining of the brain slices from the animals untreated and treated with 10 mg kg−1 day−1 nefiracetam for 9 days were performed according to the method described previously (Miyake et al., 1993). In brief, coronal sections with a 2-mm width were made from the brains of ME or drug-treated ME rats, and the brain slices were incubated for 30 min with 2% TTC in physiological saline. TTC-unstained areas were analysed by the Image analyser (NIH image).
Brain membrane and cytosolic preparation
To determine the level of GTP-binding protein and AC activity in the parietal cortex and hippocampus of the right hemisphere, we prepared membrane fractions from these areas. Both brain regions were selected because these regions are believed to be involved in learning and memory function (DiMattia & Kesner, 1988; Save et al., 1992) and are quite sensitive to ischaemia (Kirino, 1982; Smith et al., 1984). The isolated brain regions were homogenized in buffer A (in mM): HEPES (pH 7.4) 20, sucrose 250, phenylmethylsulfonyl fluoride (PMSF) 0.3, dithiothreitol 1, ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetra-acetic acid (EGTA) 1, MgCl2 1. The homogenates were centrifuged at 1000×g for 10 min. The supernatant fluid was collected and centrifuged at 100,000×g for 20 min to pellet the membrane fraction. The pellets were then resuspended in the same buffer as above and stored at −80°C until used. The supernatant obtained after the second centrifugation was used as the cytosolic preparation. Determination of protein concentrations was conducted with a protein assay kit according to the method of Bradford (1976).
AC activity
AC activity was assayed by the method of Salomon et al. (1974) with some modification. The basal AC activity was determined by incubating the membrane protein (50 μg) in buffer B (in mM): glycylglycine (pH 7.5) 50, MgCl2 5, Mg·ATP 0.5, isobutylmethylxanthine 1, NaCl 100, EGTA 2, and an ATP-regenerating system consisting of 2 mM creatine phosphate, 20 μg ml−1 creatine kinase, and 0.1 mg ml−1 myokinase in a final volume of 200 μl. The reaction was carried out for 5 min at 37°C and terminated by the addition of 60% HClO4. Then the mixture was neutralized with 2.5 M K2CO3. The generated cAMP was measured with a commercially available kit (Amersham cAMP EIA system, RPN. 225; Amersham, London, U.K.). 5′-Guanylyl imidodiphosphate (Gpp(NH)p)- and Ca2+/CaM-sensitive AC activities were assessed in the presence of 10 μM Gpp(NH)p and 2.4 μM CaM/1 μM free Ca2+, respectively, i.e., conditions that gave optimal stimulation of AC activity. The free Ca2+ concentration in buffer B was estimated by using a Ca2+/EGTA buffer system.
Measurement of cAMP content
The procedure for rapid fixation of the brain to determine the cAMP content was exactly the same as that reported elsewhere (Jones et al., 1974). In brief, the head of a rat was focally irradiated (5 kw, 0.85 s) by a microwave applicator (model TMW-6402C, Muromachi Kikai Co., Tokyo). After decapitation, the head of the animal was immersed into liquid nitrogen for 10 s, and the right parietal cortex and hippocampus were isolated from the cerebral hemispheres. Each brain tissue was homogenized in 0.2 M HClO4 and 0.01% ethylenediamine-N,N,N′,N′-tetra-acetic acid (EDTA) with a Polytron homogenizer (PT-10, Kinematica, Littau/Luzern, Switzerland). The homogenates were centrifuged at 10,000×g for 15 min. The supernatant fluid was neutralized with Na2CO3, and centrifuged 10,000×g for 15 min. The cAMP content in the supernatant fluid was assessed with a commercially available kit (Amersham cAMP EIA system, RPN.225; Amersham, London, U.K.). The pellets from the homogenate obtained after centrifugation were digested with 2 N NaOH and then heated at 60°C. Protein concentrations in the extract were determined by the method of Lowry et al. (1951).
Preparation of nuclear extracts
Nuclear extracts were prepared by the method of Schreiber et al. (1989) with minor modification. In brief, isolated parietal cortex and hippocampus of the right hemisphere were homogenized in 10 volumes of buffer C (in mM): HEPES-NaOH (pH 7.9) 10, containing KCl 10, EDTA 1, EGTA 1, DTT 5, sodium fluoride (NaF) 10, sodium pyrophosphate 10, Na3VO4 1, sodium β-glycerophosphate 10, PMSF 1, 1.25 μg ml−1 pepstatin A, 10 μg ml−1 leupeptin and 2.5 μg ml−1 aprotinin. Following the addition of 10% Nonidet P-40 for a final concentration of 1%, the homogenates were centrifuged at 15,000×g for 5 min. Pellets were washed in three volumes of buffer C, and centrifuged at 15,000×g for 5 min. The pellets were then suspended in one volume of buffer D (in mM): HEPES NaOH (pH 7.9) 20, NaCl 400, EDTA 1, EGTA 1, DTT 5, NaF 10, sodium pyrophosphate 10, Na3VO4 1, sodium β-glycerophosphate 10, 1.25 μg ml−1 pepstatin A, 10 μg ml−1 leupeptin, 2.5 μg ml−1 aprotinin, and PMSF 1, and centrifuged at 15,000×g for 5 min. The supernatant was stored at −80°C as the nuclear extract. The protein concentrations of the samples were determined by the method prescribed by Bradford (1976).
Western immunoblot analysis
Samples containing 5–50 μg of protein were separated on 8 or 10% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Immobilon PVDF, Millipore Co., Bedford, MA, U.S.A.). The following primary antibodies were used: anti-Gαs, Gαi1,2, or Gβ antibodies (NENTM Life Science Products, Inc.; 1 : 10000), anti-AC-I, V/VI, or VIII antibodies (Santa Cruz Biotechnology; 1 : 1000), anti-PKA RIIα, Cα, or Cβ antibodies (Santa Cruz Biotechnology: 1 : 1000), and anti-pCREB or CREB antibodies (Cell Signaling Technology; 1 : 1000). After incubation with the corresponding secondary antibodies, the blots were developed by the enhanced chemiluminescence (ECL) detection method (Amersham Pharmacia Biotech, Buckinghamshire, U.K.). Quantification of the immunoreactive bands was performed by using an Image Analysis System (NIH 1.61). Care was taken to ensure that bands to be quantified were in the linear range of response. To minimize between-blot variability, we applied an aliquot of pooled ‘control' membranes to one lane of every gel, and calculated the immunolabelling of samples relative to this standard. Immunoblotting of AC-I, AC-V/VI, AC-VIII, RIIα, Cα, and Cβ was also performed after pre-absorption of these antibodies with their respective synthetic complementary peptides according to the method of Yamamoto et al. (1996). These synthetic peptides completely abolished each corresponding band (data not shown).
Electrophoretic mobility shift assay (EMSA)
Double-stranded oligonucleotide (Promega; 5′-AGAGATTGCCTGACGTCAGAGAGCTAG-3′) containing the cAMP response element (CRE) consensus sequence, as indicated in underline, was labelled with [γ-32P]ATP by use of T4 polynucleotide kinase. The assays were performed as reported previously (Kornhauser et al., 1992). In brief, nuclear extracts were incubated with 35 fmol of 32P-labelled probe for 20 min at room temperature before electrophoresis on a 4% nondenaturing polyacrylamide gel in 0.5 X TBE buffer containing 45 mM Tris-borate (pH 8.0) and 1 mM EDTA. In competition assays, a 25 X excess of unlabelled CRE or AP-1 was added to the reaction mixture 10 min prior to the addition of the labelled probe. Supershift experiments were performed by preincubating the extracts for 30 min at room temperature with anti-CREB antibody. After electrophoresis, the gels were dried and exposed to X-ray film. Radioactive bands were quantified by using a liquid scintillation counter (LSC-3500, Aloka, Tokyo, Japan).
Statistical analysis
The results were presented as means±s.e.mean. The data of biochemical variables of nefiracetam-untreated sham-operated (S), nefiracetam-treated sham-operated (SN), nefiracetam-untreated microsphere-embolized (ME), and nefiracetam-treated microsphere-embolized (MN) animals were evaluated by using two-way analysis of variance (ANOVA) followed by post-hoc Fisher's protected least significant difference (PLSD) t-test. The escape latency of the water maze task was analysed by using two-way ANOVA for repeated measures followed by post-hoc Fisher's PLSD. Differences with a probability of 5% or less were considered to be significant (P<0.05).
Results
Operation
In the present study, microspheres were injected into 112 rats. Eighty-five of the surviving rats (76%) showed cerebral ischaemia-like symptoms with a total score of 7–9 points and 10 animals (9%) showed the symptoms of cerebral ischaemia with one of less than 7 points. Seventeen rats (15%) died before all examinations were completed. Twenty-three microsphere-embolized rats and 20 sham-operated rats were used in the first series of experiments for water maze test. Among the 23 ME rats, three animals (one in the microsphere-embolized group and two in nefiracetam-treated microsphere-embolized group) were eliminated due to a failure in swimming in the visible platform test. Seven ME and seven nefiracetam-treated ME rats were used for determination of infarct size. Forty-eight microsphere injected rats and 44 sham-operated rats were used in the second series of experiments for alterations in the signal transduction. Sixty-four sham-operated rats (20 for the first series of experiments and 44 for the second series of experiments) did not die throughout the experiment.
Effects on neurological deficits
Figure 1 shows the effects of the delayed treatment with nefiracetam on neurological deficits of ME rats in the first series of experiments. The neurological deficits were gradually decreased with time after the operation. A significant difference in the neurological deficits between nefiracetam-treated and -untreated ME rats was observed. There were significant differences in the neurological deficits by groups (F[1,24]=6.9, P<0.05) and by days (F[9,216]=157.3, P<0.0001)(n=10 each). Significant interaction effects were not noted. Fisher's PLSD test revealed that the neurological deficits of ME rats on day 1 was not significantly different from those of the nefiracetam-treated ME rats (P=0.1). There were no neurological deficits in the sham-operated animals irrespective of treatment with or without nefiracetam. In the second series of experiments and in the determination of the infarct size, changes in the neurological deficits of the operated animals were similar to those observed in the first series of experiments up to day 3 after ME (data not shown).
Figure 1.
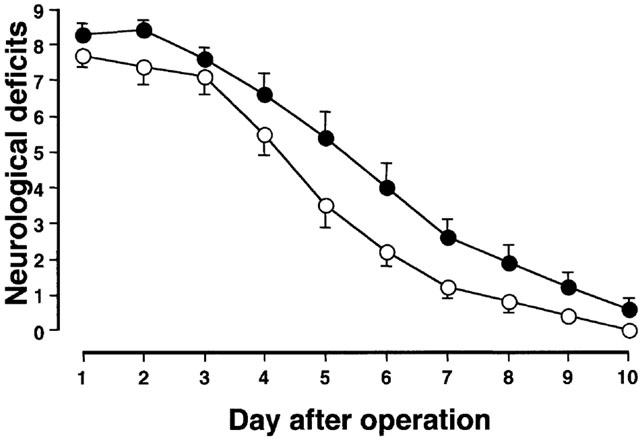
The time course of changes in neurological deficits of nefiracetam-untreated (closed circles) and nefiracetam-treated microsphere-embolized rats (open circles). The neurological deficits were scored on the basis of paucity of movement, truncal curvature, and forced circling during locomotion. The rats that had a total score of 7–9 points on day 1 after the operation were used in the present study. No neurological deficits of the nefiracetam-untreated and nefiracetam-treated sham-operated rats were observed. Each value represents the mean±s.e.mean (n=10 each).
Effects on the water maze task
In the first series of experiments, the effect of nefiracetam on learning and memory function after ME was examined. The examination on day 7 to 9 was selected on the basis of the finding that neurological deficits that might interfere with the swimming ability in the water maze task had almost disappeared by this time. At first, we determined whether nefiracetam per se would affect the spatial memory function of non-ischaemic animals. The escape latency of untreated sham-operated rats (S) was similar to that of the nefiracetam-treated sham-operated rats (SN; F[1,18]=0.604, P=0.4472, Table 1), indicating that there was no effect of nefiracetam on learning and memory function of non-ischaemic animals (n=10 each). A significant difference in the escape latency among S, ME, and MN groups was observed. There were significant differences in the escape latency by groups (F[2,27]=29.4, P<0.0001) and by days (F[8,216]=10.1, P<0.0001, n=10 each). Fisher's PLSD revealed that the escape latency of ME rats was significantly lengthened compared with that of sham-operated rats from the second trial of day 7 to the third trial of day 9 (P<0.01, Figure 2). Nefiracetam-treated ME rats showed a significant attenuation in the prolongation of the escape latency compared with ME rat at the first and third trial of day 8, and the first and third trial of day 9 (P<0.05, Figure 2), whereas the escape latency of MN group was significantly different from that of S group at the second and third trial of day 8, and the first trial of day 9 (P<0.05, Figure 2). The swimming speed (the distance that the animals swam divided by escape time) in the water maze test (hidden platform test) was similar in all groups examined (Table 2). Furthermore, the escape latency (F[3,36]=2.0, P=0.13) and swimming speed of the visible platform test (F[3,36]=0.8, P=0.50), when determined on day 10, did not differ among all of the four groups.
Table 1.
Effects of nefiracetam treatment on the escape latency of sham-operated animals in the water maze test

Figure 2.
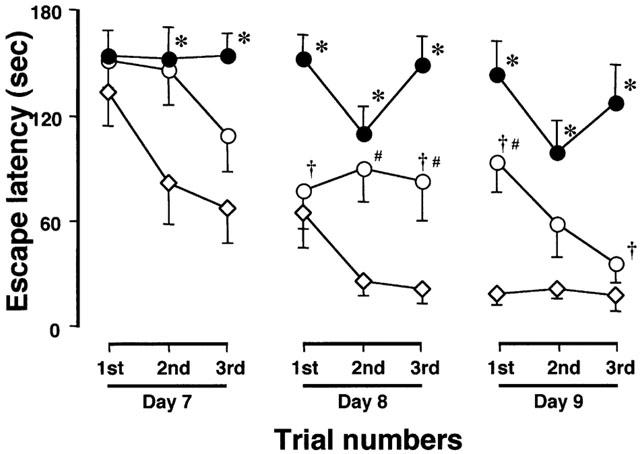
Changes in the escape latency in the water maze task of the nefiracetam-untreated (closed circles) and nefiracetam-treated (open circles) rats with ME, and nefiracetam-untreated sham-operated rats (open diamonds). Changes in the escape latency of the nefiracetam-treated sham-operated animals were quite similar to those of the corresponding untreated animals, as described in Table 1. Thus, the former changes are not shown in this figure. The escape latency was determined on day 7 to 9 after the ME. Each value represents the mean±s.e.mean of 10 animals. Two-way ANOVA of the data revealed significant differences in the escape latency between the sham-operated and ME rats (P<0.0001), and between nefiracetam-untreated and nefiracetam-treated rats with ME (P<0.0001). *Significantly different (P<0.05) from the sham-operated group when estimated by Fisher's post-hoc PLSD. †Significantly different (P<0.05) from the corresponding untreated ME group when estimated by Fisher's PLSD. #Significantly different (P<0.05) from the sham-operated group when estimated by Fisher's PLSD.
Table 2.
Swimming speed in the water maze test using a hidden platform (a) and in the visible platform test (b) of the sham-operated and microsphere-embolized animals with and without nefiracetam treatment

Effects on infarct size
The effects of delayed treatment with nefiracetam on ME-induced cerebral infarction on day 9 were examined in another series of experiments. As shown in Figure 3, there were no significant differences in the TTC-unstained areas of the brain slices at 2, 4, 6, 8, 10, 12, and 14 mm from the forebrain between the ME and nefiracetam-treated ME animals. No infarct areas were seen in the brain slices of sham-operated and nefiracetam-treated sham-operated animals.
Figure 3.
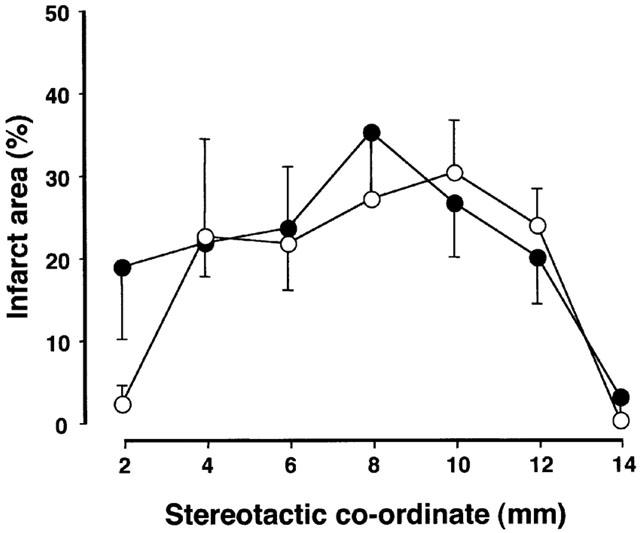
TTC-staining of the brain slices at 2, 4, 6, 8, 10, 12, and 14 mm from the forebrain of the microsphere-embolized and nefiracetam-treated microsphere embolized rats on day 10. There was no significant difference in the infarct areas between the two groups (P=0.636).
Effects on GTP-binding protein subunits
In the second series of experiments, we first examined the level of GTP-binding proteins (Gαs, Gαi1,2, and Gβ) by Western immunoblotting. There were no significant alterations in the immunoreactivity of Gαs, Gαi1,2, and Gβ in the right parietal cortex or hippocampus of S, SN, ME, and MN rats on day 3 after the operation (Figure 4A,B).
Figure 4.
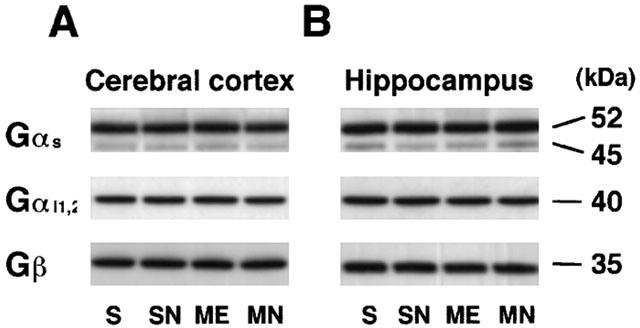
Representative Western immunoblots of GTP-binding protein subunits (Gαs, Gααi1,2, and Gβ) indicating the specific 52, 45-kDa band for Gαs, 40-kDa one for Gαi1,2, and 35-kDa one for Gβ of the right parietal cortex (A) and hippocampus (B) from the right hemisphere of the nefiracetam-untreated (ME) and nefiracetam-treated (MN) rats with ME, and from that of the nefiracetam-untreated (S) and nefiracetam-treated (SN) sham-operated rats on day 3 after the operation. There were no differences in Gαs, Gαi1,2, and Gβ protein levels among these groups.
Effects on AC activities
We examined AC activity of S, SN, ME, and MN rats on day 3 after the operation (Table 3). There were no significant changes in the basal AC activity of either the right parietal cortex or hippocampus of any group. Furthermore, there were also no significant differences in Gpp(NH)p-sensitive AC activity in the brain regions among these groups. Thus the delayed treatment with nefiracetam did not alter the basal or Gpp(NH)p-sensitive AC activities of sham-operated and ME rats. In contrast, Ca2+/CaM-sensitive AC activity in the right parietal cortex and hippocampus of the ME rats was significantly decreased. The delayed treatment with the drug partially reversed this decrease in the parietal cortex and completely in the hippocampus.
Table 3.
AC activities in the right parietal cortex and hippocampus of the sham-operated and microsphere-embolized animals with and without nefiracetam treatment on day 3 after the operation

Effects on AC subtypes
We performed immunoblotting analysis to examine the subtypes of AC (Figure 5A,B). Immunoreactivity of AC-I in the right parietal cortex and hippocampus of the ME animals was significantly decreased. The delayed treatment with nefiracetam partially reversed this decrease in both regions. No significant alterations in the immunoreactivity of AC-V/VI and AC-VIII were seen. The delayed treatment did not alter the immunoreactivity of AC-V/VI and AC-VIII in sham-operated and ME rats irrespective of treatment with or without nefiracetam.
Figure 5.
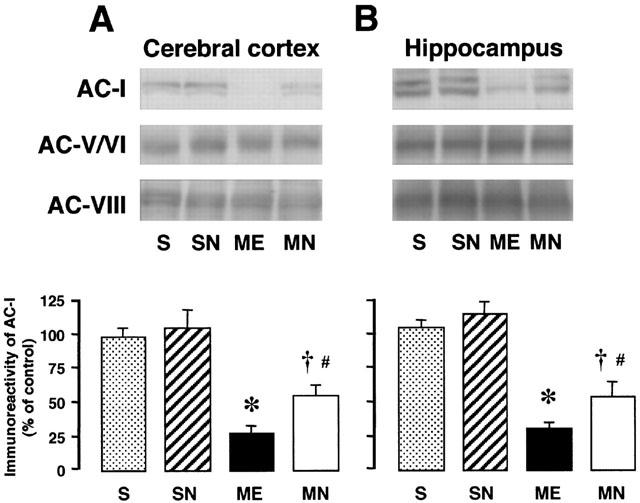
Representative Western immunoblots of AC-I, AC-V/VI, and AC-VIII in the parietal cortex (A) and hippocampus (B) from the right hemisphere of the nefiracetam-untreated (ME) and nefiracetam-treated (MN) rats with ME, and from that of the nefiracetam-untreated (S) and nefiracetam-treated sham-operated (SN) rats on day 3 after the operation (upper panel). Quantified data for the levels of AC-I protein in the right parietal cortex and hippocampus are shown in the lower panels. Each value represents the mean percentage of the control±s.e.mean of eight animals. *Significantly different from the sham-operated group (P<0.05). †Significantly different from the corresponding untreated ME group (P<0.05). #Significantly different from the nefiracetam-treated sham-operated (SN) group (P<0.05).
Effects on cAMP content
In another set of experiments, the effects of nefiracetam on the cAMP content of the right parietal cortex and hippocampus of the ME rats on day 3 after the operation were examined (Figure 6). A significant decrease in cAMP content was seen in both regions of the ME rats. The delayed treatment with nefiracetam partially reversed the decrease in the cAMP content in the parietal cortex and completely in the hippocampus. The delayed treatment did not affect the cAMP content of the sham-operated and nefiracetam-treated sham-operated rats.
Figure 6.
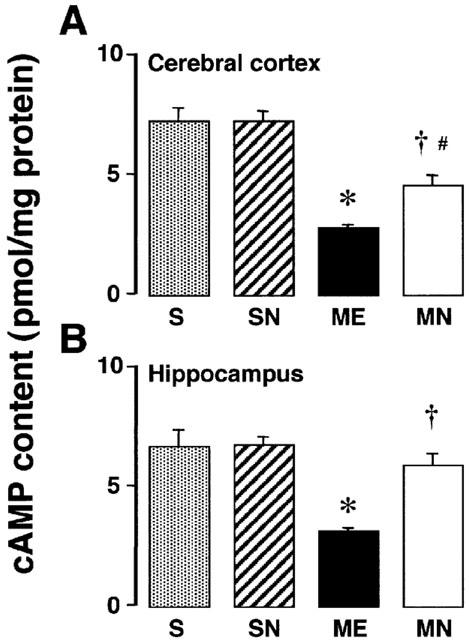
cAMP content of the parietal cortex (A) and hippocampus (B) in the right hemisphere of the nefiracetam-untreated (ME) and nefiracetam-treated (MN) rats with ME, and of the nefiracetam-untreated (S) and nefiracetam-treated (SN) sham-operated rats on day 3 after the operation. Each value represents the mean±s.e.mean of six (S and SN) or eight (ME and MN) animals. *Significantly different from the sham-operated group (P<0.05). †Significantly different from the corresponding untreated ME group (P<0.05). #Significantly different from the nefiracetam-treated sham-operated (SN) group (P<0.05).
Effects on PKA subunits in cytosolic and nuclear fractions
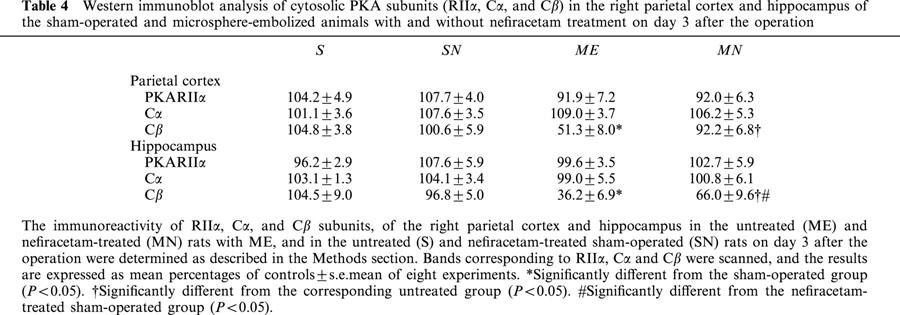
Changes in PKA subunits (RIIα, Cα, and Cβ) in the cytosol from the right parietal cortex and hippocampus of the ME and sham-operated rats were examined on day 3 after the operation (Table 4). The immunoreactivity of cytosolic Cβ in the right parietal cortex and hippocampus decreased after ME. The delayed treatment with nefiracetam completely reversed this decrease in the parietal cortex and partially in the hippocampus. No significant alterations in the immunoreactivity of cytosolic RIIα and Cα were seen in the ME and MN animals. Also there was no change in these protein contents of sham-operated rats by treatment with nefiracetam.
Table 4.
Western immunoblot analysis of cytosolic PKA subunits (RIIα, Cα, and Cβ) in the right parietal cortex and hippocampus of the sham-operated and microsphere-embolized animals with and without nefiracetam treatment on day 3 after the operation
Representative immunoreactivity of PKA Cα and Cβ in the nuclear extract from S, SN, ME, and MN rats are shown in Figure 7. The immunoreactivity of nuclear Cα and Cβ in the right parietal cortex and hippocampus decreased after ME. The delayed treatment with nefiracetam completely reversed these decreases in the parietal cortex and partially in the hippocampus, while it did not affect the nuclear PKA Cα and Cβ of sham-operated rats.
Figure 7.
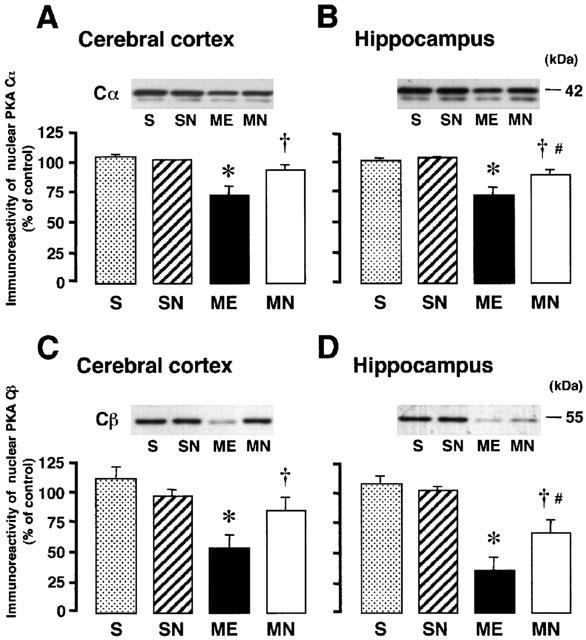
Representative Western immunoblots of nuclear PKA subunits (Cα and Cβ) in the upper panels indicate the specific 42-kDa band for Cα in the right parietal cortex (A) and hippocampus (B), and the 55-kDa band for Cβ in the parietal cortex (C) and hippocampus (D) from the right hemisphere of the nefiracetam-untreated (ME) and nefiracetam-treated (MN) rats with ME, and from that of the nefiracetam-untreated (S) and nefiracetam-treated (SN) sham-operated rats on day 3 after the operation. Quantified data for the levels of Cα and Cβ proteins in the right parietal cortex and hippocampus are shown in the lower panels. Each value represents the mean percentage of the control±s.e.mean of eight animals. *Significantly different from the sham-operated group (P<0.05). †Significantly different from the corresponding untreated ME group (P<0.05). #Significantly different from the nefiracetam-treated sham-operated (SN) group (P<0.05).
Effects on CREB in nuclear fractions
The effects of the delayed treatment with nefiracetam on the immunoreactivity of pCREB and total CREB of the right parietal cortex and hippocampus from ME and sham-operated rats on day 3 were examined by Western immunoblotting. A significant decrease in the immunoreactivity of pCREB was seen in both regions after ME. The delayed treatment with nefiracetam partially attenuated this decrease in both regions (Figure 8). Treatment with nefiracetam did not alter the immunoreactivity of pCREB of the sham-operated animal. The total CREB in both regions of the rat remained the same in all four groups.
Figure 8.
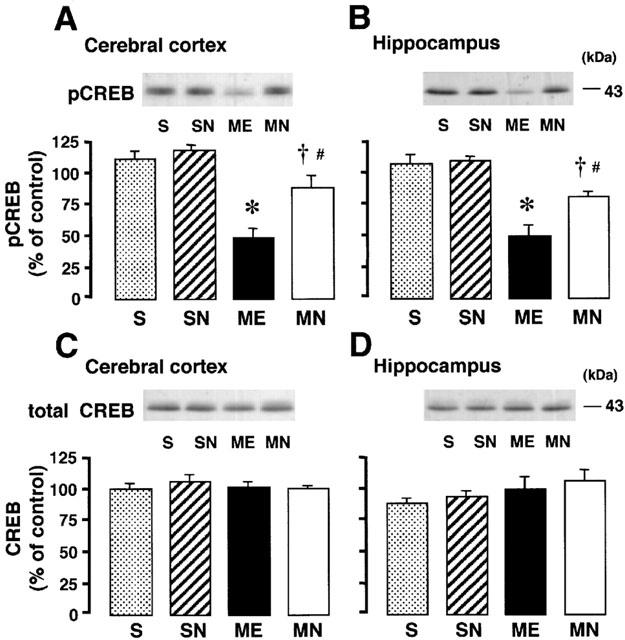
Representative Western immunoblots of pCREB and total CREB in the upper panels indicate the specific 43-kDa band for pCREB in the parietal cortex (A) and hippocampus (B), and total CREB of the parietal cortex (C) and hippocampus (D) in the right hemisphere of the nefiracetam-untreated (ME) and nefiracetam-treated (MN) rats with ME and in that of the nefiracetam-untreated (S) and nefiracetam-treated (SN) sham-operated rats on day 3 after the operation. Quantified data for the levels of pCREB protein and total CREB protein of the right parietal cortex and hippocampus are shown in the lower panels. Each value represents the mean percentage of the control±s.e.mean of eight animals. *Significantly different from the sham-operated group (P<0.05). †Significantly different from the corresponding untreated ME group (P<0.05). #Significantly different from the nefiracetam-treated sham-operated (SN) group (P<0.05).
Effects on DNA-binding activity of CREB
Figure 9A,D shows a retardation of the probe mobility caused by CREB binding to the CRE oligonucleotide in the absence of an antibody or any competitor oligonucleotide (the first lanes in Figure 9A,D). The addition of CREB antibody supershifted the entire retarded band (the second lanes in Figure 9A,D as marked by arrowheads). In addition, inclusion of excess cold CRE oligonucleotides blocked the binding of the CREB protein to the radioactive probe, whereas excess AP-1 oligonucleotide had no effect on the CREB binding (the third and fourth lanes in Figure 9A,D, as marked by arrows). These results indicate that the shift was caused by binding of CREB-related proteins. The effects of nefiracetam on CRE-binding activity of CREB of the right parietal cortex and hippocampus of ME rats on day 3 were examined (Figure 9B,C,E,F). A significant decrease in radioactivity representing CREB-DNA binding was seen in the right parietal cortex and hippocampus after ME. The delayed treatment with nefiracetam completely reversed this decrease in both regions. No significant difference in the CRE-DNA binding between sham-operated and drug-treated sham-operated rats was seen.
Figure 9.
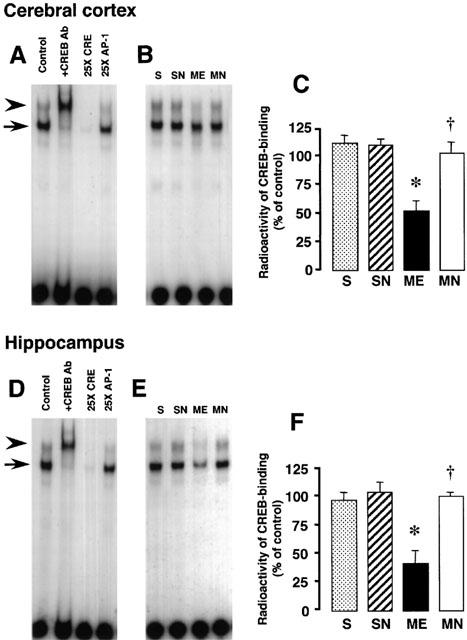
EMSA of CREB-binding activity in the right parietal cortex and hippocampus. Representative autoradiograms on the left show CREB-binding in the right parietal cortex (A) and hippocampus (D). The first lanes in Figure 9A,D (control) show a retardation caused by CREB binding to the CRE oligonucleotide in the absence of an antibody or competitor oligonucleotides. The addition of CREB antibody supershifted entirely the shifted band (the second lanes), as marked by arrowheads. In addition, the presence of excess cold CRE oligonucleotide blocked the binding of the CREB protein to the radioactive CREB band (the third lanes), whereas excess AP-1 oligonucleotide had no effect on the CREB binding (the fourth lanes), as marked by arrows. The finding indicates that the shift was caused by binding of CREB-related proteins. Representative autoradiograms in the middle indicate the specific band for CREB-CRE binding in samples from the right parietal cortex (B) and hippocampus (E) from the right hemisphere of the nefiracetam-untreated (ME) and nefiracetam-treated (MN) ME rats, and from that of the nefiracetam-untreated (S) and nefiracetam-treated (SN) sham-operated rats on day 3 after the operation. Quantified data for the level of CREB binding activity of the right parietal cortex (C) and hippocampus (F) are shown at the right. Each value represents the mean percentage of the control±s.e.mean of eight animals. *Significantly different from the sham-operated group (P<0.05). †Significantly different from the corresponding untreated ME group (P<0.05).
Discussion
In the present study, we examined the possible therapeutic effect of nefiracetam on sustained ischaemia-induced learning and memory dysfunction and a plausible mechanism underlying the effect. Cerebral ischaemia induced by middle cerebral artery occlusion (Yonemori et al., 1999) or four-vessel ligation/reperfusion in rodents (Imanishi et al., 1997) has been shown to induce learning and memory dysfunction. As far as we know, the ME-induced memory dysfunction in water maze task appears to be more severe with respect to prolongation of the escape latency than that induced by other ischaemic interventions as above, probably due to permanent embolism of cerebral arterioles caused by the microspheres. We found a partial improvement of the escape latency in the ME animals upon the delayed treatment with nefiracetam, suggesting that this agent may have a potential to improve spatial memory dysfunction induced by sustained cerebral ischaemia. The infarct sizes of the ME and drug-treated ME animal brains were not different from each other, suggesting that the improvement of memory function by nefiracetam is unlikely attributed to reduction in the infarct of the ME animal. The effective doses of nefiracetam for the cognition-enhancing effects in the present study were comparable to those of others obtained in the passive avoidance test (Nabeshima et al., 1990; 1991; Yamada et al., 1999).
Several studies and our previous data have suggested a possible connection between the cAMP/PKA/CREB signal transduction system and learning and memory function, as was described in the Introduction. Thus, we examined the effects of nefiracetam on the alterations in the cAMP/PKA/CREB signal transduction system of the right parietal cortex and hippocampus of ME rats. To determine whether the effect of nefiracetam may be associated with neurochemical alterations, the examination on day 3 after ME was selected on the basis of our findings in previous studies on cerebral blood flow, metabolic parameters and AC-I activity as described in the Methods section, and the examination on the parietal cortex and hippocampus was chosen based on data indicating that these regions are involved in learning and memory function (DiMattia & Kesner, 1988; Save et al., 1992). In the present study, there were no alterations in the immunoreactivity of Gαs, Gαi, and Gβ in the right parietal cortex and hippocampus of the ME rat, suggesting that GTP-binding proteins appear to be substantially resistant to ischaemia. AC activity was earlier shown to be regulated by neurotransmitter receptors coupled to the GTP-binding proteins (Ross & Gilman, 1980), and by intracellular Ca2+ and CaM (Wu et al., 1995). Recently, we characterized alterations in AC activity in the ME animal (Nagakura et al., 2002a). We found a decrease in the Ca2+/CaM-stimulated AC-I activity without alterations in the basal and Gpp(NH)p-stimulated AC activities and reduction in the protein level of AC-I without alterations in the protein levels of AC-VIII and AC-V/VI, as consistent with the results previously reported (Nagakura et al., 2002a). One can claim that the decrease in AC-I level might be due to a reduction in the number of neurons or an increase in the proliferation of glial cells after cerebral ischaemia. We cannot, however, simply attribute the decline in AC-I protein to ischaemia-induced neuronal loss or gliosis, since the levels of AC-V/VI and AC-VIII were not altered. These results suggest that ME results in an impairment of Ca2+/CaM-sensitive AC, particularly AC-I.
To determine pathological consequences of the impairment of Ca2+/CaM-sensitive AC-I in the sustained cerebral ischaemia, we examined the cAMP content after ME. We observed a decrease in cAMP content in the right parietal cortex and hippocampus of the ME rat. In a previous study, we demonstrated a marked reduction in tissue high-energy phosphates after microsphere embolism (Takeo et al., 1992). Therefore, the decrease in the cAMP content after ME may be, in part, attributable to a lack of the substrate for AC. Taken together, our data suggest that not only the dysfunction of Ca2+/CaM-sensitive AC-I but also the lack of the substrate ATP may contribute to the decrease in cAMP content after ME.
We found that the level of cytosolic Cβ was reduced after ME without alterations in the levels of Cα and RIIα. Although further studies will be required to determine the mechanisms and pathophysiologic consequences, the decrease in cytosolic Cβ may be, in part, due to a specific degradation of this catalytic subunit after sustained cerebral ischaemia. Since cytosolic PKA activated by cAMP undergoes dissociation of the regulatory subunit from the catalytic one with the subsequent translocation of the catalytic subunit into the nucleus (Hagiwara et al., 1993), we focused on changes in nuclear Cα and Cβ levels. We observed the reduction in the nuclear Cα and Cβ levels after ME, whereas a very small amount of RIIα was detected in the nuclear fraction (data not shown), which is consistent with the earlier observation that RII subunits did not translocate to the nucleus (Ventra et al., 1996). Our results suggest that cytosolic PKA catalytic subunits can hardly dissociate and/or be translocated to the nucleus after ME, possibly due to the decrease in the cAMP level.
The delayed treatment with nefiracetam attenuated the ME-induced reduction in AC-I level and Ca2+/CaM-stimulated AC activity. Although the mechanisms for the attenuation of these ME-induced reductions remain unclear, these effects may contribute to the attenuation of reduction in the nuclear Cα and Cβ levels as a result of restoration of the cAMP content in the ischaemic brain. Consequently, by these means nefiracetam may thus partially improve the cAMP signalling in the nucleus of ME animals. In this sense, we found a decrease in phosphorylation of CREB, which is one of the substrates for the PKA catalytic subunit in the nucleus (Hagiwara et al., 1993) and a decrease in the CRE-DNA binding of CREB after ME. The decrease in this binding was lessened by the delayed treatment with nefiracetam, and the degree of attenuation was comparable to that of the reduced level of pCREB. Recently, we reported that treatment with rolipram, a specific phosphodiesterase IV inhibitor, ameliorated ME-induced impairment of learning and memory function, and that the drug effect might be partly attributed to activation of the cyclic AMP/PKA/CREB signaling (Nagakura et al., 2002b). Although other molecular and cellular disturbances may also contribute to learning and memory deficits after ME (Takagi et al., 1997; 2002), our present results suggest that ME may impair CREB-mediated transcription of new proteins for learning and memory and that the delayed treatment with nefiracetam can partially improve this process, which possibly leads to amelioration of spatial memory function of the ME animal.
In summary, we demonstrated a potential for the anti-amnesic effects of the delayed treatment with nefiracetam in ME rats with defective spatial memory. The delayed treatment with nefiracetam partially attenuated the ME-induced decreases in Ca2+/CaM-sensitive AC-I activity, cAMP content, levels of nuclear PKA Cα and Cβ and pCREB proteins, and CRE-binding activity. Our study thus demonstrated substantial effects of nefiracetam on learning and memory dysfunction and on impairment of the cAMP/PKA/CREB signal transduction system that occurred in sustained cerebral ischaemia.
Acknowledgments
We gratefully acknowledge Dr Shigeo Watabe, Daiichi Pharmaceutical Co., Ltd., Tokyo, Japan, for a kind gift of nefiracetam (DM-9384), for physicochemical and biochemical information about nefiracetam, and for helpful advice.
Abbreviations
- AC
adenylyl cyclase
- ANOVA
analysis of variance
- CaM
calmodulin
- cAMP
cyclic AMP
- CRE
cAMP response element
- CREB
cAMP-responsive element binding protein
- ECL
enhanced chemiluminescence
- EDTA
ethylenediamine-N,N,N′,N′-tetra-acetic acid
- EGTA
ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetra-acetic acid
- EIA
enzyme immunoassay
- EMSA
electrophoretic mobility shift assay
- GTP-binding protein
guanine nucleotide-binding protein
- Gpp(NH)p
5′-guanylyl imidodiphosphate
- HEPES
2-[4-2(-hydroxyethyl)-1-pioperazinyl]ethanesulphonic acid
- HRP
horseradish peroxidase
- LTP
long-term potentiation
- ME rat
microsphere-embolized rat
- PKA
protein kinase A
- PVDF
polyvinylidene difluoride
References
- BERNABEU R., BEVILAQUA L., ARDENGHI P., BROMBERG E., SCHMITZ P., BIANCHIN M., IZQUIERDO I., MEDINA J.H. Improvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7041–7046. doi: 10.1073/pnas.94.13.7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOURTCHULADZE R., FRENGUELLI B., BLENDY J., CIOFFI D., SCHUTZ G., SILVA A.J. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CALI J.J., ZWAAGSTRA J.C., MONS N., COOPER D.M.F., KRUPINSKI J. Type VIII adenylyl cyclase; a Ca2+/calmodulin-stimulated enzyme expressed in discrete regions of rat brain. J. Biol. Chem. 1994;269:12190–12195. [PubMed] [Google Scholar]
- CHUTE D.L., VILLIGER J.W., KIRTON N.F. Testing cyclic AMP mediation of memory: reversal of alpha-methyl-p-tyrosine-induced amnesia. Psychopharmacology. 1981;74:129–131. doi: 10.1007/BF00432678. [DOI] [PubMed] [Google Scholar]
- DIMATTIA B.C., KENSER R.P. Spatial cognitive maps; differential role of parietal cortex and hippocampal formation. Behav. Neurosci. 1988;102:471–480. doi: 10.1037//0735-7044.102.4.471. [DOI] [PubMed] [Google Scholar]
- DOYLE E., REGAN C.M., SHIOTANI T. Nefiracetam (DM-9384) preserves hippocampal neural cell adhesion molecule-mediated memory consolidation processes during scopolamine-disruption of passive avoidance training in the rat. J. Neurochem. 1993;61:266–272. doi: 10.1111/j.1471-4159.1993.tb03564.x. [DOI] [PubMed] [Google Scholar]
- GUILLOU J.L., MICHEAU J., JAFFARD R. The opposite effects of cysteamine on the acquisition of two different tasks in mice are associated with bidirectional testing-induced changes in hippocampal adenylyl cyclase activity. Behav. Neurosci. 1998;112:900–908. doi: 10.1037//0735-7044.112.4.900. [DOI] [PubMed] [Google Scholar]
- GUILLOU J.L., ROSE G.M., COOPER D.M. Differential activation of adenylyl cyclases by spatial and procedural learning. J. Neurosci. 1999;19:6183–6190. doi: 10.1523/JNEUROSCI.19-14-06183.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUZOWSKI J.F., MCGAUGH J.L. Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impaired consolidation of memory for water maze training. Proc. Natl. Acad. Sci. U.S.A. 1997;94:2693–2698. doi: 10.1073/pnas.94.6.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HABENER J.F. Cyclic AMP response element binding proteins: a cornucopia of transcription factors. Mol. Endocrinol. 1990;4:1087–1094. doi: 10.1210/mend-4-8-1087. [DOI] [PubMed] [Google Scholar]
- HAGIWARA M., BRINDLE P., HAROOTUNIAN A., ARMSTRONG R., RIVIER J., VALE W., TSIEN R., MONTMINY M.R. Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol. Cell Biol. 1993;13:4852–4859. doi: 10.1128/mcb.13.8.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IMANISHI T., SAWA A., ICHIMARU Y., MIYASHIRO M., KATO S., YAMAMOTO T., UEKI S. Ameliorating effects of rolipram on experimentally induced impairments of learning & memory in rodents. Eur. J. Pharmacol. 1997;321:273–278. doi: 10.1016/s0014-2999(96)00969-7. [DOI] [PubMed] [Google Scholar]
- IYENGAR R. Molecular and functional diversity of mammalian Gs-stimulated adenylyl cyclases. FASEB J. 1993;7:768–775. doi: 10.1096/fasebj.7.9.8330684. [DOI] [PubMed] [Google Scholar]
- JONES D.J., MEDINA M.A., ROSS D.H., STAVINOHA W.B. Rate of inactivation of adenylyl cyclase and phosphodiesterase: Determinants of brain cyclic AMP. Life Sci. 1974;14:1577–1585. doi: 10.1016/0024-3205(74)90168-4. [DOI] [PubMed] [Google Scholar]
- KIRINO T. Delayed neuronal death in the gerbil hippocampus following ischaemia. Brain Res. 1982;39:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- KORNHAUSER J.M., NELSON D.E., MAYO K.E., TAKAHASHI J.S. Regulation of jun-B messenger RNA and AP-1 activity by light and a circadian clock. Science. 1992;255:1581–1584. doi: 10.1126/science.1549784. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- LYDEN P.D., ZIVIN J.A., CHABOLLA D.R., JACOBS M.A., GAGE F.H. Quantitative effects of cerebral infarction on spatial learning in rats. Exp. Neurol. 1992;116:122–132. doi: 10.1016/0014-4886(92)90160-r. [DOI] [PubMed] [Google Scholar]
- MIYAKE K., TAKEO S., KAJIHARA H. Sustained decrease in brain regional blood flow after microsphere embolism in rats. Stroke. 1993;24:415–420. doi: 10.1161/01.str.24.3.415. [DOI] [PubMed] [Google Scholar]
- MONS N., COOPER D.M.F. Adenylate cyclases: critical foci in neuronal signaling. Trends Neurosci. 1995;18:536–542. doi: 10.1016/0166-2236(95)98375-9. [DOI] [PubMed] [Google Scholar]
- MORRIS R.G.M. Spatial localization does not require the presence of local cues. Learn. Motiv. 1981;12:239–260. [Google Scholar]
- NABESHIMA T., TOHYAMA K., KAMEYAMA T. Effects of DM-9384, a pyrrolidone derivative, on alcohol- and chlordiazepoxide-induced amnesia in mice. Pharmacol. Biochem. Behav. 1990;36:233–236. doi: 10.1016/0091-3057(90)90396-y. [DOI] [PubMed] [Google Scholar]
- NABESHIMA T., TOHYAMA K., MURASE K., ISHIHARA S., KAMEYAMA T., YAMASAKI T., HATANAKA S., KOJIMA H., SAKURAI T., TAKASU Y., SHIOTANI T. Effects of DM-9384, a cyclic derivative, on alcohol- and chlordiazepoxide-induced amnesia in mice. J. Pharmacol. Exp. Ther. 1991;257:271–275. [PubMed] [Google Scholar]
- NAGAKURA A., MIYAKE-TAKAGI K., TAKAGI N., FUKUI M., TAKEO S. Improvement of adenylyl cyclase and of spatial memory function after microsphere embolism in rats. J. Neurosci. Res. 2002a;68:363–372. doi: 10.1002/jnr.10238. [DOI] [PubMed] [Google Scholar]
- NAGAKURA A., NIIMURA M., TAKEO S. Effects of a phosphodiesterase IV inhibitor rolipram on microsphere embolism-induced defects in memory function and cerebral cyclic AMP signal transduction system in rats. Br. J. Pharmacol. 2002b;135:1783–1793. doi: 10.1038/sj.bjp.0704629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARITOMI H. Experimental basis of multi-infarct dementia: memory impairments in rodent models of ischemia. Alzheimer Dis. Assoc. Disord. 1991;5:103–111. doi: 10.1097/00002093-199100520-00007. [DOI] [PubMed] [Google Scholar]
- NISHIZAKI T., MATSUOKA T., NOMURA T., MATSUYAMA S., WATABE S., SHIOTANI T., YOSHII M. A long-term-potentiation-like facilitation of hippocampal synaptic transmission induced by the nootropic nefiracetam. Brain Res. 1999;826:281–288. doi: 10.1016/s0006-8993(99)01312-8. [DOI] [PubMed] [Google Scholar]
- PARNETTI L., SENIN U., MECOCCI P. Cognitive enhancement therapy for Alzheimer's disease. The way forward. Drugs. 1997;53:752–768. doi: 10.2165/00003495-199753050-00003. [DOI] [PubMed] [Google Scholar]
- ROSS E.M., GILMAN A.G. Biochemical properties of hormone-sensitive adenylate cyclase. Annu. Rev. Biochem. 1980;49:533–564. doi: 10.1146/annurev.bi.49.070180.002533. [DOI] [PubMed] [Google Scholar]
- SALOMON Y., LONDOS C., RODBELL M. A highly sensitive adenylate cyclase assay. Anal. Biochem. 1974;58:541–548. doi: 10.1016/0003-2697(74)90222-x. [DOI] [PubMed] [Google Scholar]
- SAVE E., POUCET B., FOREMANN, BUHOT M.C. Object exploration and reactions to spatial and nonspatial changes in hooded rats following damage to parietal cortex or hippocampal formation. Behav. Neurosci. 1992;106:447–456. [PubMed] [Google Scholar]
- SCHREIBER E., MATTHIAS P., MULLER M.M., SCHAFFNER W. Rapid detection of octamer binding proteins with ‘mini-extracts', prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH M.L., AUER R.N., SIESJO B.K. The density and distribution of ischemic brain injury in the rat following 2–10 min of forebrain ischemia. Acta. Neuropathol. 1984;64:319–332. doi: 10.1007/BF00690397. [DOI] [PubMed] [Google Scholar]
- TAGUCHI T., MIYAKE K., TANONAKA K., OKADA M., TAKAGI N., FUJIMORI K., TAKEO S. Sustained changes in acetylcholine and amino acid contents of brain regions following microsphere embolism in rats. Jpn. J. Pharmacol. 1993;62:269–278. doi: 10.1254/jjp.62.269. [DOI] [PubMed] [Google Scholar]
- TAKAGI N., MIYAKE K., TAGUCHI T., TAMADA H., TAKAGI K., SUGITA N., TAKEO S. Failure in learning task and loss of cortical cholinergic fibres in microsphere-embolized rats. Exp. Brain Res. 1997;114:279–287. doi: 10.1007/pl00005636. [DOI] [PubMed] [Google Scholar]
- TAKAGI N., MIYAKE-TAKAGI K., TAKAGI K., TAMURA H., TAKEO S. Altered extracellular signal-regulated kinase signal transduction by the muscarinic acetylcholine and metabotropic glutamate receptors after cerebral ischemia. J. Biol. Chem. 2002;277:6382–6390. doi: 10.1074/jbc.M108081200. [DOI] [PubMed] [Google Scholar]
- TAKEO S., TAGUCHI T., TANONAKA K., MIYAKE K., HORIGUCHI T., TAKAGI N., FUJIMORI K. Sustained damage to energy metabolism of brain regions after microsphere embolism in rats. Stroke. 1992;23:62–68. doi: 10.1161/01.str.23.1.62. [DOI] [PubMed] [Google Scholar]
- VENTRA C., PORCELLINI A., FELICIELLO A., GALLO A., PAOLILLO M., MELE E., AVVEDIMENTO V.E., SCHETTINI G. The differential response of protein kinase A to cyclic AMP in discrete brain areas correlates with the abundance of regulatory subunit II. J. Neurochem. 1996;66:1752–1761. doi: 10.1046/j.1471-4159.1996.66041752.x. [DOI] [PubMed] [Google Scholar]
- WU Z.L., THOMAS S.A., VILLACRES E.C., XIA Z., SIMMONS M.L., CHAVKIN C., PALMITER R.D., STORM D.R. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc. Natl. Acad. Sci. U.S.A. 1995;92:220–224. doi: 10.1073/pnas.92.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIA Z., REFSDAL C.D., MERCHANT K.M., DORSA D.M., STORM D.R. Distribution of mRNA for the calmodulin-sensitive adenylate cyclase in rat brain: Expression in areas associated with learning and memory. Neuron. 1991;6:431–443. doi: 10.1016/0896-6273(91)90251-t. [DOI] [PubMed] [Google Scholar]
- YAMADA K., TANAKA T., MAMIYA T., SHIOTANI T., KAMEYAMA T., NABESHIMA T. Improvement by nefiracetam of β-amyloid-(1-42)-induced learning and memory impairments in rats. Br. J. Pharmacol. 1999;126:235–244. doi: 10.1038/sj.bjp.0702309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAMOTO M., OZAWA H., SAITO T., FRÖHLICH L., RIEDERER P., TAKAHATA N. Reduced immunoreactivity of adenylyl cyclase in dementia of the Alzheimer type. NeuroReport. 1996;7:2965–2970. doi: 10.1097/00001756-199611250-00033. [DOI] [PubMed] [Google Scholar]
- YOSHII M., WATABE S., SAKURAI T., SHOTANI T. Cellular mechanisms underlying cognition-enhancing actions of nefiracetam (DM-9384) Behav. Brain Res. 1997;83:185–188. doi: 10.1016/s0166-4328(97)86066-4. [DOI] [PubMed] [Google Scholar]
- YOSHII M., WATABE S., MURASHIMA Y., NUKADA T., SHIOTANI T., NISHIZAKI T. Psychogeriatrics. 2001;1:39–49. [Google Scholar]
- YONEMORI F., YAMAGUCHI T., YAMADA H., TAMURA A. Spatial cognitive performance after chronic focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 1999;19:483–494. doi: 10.1097/00004647-199905000-00002. [DOI] [PubMed] [Google Scholar]
- ZHAO W.Q., POLYA G.M., WANG B.H., GIBBS M.E., SEDMAN G.L., NG K.T. Inhibitors of cAMP-dependent protein kinase impair long-term memory formation in day-old chicks. Neurobiol. Learn Mem. 1995;64:106–118. doi: 10.1006/nlme.1995.1049. [DOI] [PubMed] [Google Scholar]