

Abstract
Group III metabotropic glutamate receptors (mGluRs) of the subtype 4a are localized within presynaptic active zones of cerebellar parallel fibre (PF)-Purkinje cell (PC) synapses. In order to investigate the conditions necessary for group III mGluR autoreceptor-activation by synaptically released glutamate, we characterized the effects of selective agonists and antagonists on excitatory postsynaptic currents (EPSCs) evoked by several distinct PF stimulation patterns.
The group III mGluR-selective agonist L-AP4 depressed evoked EPSCs at PF-PC synapses in rat brain slices with an EC50 of 2.4 μM and maximum inhibition of 80%. This L-AP4-induced depression was antagonized by the group III mGluR-selective antagonist MSOP with an estimated equilibrium dissaciation constant of 12.5 μM.
Paired-pulse or four-pulse PF stimulations did not activate presynaptic group III mGluRs as revealed by the lack of effect of 1 mM MSOP on relative test EPSC amplitudes with latencies of 250–500 ms. The potentiation of a test EPSC evoked 200–500 ms after a short tetanic burst (100 Hz for 60 ms), was also unchanged in the presence of MSOP.
Endogenous autoreceptor-activation was revealed only during prolonged stimulation trains (10 Hz for 4.4 s), where, in the presence of 1 mM MSOP, the EPSC amplitudes were enhanced by 15%.
These observations support an autoreceptor function of group III mGluRs and a role in short-term synaptic plasticity at PF synapses. However, the low to moderate activation levels observed, despite the close spatial relation with glutamate release sites, suggests that additional mechanisms regulate receptor activation.
Keywords: Metabotropic glutamate receptor, autoreceptor, presynaptic inhibition, Purkinje cell, cerebellum, brain slice, L-AP4, MSOP
Introduction
To date, eight G-protein coupled metabotropic glutamate receptors (mGluRs) have been cloned (de blasi et al., 2001; Schoepp, 2001). These have been subdivided into group I (mGluR1 and 5), group II (mGluR2 and 3) and group III (mGluR4, 6, 7 and 8) on the basis of sequence similarity, transduction properties and agonist rank order of potency. Amongst the group III mGluRs, alternative splicing generates at least two isoforms, a and b, for each of mGluR4, 7, 8 (Corti et al., 1998; Thomsen et al., 1997). Group III mGluRs are expressed presynaptically in many brain regions and pharmacological activation by agonists generally leads to inhibition of transmitter release (Anwyl, 1999). Thus, they possibly serve a role as inhibitory autoreceptors similar to presynaptic group II mGluRs (Schoepp, 2001). The physiological conditions required for their activation are not well investigated. These might differ significantly from group II mGluRs due to their predominant localization within presynaptic active zones rather than in peri- or extrasynaptic membrane regions (Corti et al., 2002; Kinoshita et al., 1996; Mateos et al., 1998; 1999). Reports demonstrating group III mGluR activation by synaptically released glutamate are rare and generally reveal low levels of receptor recruitment (Chen et al., 2002; Semyanov & Kullmann, 2000; von gersdorff et al., 1997). The physiological significance of group III mGluR-mediated modulation of synaptic strength at excitatory terminals during normal brain function is thus still unresolved.
We have utilized exogenous agonists, antagonists and glutamate uptake blockers and have manipulated the stimulation protocol to alter transmitter release, in order to investigate the conditions under which endogenous activation of presynaptic group III mGluRs might occur. Synapses of cerebellar parallel fibres (PFs) onto Purkinje cells (PCs) offer important advantages for this study. There are no recurrent excitatory connections within the cerebellar cortex which might confound the interpretation of results (reviewed by Llinas & Walton, 1998). Also, by taking advantage of the anatomy of PFs, it is possible to measure the excitability of afferent fibres. More importantly, PF endings express group III mGluRs (mGluR4a) in very high density (Corti et al., 2002; Kinoshita et al., 1996; Mateos et al., 1998; 1999) and their subcellular localization is well established. In addition, group II mGluRs do not contribute to the modulation of transmission at this synapse (Neale et al., 2001). The group III specific agonist L-AP4 has been shown to depress PF synaptic strength at PCs by 50–70% (Conquet et al., 1994; Neale et al., 2001) via inhibition of presynaptic Ca2+ influx (Daniel & Crepel, 2001). Mutant mice, deficient in mGluR4, exhibited an impairment in short-term synaptic plasticity at PF to PC synapses but this was not observed in rat cerebellar cortex on exposure to a group III selective antagonist (Pekhletski et al., 1996). Furthermore, the difference in short term plasticity in mGluR4-/- mice suggested a tonic, stimulation-independent receptor activity in wild-type mice, indicating the need for further investigation of the role of group III mGluRs at this synapse.
Here, we demonstrate activation of group III mGluRs by synaptically released glutamate during long trains of afferent stimulation (4.5 s, 10 Hz) amounting to a 15% reduction of synaptic strength. Paired-pulse and tetanic stimulations (60 ms, 100 Hz) were ineffective suggesting that group III mGluRs are not activated during single synaptic events or after short bursts of high-frequency activity.
Thus, despite the considerable potential of group III mGluRs to depress PF to PC transmission when activated with exogenous agonists, and their close association with glutamate release sites, the observed inhibition by synaptically released glutamate appears limited, perhaps indicating the existence of as yet uncharacterized regulatory mechanisms.
Methods
Slice preparation
Wistar rats, 2–3 weeks postnatal, were deeply anaesthetized with halothane and decapitated. The brain was rapidly removed and placed in ice-cold (1°C) oxygenated (95% O2; 5% CO2) artificial cerebrospinal fluid (aCSF) containing (in mM) NaCl 124, KCl 2.5, KH2PO4 1.25, MgSO4 2, CaCl2 2.5, NaHCO3 26, glucose 15. During preparation, aCSF was supplemented with 4 mM MgCl2. A tissue block containing the cerebellar vermis was glued to the tray of a vibrating tissue slicer (Vibratome 1000; Ted Pella Inc.) and submerged in ice-cold aCSF. Transversal slices of 350 μM thickness were sectioned and incubated for 1 h in 35°C oxygenated aCSF and thereafter for up to 8 h at room temperature.
Electrophysiology and data analysis
Patch electrodes (2–4 MΩ) were pulled from borosilicate glass capillaries (1.5 mm o.d., 1.17 mm i.d.; Harvard Apparatus, Edenbridge, U.K.) using the DMZ-Universal puller (Zeitz-Instrumente GmbH, München, Germany). The pipette solution contained (in mM) K-D-gluconate 145, CaCl2 0.1, MgCl2 2, N-(2-hydroxyethyl) piperazine-N′-(ethanesulfonic acid) (HEPES) 10, ethyleneglycol-bis(β-aminoethyl ether)-N,N,N′N′-tetraacetic acid (EGTA) 0.75, Mg-ATP 2, Na-GTP 0.3 and QX314 5; pH 7.30 adjusted with KOH, 290 mOsm kg−1. In all experiments, aCSF was supplemented with 10 μM D-AP5 and 50 μM picrotoxin in order to block NMDA- and GABAA-receptors, respectively. Slices were placed into the recording chamber (∼1 ml volume) and continuously perfused with oxygenated aCSF at a rate of 1.5 ml min−1.
Whole cell voltage clamp recordings from Purkinje cell somata were performed at room temperature with the Axoclamp 1D amplifier (Axon Instruments, Foster City, CA, U.S.A.) and low-pass Bessel filter set at 5 kHz (−3 dB). The recordings were digitized at 10 kHz with the DigiData 1200 interface and Clampex 8.0 software (both Axon Instruments). Only experiments with uncompensated series resistances <20 MΩ and <30% change during the course of an experiment were used for data analysis. Parallel fibres of the cerebellar cortex were stimulated through bipolar tungsten electrodes (25 μM tips) using 100 μs current steps of 20–500 μA. Parallel fibre volleys and field excitatory postsynaptic potentials (fEPSPs) of Purkinje cells were recorded extracellularly with glass micropipettes (1–3 MΩ) containing 2 M NaCl, low-pass filtered with 2.4 kHz. They were placed into the lower third of the molecular layer in transverse slices. Amplitudes were determined relative to the pre-stimulation baseline.
Data were analysed off-line using Clampfit 8.0 (Axon Instruments) and MiniAnalysis software (Synaptosoft, Leonia, NJ, U.S.A.). Data are expressed as mean±s.e.m. unless otherwise indicated. Dose-response relationships were investigated by washing in increasing agonist concentrations, followed by recovery during wash-out. The data points were fitted using the Hill equation:
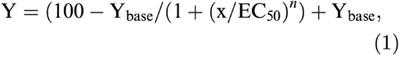 |
where Ybase is the maximum effect and n is the slope factor. The Gaddum equation was used to estimate the equilibrium dissociation constant of the antagonist MSOP, assuming a slope of 1 in the Schild plot:
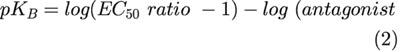 |
The glutamate response in the presence of 1 mM MSOP was estimated using:
where A is the glutamate concentration, B the MSOP concentration, KA,B are the corresponding dissociation constants. Recordings of mEPSCs were low-pass filtered at 1 kHz and a detection threshold of five times the baseline noise (Irms 1.7–2.2 pA) was applied. In the experiments using tetanic stimulation, the aCSF was supplemented with CPCCOEt (50 μM) to inhibit mGluR1. In the experiments using stimulation trains, the aCSF was supplemented with CGP54626 (80 nM) and CPCCOEt (50 μM) to prevent GABABR and mGluR1 activation. The first EPSC amplitudes in trains showed a trend to decrease during the course of an experiment which enhanced the amount of facilitation within the trains. The analysis has thus been limited to experiments where this decrease was <35%. The time of maximal synaptic strength within trains (‘Peak') was determined as the maximal running average of five consecutive EPSCs. In the experiments with direct application of glutamate, aCSF was supplemented with 50 μM D-AP5, 50 μM CPCCOEt and 80 nM CPG54626 in order to block NMDA, mGluR1 and GABAB receptors, respectively. The AMPA receptor antagonist NBQX was used at 10 μM.
Drugs
The mGluR agonist L-AP4 (L-(+)-2-amino-4-phosphonobutyric acid), the mGluR antagonists MSOP ((RS)-α-methylserine-O-phosphate), CPCCOEt (7-(hydroxyimino)cyclopropa(b)chromen-1a-carboxylate ethyl ester), the non-transportable glutamate uptake blocker D,L-TBOA (DL-threo-β-benzyloxyaspartic acid), the GABAB antagonist CGP-54626 (S-(R*, R*)) - (3) - ((1- (3,4 - dichlorphenyl)ethyl)amino)-2-hydroxypropyl)(cyclohexylmethyl)phosphinic acid hydrochloride), the AMPA receptor antagonist NBQX (2,3-dioxo-6-nitro-1,2,3,4 - tetrahydrobenzo(f)quinoxaline - 7 - sulfonamide), the NMDA receptor antagonist D-AP5 (D-(-)-2-amino-5-phosphonopentanoic acid) were obtained from Tocris Cookson (Bristol, U.K.). TTX, tetrodotoxin, from Fugu fish organs was obtained from Latoxan (Rosans, France).
Statistics
Wilcoxon signed rank tests were carried out using GraphPad InStat v2 (San Diego, CA, U.S.A.) and two-way ANOVA was carried out using the VassarStats web site (http://faculty.vassar.edu/lowry/VassarStats.html) by Prof Richard Lowry (Vassar College, Poughkeepsie, NY, U.S.A.) and StatView v5 (SAS Institute Inc., Cary, U.S.A.).
Results
Excitatory postsynaptic currents (EPSCs), evoked by stimulation of parallel fibres (PFs) in the molecular layer of the cerebellar cortex, were recorded from Purkinje cells (PCs), at −65 mV holding potential using the whole-cell configuration of the patch-clamp technique.
Pharmacology of group III mGluR-mediated suppression of synaptic transmission
L-AP4 reversibly suppressed evoked synaptic transmission (0.05 Hz) between PFs and PCs in a concentration-dependent manner. The half-maximal effective concentration (EC50) of L-AP4 was 2.4 μM (mean pEC50=5.62±0.03), the maximal suppression was 81.2±1.6% and the Hill coefficient 1.3±0.1 (n=9) (Figure 1a). The 90-10% current decay time of EPSCs was not affected by L-AP4 (predrug: 20.1±2.0 ms; 100–300 μM L-AP4: 18.6±1.7 ms; P=0.3, Wilcoxon signed rank test on paired values; n=6). L-AP4 enhanced paired-pulse facilitation (PPF; latency of 80 ms) of EPSCs to maximally 152±3% of control (determined by the fit to population means; n=3-5 per data point) (Figure 1b). Enhanced facilitation indicates that a reduction in presynaptic release probability is involved in the L-AP4 effect but it does not exclude the contribution of other mechanisms (Dittman & Regehr, 1996). If other, possibly non-synaptic mechanisms contribute to the L-AP4 effect, they may not be reproducible with receptor activation by synaptically released glutamate, thereby complicating experimental interpretation. One such possibility is changes in impulse initiation in cerebellar granule cells and/or conduction properties of PFs. L-AP4 has been shown to activate 4-AP sensitive presynaptic K+-channels in PFs (Daniel & Crepel, 2001), a mechanism which may cause action-potential propagation failures (Debanne et al., 1997). PF properties were examined by measuring fibre volleys. There was no significant change in the amplitude or the time course of PF volleys (volley amplitude in 100 μM L-AP4: 94.7±3.5% of control, n=9) while, at the same time, field excitatory postsynaptic potentials (field-epsps) were reduced (Figure 2a,b). Other possible sites of action for L-AP4 include the postsynaptic ion channels. There is evidence that group III mGluRs are expressed postsynaptically as well as presynaptically in parts of the basal ganglia (Bradley et al., 1999). We recorded AMPA receptor-mediated miniature EPSCs (mEPSCs) in the presence of 0.5 μM TTX. L-AP4 did not affect the amplitudes or the frequency of occurrence of mEPSCs (mean amplitude predrug: 19.1±2.0 pA; in 100 μM L-AP4: 19.2±2.1 pA; mean event interval predrug: 1856±454 ms; in 100 μM L-AP4: 2025±533 ms; n=6; P>0.1 for amplitudes and intervals, Wilcoxon test on paired values) (Figure 3a,b).
Figure 1.
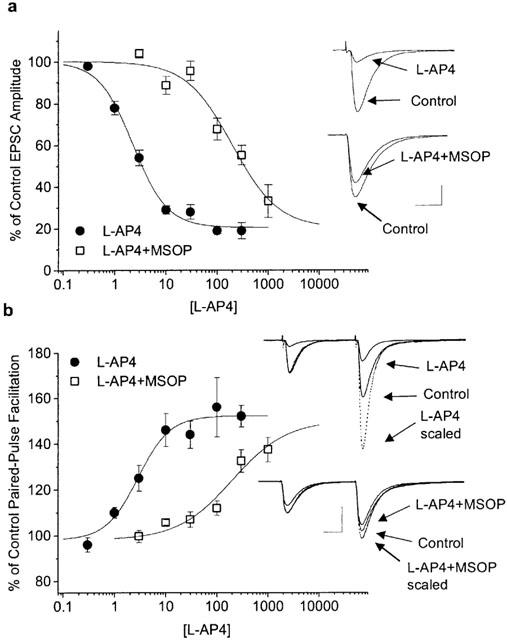
Pharmacological properties of synaptic transmission at PF-PC synapses. (a) Concentration-dependent inhibition of EPSC amplitude by L-AP4, constructed from nine cells (n=3-5 per conc.). The EC50 value was 2.4 μM. In the presence of the group III mGluR selective competitive antagonist MSOP (1 mM) the L-AP4 EC50 was shifted to 192 μM (data from eight cells; n⩾4 per conc.). The upper inset shows the inhibition induced by 10 μM L-AP4 and the inset below, the reduction in L-AP4 inhibition (10 μM) in the presence of MSOP in a different cell. Scale bars: 200 pA, 20 ms. (b) Concentration-response relationship between L-AP4 and the percentage increase in paired-pulse facilitation (PPF), inter-stimulus interval=80 ms. Control PPF was 1.92±0.08 (16 cells). The EC50 of the L-AP4-induced increase in PPF was 2.9 μM and in the presence of 1 mM MSOP it was 217 μM (values derived from fit to group data). The increased facilitation concomitant to L-AP4 inhibition is exemplified in the upper inset. The paired-pulse response in 10 μM L-AP4 is shown with true amplitudes and with amplitudes scaled to match the 1st EPSC in control in order to show the increased facilitation. The inset below shows corresponding traces from a different cell in the presence of 1 mM MSOP. Scale bars: 500 pA, 20 ms.
Figure 2.
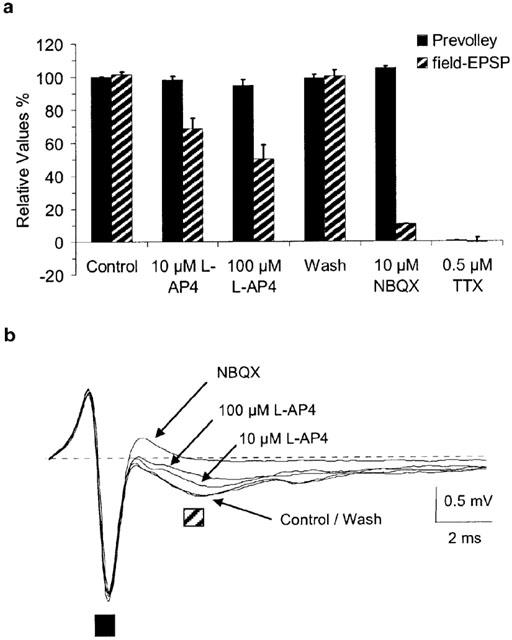
Axonal properties of PFs are not affected by L-AP4. Parallel fibre prevolleys and field excitatory postsynaptic potentials of Purkinje cells were recorded extracellularly with glass micropipettes placed into the lower ⅓ of the molecular layer in transverse slices. PFs were stimulated with 0.05 Hz. (a) Group data for nine experiments showing that the amplitude and the time-course of PF prevolleys are not affected by L-AP4 while PC field EPSP amplitudes are reduced. (b) Superimposed extracellular recordings from one experiment. Each trace represents an average of three sweeps. The pre-stimulation baseline is shown as dashed line. The prevolley is marked with the solid black square, the field EPSP is marked with the striped square. NBQX 10 μM.
Figure 3.
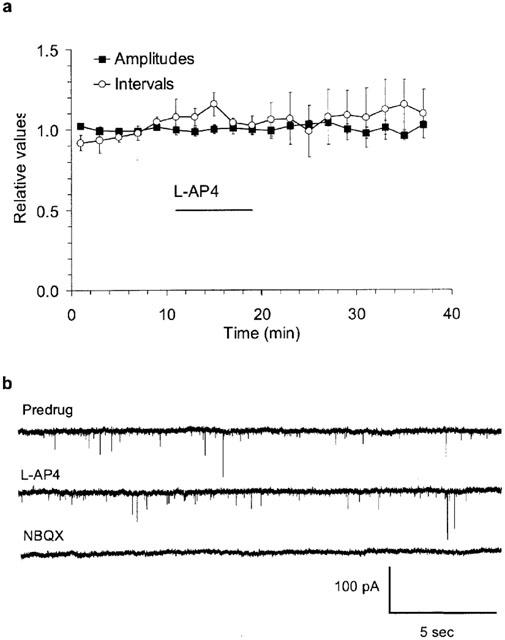
Miniature EPSCs of PCs are not affected by L-AP4. Spontaneously occurring AMPA receptor-mediated miniature EPSCs were recorded in the presence of 0.5 μM TTX. (a) Means±s.e.m from six experiments showing that neither the amplitude nor the frequency of occurrence of miniature EPSCs were affected by 100 μM L-AP4. Mean amplitude predrug was 19±2 pA and mean event interval predrug was 1.9±0.5 s. (b) Example traces from one experiment on miniature EPSCs, low-pass filtered 1 kHz. L-AP4 100 μM, NBQX 10 μM.
The Group III mGluR selective antagonist MSOP strongly reduced the L-AP4-mediated inhibition. In the presence of 1.0 mM MSOP, the potency of L-AP4 was reduced from an EC50 of 2.4 to 192.0 μM (mean pEC50=3.72±0.06; n=8) (Figure 1a). There was no change of the maximal L-AP4 effect or the Hill coefficient in the presence of MSOP (76.1±2.8% and 1.2±0.2, respectively; n=8). MSOP induced a similar rightward shift of L-AP4 potency in the PPF concentration-response relation from EC50=2.9 μM to 217 μM (fit to population means; n=3–5 per data point) (Figure 1b). The right-shift in EC50 values of EPSC amplitude measurements was used to estimate the equilibrium dissociation constant (KB) for MSOP and the L-AP4 receptor which amounted to 12.5 μM (see Methods).
These observations indicate that group III mGluR activation via L-AP4 greatly reduces synaptic release of glutamate from PF terminals which can be reversed by the competitive antagonist MSOP.
Test of endogenous autoreceptor activation during distinct stimulation patterns
We utilized the selective antagonist MSOP to probe the endogenous activation of group III mGluRs by synaptically released glutamate. For endogenous mGluR activation to be detected, when transmitter concentrations rise briefly to high levels after release, it is critical that the antagonist is present at sufficiently high concentrations. EC50 values of glutamate at recombinant rat mGluR4a or mGluR8a have been reported to range from 3–39 μM (Pin et al., 1999; Schoepp et al., 1999). Using the above derived KB for MSOP of 12.5 μM, a steady-state synaptic concentration of 1 mM glutamate would be expected to be 20–70% less potent in the presence of 1 mM MSOP (see Methods). Due to the fast removal of synaptically released glutamate, the reduction in glutamate potency by MSOP is expected to be even larger, depending on the dissociation rate of MSOP from its receptor. We concluded that 1 mM MSOP should effectively antagonize endogenous group III mGluR autoreceptor activation. In the following experiments, the levels of group III mGluR activation were compared during paired-pulse or four-pulse stimulation, after tetanic bursts and during long-duration train stimulation.
Paired-pulse facilitation (PPF)
Due to the reported localization of mGluR4a in the presynaptic active zone, we tested whether the receptors are recruited during minimal afferent activity using paired stimulations with 200–500 ms latency and at 20–30 s intervals. We repeated these experiments in the presence of glutamate uptake inhibitor D,L-TBOA in order to enhance and prolong receptor exposure.
During paired-pulse stimulations, neither the amplitude of EPSC1 nor the degree of facilitation of EPSC2 at 250 or 500 ms latency was affected by MSOP (Figure 4a). The mean amplitude of EPSC1 was 544.8±74.9 pA (control amplitudes determined during min -5 to 0; n=7) and 0.96±0.04 relative to control in the presence of MSOP (min 5 to 10; this represents the time period when MSOP maximally reversed L-AP4-effects; data not shown). The facilitation of EPSC2 at 250 ms latency was 1.42±0.03 (control) and 1.45±0.02 (+MSOP; n=4). The facilitation at 500 ms latency was 1.24±0.03 and 1.26±0.03, respectively (n=3). The EPSC1 decay kinetics were not affected (Figure 4b). The time of decay to 1/e of the peak amplitude (estimated time constant) was 11.0±1.9 ms (control) and 10.9±1.9 ms (+MSOP) (n=7).
Figure 4.
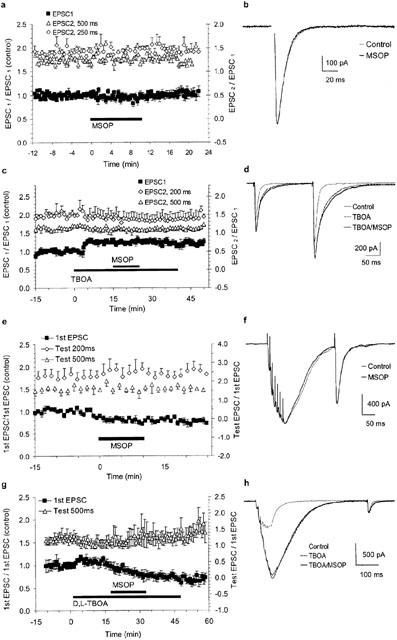
Paired-pulse and tetanic stimulations did not activate group III mGluRs. (a) Group data for seven experiments using paired-pulse stimulation (latencies 250 ms in four cells and 500 ms in three cells). Closed symbols are EPSC1 amplitudes relative to the pretreatment phase (left-hand y-axis). Open symbols correspond to facilitation of EPSC2 vs. EPSC1 (right-hand y-axis). (b) Superimposed EPSC1 averages (seven traces) show that single EPSCs were not affected in the presence of MSOP (stimulus artefact blanked). (c) Group data for three experiments using stimulation protocols similar to (a) but with application of D,L-TBOA prior to MSOP. Note the increase in EPSC1 amplitude without change in facilitation of EPSC2 after D,L-TBOA. (d) Superimposed averages of six paired-pulse traces under control conditions and after addition of TBOA and TBOA/MSOP. (e) Group data for six experiments using tetanic bursts followed by single test pulses. Closed symbols are the amplitude of the 1st EPSC of the burst relative to pretreatment phase (left-hand y-axis). Open symbols show the potentiation of testpulses with 200 and 500 ms latency (right-hand y-axis). (f) Averages of four traces in control and MSOP conditions show that the post-tetanic potentiation of a test pulse elicited 200 ms after burst stimulation was not different. A scaling factor of 1.1 was applied to the trace in MSOP to compensate for the run-down in 1st EPSC amplitudes. (g) Group data of five experiments using tetanic bursts as in (e) and applying D,L-TBOA prior to MSOP. (h) Superimposed averages of six traces under the indicated conditions (stimulations artefacts deleted and traces in TBOA and TBOA/MSOP scaled by 1.5 and 1.8, respectively, to compensate for the run-down in 1st EPSC amplitudes.
Application of the glutamate uptake inhibitor D,L-TBOA (50 μM) prior to MSOP enhanced EPSC amplitudes and slowed down the current decay without any change to the degree of facilitation (Figure 4c,d). The absence of effect on the facilitation suggested a lack of autoreceptor activation which was confirmed by the absence of change in these parameters after addition of MSOP. The control amplitude of EPSC1 was 1347±222 pA with amplitudes relative to control of 1.26±0.01 or 1.25±0.06 during TBOA or TBOA+MSOP, respectively (control: -5–0 min; TBOA: 10–15 min; TBOA+MSOP: 20–25 min; n=3). The facilitation of EPSC2 relative to EPSC1 at 200 ms latency was 1.50±0.08, 1.42±0.19 and 1.38±0.18 in control, TBOA and TBOA+MSOP, respectively. At 500 ms latency it was 1.15±0.06, 1.11±0.10 and 1.09±0.07 in control, TBOA and TBOA+MSOP, respectively. D,L-TBOA slowed down EPSC decay by a factor of 2.0±0.1 (n=3) consistent with the observations of Barbour et al. (1994) and Takahashi et al. (1995) using different glutamate uptake blockers. MSOP had no influence on current decay kinetics in the presence of TBOA (Figure 4d).
Four-pulse stimulation
We employed four stimulations at 250 ms intervals, repeated every 30 s. Neither the amplitude of the EPSC1 nor the facilitation (EPSC4/EPSC1) or current decay of EPSC4 was effected by MSOP (data not shown; mean EPSC1 amplitude was 495±97 pA (control) and 472±104 pA (+MSOP); the facilitation was 1.6±0.1 (control) and 1.7±0.2 (+MSOP); decay time constant of EPSC4 was 14.4±2.9 ms (control) and 14.5±3.2 ms (+MSOP); n=4). These experiments were repeated with 50 μM D,L-TBOA applied prior to MSOP (data not shown). TBOA increased the amplitudes of EPSC1 by a factor of 1.2±0.01 and slowed their decay time constants from 10.9±0.1 ms to 18.4±0.6 ms (n=5). TBOA alone did not affect the degree of facilitation of EPSC4. Addition of MSOP in the presence of D,L-TBOA did not significantly affect any of the parameters listed.
Post-tetanic potentiation (PTP)
Next, we examined the possibility that group III mGluRs are endogenously activated after tetanic burst stimulation which has been shown to activate presynaptic group II mGluRs in the hippocampus (Kew et al., 2001; 2002; Scanziani et al., 1997). Bursts consisted of seven stimulations at 100 Hz, followed by a test stimulation with latency 200 ms or 500 ms after the end of the burst. The pattern was repeated every 45 s. To prevent the activation of postsynaptic mGluR1 during high frequency stimulation (Batchelor & Garthwaite, 1993), the non-competitive antagonist CPCCOEt (50 μM) was added to the extracellular solution (Litschig et al., 1999). A slow run-down of EPSC amplitudes usually accompanied the use of burst stimulation.
Addition of MSOP did not affect the relative amplitude of the first EPSC in the burst and the test EPSCs at latencies of 200 or 500 ms (Figure 4e). EPSCs did not return to baseline during bursts and a baseline current developed (Figure 4f). Because of possible series resistance errors and accumulation of glutamate during bursts which may cause desensitization of postsynaptic receptors, we did not attempt to analyse within-burst EPSC2-7. The control amplitude of the first EPSC within bursts was 397.5±89.1 pA with amplitudes relative to controls of 0.81±0.01 or 0.76±0.01 after MSOP or wash-out, respectively (wash-out: 20–25 min; n=6). Control facilitations at 200 ms and 500 ms were 2.27±0.04 and 1.69±0.04, respectively. In MSOP they were 2.47±0.06 and 1.64±0.06, after wash-out they were 2.50±0.06 and 1.65±0.04, respectively (n=6). The presence of D,L-TBOA greatly enhanced the burst-induced baseline current (Figure 4h) and only test EPSCs at 500 ms latency after the burst were analysed. Also under these conditions, MSOP did not affect the degree of facilitation or the time course of the test EPSC (Figure 4g,h).
Train stimulations
We employed trains consisting of 45 stimulations at 10 Hz, repeated three times with intervals of 2.5 min, to represent either pre- or post-treatment situations. Post-treatment stimulation started 5 min after wash-in of the test solution. We limited stimulation strength and repetitions to minimize the possible contribution of long-term changes in synaptic strength (Hartell, 1996; Salin et al., 1996). Experiments were performed in the presence of CGP54626 (80 nM) and CPCCOEt (50 μM). In order to better separate long-term changes in synaptic transmission induced by train stimulation from autoreceptor inhibition, the group of MSOP-treated cells (n=8) was compared with cells that were sham-treated with aCSF (aCSF group; n=8).
In general, EPSCs facilitated during the initial phase of the train which was followed by a phase of synaptic depression (Figure 5a). Trains were characterized by three parameters: the initial amplitude (Start), the average of five EPSC amplitudes during the apex (Peak) and of the last five EPSCs (Tail). The time-to-Peak within trains averaged 1.32±0.14 s (n=15) which represents the 12th±2 EPSC. In the MSOP-group, relative Peak- and Tail-values (post- vs pre-treatment) were significantly larger as compared to corresponding values in the aCSF treated group of cells (Figure 5b; two-way ANOVA with MSOP or aCSF as treatment factor A and repeated measures of the synaptic strength (Peak and Tail) as factor B: P(A)=0.04, P(B)=0.33, P(A×B)=1.0). The relative Start-values decreased during the experiment in both groups (aCSF: −0.17±0.07; MSOP: −0.17±0.06; n=8 for both). The increase in relative Peak-amplitudes was +0.04±0.05 and +0.19±0.06 for aCSF- and MSOP-groups, respectively. The change in relative tail amplitudes was −0.01±0.03 and +0.16±0.07 in aCSF- and MSOP-groups, respectively. These changes in train parameters are exemplified in the superimposed trains in Figure 5a, the experimental data pairs for Peak and Tail parameters are shown in Figure 5c.
Figure 5.
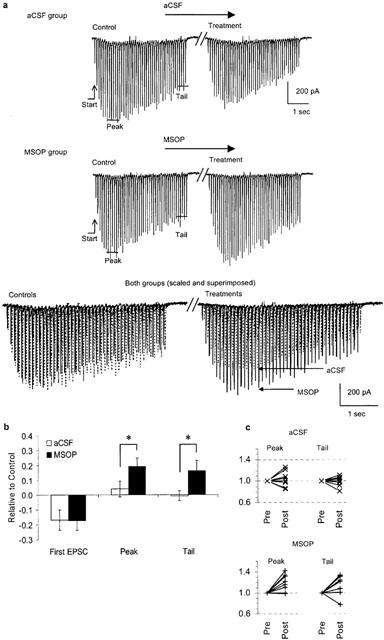
MSOP reveals endogenous activation of group III mGluRs during EPSC trains. (a) Trains (10 Hz, 4.5 s) were characterized by their ‘Start' EPSC amplitude and the mean amplitude of five EPSCs during ‘Peak' and ‘Tail'. The upper example illustrates sham-treatment with aCSF, the middle example is from a neurone treated with 1 mM MSOP. Both pre- and post-treatment trains are shown below, superimposed and scaled to their respective ‘Start' values (the time resolution is enhanced and aCSF trains in dashed lines are shifted slightly along the time axis to facilitate visual comparison). The time gap between control and treatment represents 12 min. (b) Group data for all three post-treatment train parameters for aCSF- and MSOP-groups (eight cells per group). The difference in the treatment factor (aCSF/MSOP) was significant: P<0.05; two-way ANOVA. (c) All individual data pairs for relative ‘Peak' and ‘Tail' values during pre- and post-treatment.
Direct application of L-glutamate
Weak MSOP-induced relief of inhibition might indicate weak ability to compete with glutamate at group III mGluRs. Compared with L-AP4, glutamate is about 10 times less potent but 30% more efficacious at recombinant mGluR4a (Monastyrskaia et al., 1999). EC50 values for glutamate at recombinant mGluR4a and mGluR8a are reported between 3 and 39 μM (Pin et al., 1999; Schoepp et al., 1999). In order to explore the possibility that glutamate might exhibit higher potency at presynaptic group III mGluRs, we applied L-glutamate directly and assessed group III mGluR activation using paired-pulse stimulation (latency 200 ms, 0.05 Hz). To minimize unwanted glutamate-induced effects, CPCCOEt and CPG54626 were added in order to block mGluR1 and GABAB receptors, respectively. Wash-in of 25 μM L-glutamate alone did not affect the amplitude of evoked EPSCs, the holding current or the current noise (not shown). This indicated that L-glutamate might have been removed by uptake and had only limited access, if any, to synaptic AMPA/kainate-receptors and, thus, also to presynaptic mGluR4a. In order to improve access of L-glutamate to presynaptic mGluRs, we applied L-glutamate together with 50 μM D,L-TBOA. This raised holding currents (<500 pA) and the current noise (<20 pA; Irms). Coapplication of L-glutamate and D,L-TBOA increased the EPSC amplitudes without affecting the facilitation of EPSC2 relative to EPSC1 and further addition of MSOP neither effected EPSC amplitudes nor the facilitation, indicating that presynaptic mGluRs had not been activated (Figure 6a). This data argues against an increased potency of glutamate at presynaptic group III mGluRs relative to recombinant receptors as the explanation for the weak effects of the competitive antagonist MSOP.
Figure 6.
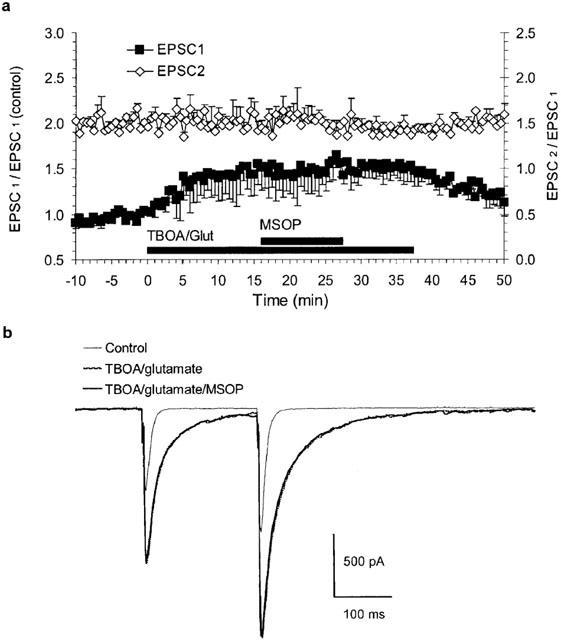
Coapplication of 25 μM L-glutamate and D,L-TBOA did not activate group III mGluRs. (a) Group data of three experiments using paired-pulse stimulation with latency 200 ms. Relative EPSC1-amplitude on the left-hand y-axis and EPSC2/EPSC1-amplitude ratio on the right y-axis. D,L-TBOA (50 μM)/L-glutamate application increased EPSC-amplitudes without affecting the facilitation. MSOP (1 mM) was without effect. (b) Averages of six traces at the indicated conditions superimposed in one experiment (a steady-state increase in I-hold of −100 pA has been subtracted for traces in TBOA/glutamate).
Discussion
The main conclusions of this study are: (1) L-AP4 strongly depressed synaptic strength at PF to PC synapses via a reduction of glutamate release probability without contribution of axonal and postsynaptic mechanisms (Figures 1, 2, 3). (2) The L-AP4- induced reduction of glutamate release probability was mediated via the Ca2+-dependent glutamate exocytosis pathway (Figure 3). (3) Endogenous group III mGluR activation by synaptically released glutamate either did not occur during single release events or short tetanic bursts (100 Hz for 60 ms) or, less likely, any inhibition was of very short duration (<200 ms) (Figure 4). (4) Group III mGluRs were activated endogenously during prolonged 10 Hz stimulation (Figure 5).
Endogenous activation of presynaptic mGluRs: group II versus group III
Localization of presynaptic receptors relative to transmitter release sites is likely of high functional importance. A striking difference between presynaptic group II and III mGluRs is the subcellular localization. While group II mGluRs are generally found at perisynaptic sites or without an obvious correlation to glutamate release sites (Lujan et al., 1997; Shigemoto et al., 1997), group III mGluRs are generally concentrated at presynaptic active zones (Corti et al., 2002; Kinoshita et al., 1996; Mateos et al., 1998; 1999). There is convincing evidence that group II mGluRs serve as autoinhibitory feedback receptors during short-term synaptic processing in mossy fibre and perforant path terminals of the hippocampus (Kew et al., 2001; 2002; Scanziani et al., 1997) and also as heterosynaptic inhibitory receptors at Golgi cell terminals in glomeruli of the cerebellum (Mitchell & Silver, 2000). These receptors are not recruited by low-frequency afferent activity but by higher-frequency activity or in the presence of uptake inhibitors. The current view assumes that group II receptor activation requires the diffusion of glutamate out of the synaptic cleft. The relatively high affinity of group II mGluRs for glutamate (EC50 4–20 μM; Pin et al., 1999) supports this interpretation.
In contrast to group II mGluRs, group III mGluRs are, due to their localization, likely to be exposed to high glutamate concentrations even at low-frequency afferent activity, as for postsynaptic receptors. Recombinant mGluR4a or mGluR8a respond to glutamate in the low μM range while mGluR7 responds in the range of several 100 μM (Pin et al., 1999). These differences point to potentially distinct physiological roles for the different group III mGluRs. We have focused on the putative role of group III mGluRs in short-term synaptic plasticity in the cerebellar cortex.
Conditions for signal transduction at PF synapses
At PF terminals, the dominant group III mGluR subtype is mGluR4a: in mGluR4-/- mice, L-AP4 remains ineffective up to 500 μM (Pekhletski et al., 1996) and mRNA levels for mGluR6, 7 or 8 are very low in cerebellar granule cells (human cerebellum: Berthele et al., 1999; mouse: Duvoisin et al., 1995; rat: Iacovelli et al., 2002; Ohishi et al., 1995).
Apart from agonist concentration, the time-course of exposure is important for receptor activation. The time constant of the glutamate transient at PF to PC synapses of about 7 ms at room temperature is 4–5 times slower compared with PF to interneurone synapses in the cerebellar cortex, most likely due to retarded transmitter diffusion around spines (Barbour et al., 1994). Still, in our experiments, group III mGluRs were apparently not activated via single release events, via four stimuli delivered at 250 ms intervals or after tetanic bursts. Similar to the present study, MSOP was without effect on paired-pulse stimulation in rat dentate gyrus (O'leary et al., 1997). The apparent lack of group III receptor activation after tetanic burst stimulation described in this study, was also observed by Dube & Marshall (2000) in rat locus coeruleus-neurones using 70 Hz/300 ms bursts and test-pulses with 200–300 ms latency (MAP4 as antagonist).
The ability of prolonged stimulation trains to activate presynaptic group III mGluRs in the cerebellar cortex as shown in this study has been demonstrated previously in other brain regions where the mGluRs involved were less well defined: Chen et al. (2002) found that MSOP relieved frequency-dependent synaptic depression in nucleus tractus solitarius-neurones only at stimulation frequencies ⩾9 Hz, most likely mediated via mGluR7. Similar, at the Calyx synapse, the group II/III mGluR-selective antagonist CPPG relieved frequency-dependent synaptic inhibition using 10 Hz stimulations (von gersdorff et al., 1997). The amount of mGluR-mediated inhibition was <15% in both cases.
It is important to consider the effect of antagonists in respect to the high concentrations of glutamate present during synaptic transmission. We estimated the antagonist potency of MSOP close to 12 μM which is higher than the KB of 51 μM calculated previously from recordings in the spinal cord (Thomas et al., 1996). With a KB of 12 μM and assuming a high potency of glutamate reported for recombinant mGluR4a (EC50 3 μM; Schoepp et al., 1999) and a synaptic glutamate-concentration of 1 mM, the expected reduction of glutamate-induced synaptic inhibition by MSOP amounts to at least 20%, (see Methods). If the potency of glutamate at presynaptic mGluR4a exceeds 3 μM, any effect of MSOP would possibly not be detected. Our data argue against such an explanation for the absence of effect of MSOP: firstly, the potency of the agonist L-AP4 of 2.4 μM was at the lower end of reported potencies at recombinant receptors (EC50 0.2–1.2 μM; Pin et al., 1999) and secondly, a steady-state glutamate concentration close to 25 μM did not activate presynaptic group III mGluRs.
Possible restrictions for presynaptic mGluR signalling
Some data is available supporting the view that group III mGluR-mediated responses might be short-lived, lasting less than 200 ms. Semyanov & Kullmann (2000) reported endogenous heterosynaptic activation of (most likely) mGluR4a expressed in CA1 interneurone terminals onto other interneurones. MSOP revealed mGluR activation if glutamate was released 100 ms before interneurone stimulation but not at latencies >200 ms, indicating that the heterosynaptic depression has decayed in the interval. However, it is unlikely that autoreceptor-activation has been missed in the present study where the test-pulse latencies were ⩾200 ms. In order to detect potentially weak and/or short-lived endogenous group III mGluR activation, we used the glutamate uptake inhibitor D,L-TBOA to increase the synaptic glutamate concentration and prolong its time-course. D,L-TBOA has been shown to inhibit both, glial and postsynaptic neuronal glutamate uptake at PF to PC synapses (Brasnjo & Otis, 2001). Increased EPSC amplitudes and decay times indicated that more AMPARs have been activated and that glutamate remained longer in the synaptic cleft permitting significant rebinding (Barbour et al., 1994). Even with prolonged exposure to glutamate following D,L-TBOA addition, no endogenous group III mGluR activation during paired-pulse and four-pulse stimulation was detected. Whether group III mGluRs were activated within tetanic bursts is less clear. Burst-induced baseline-currents prevented test-pulse measurements at very short latencies after bursts.
During tetanic stimulation, presynaptic terminals spend a large proportion of time at depolarized potentials. A number of studies have shown that depolarization per se during agonist exposure can be of relevance for GPCR signalling. Depolarization may weaken the signalling (Brody & Yue, 2000; Ilouz et al., 1999; Stefani et al., 1998) or strengthen it (Perroy et al., 2002). Modulation of pharmacological properties due to receptor localization which might involve interaction with different protein binding partners has also been described (Davare et al., 2001; Tabata et al., 2002). The localization of group III mGluRs within presynaptic active zones provides the potential for interaction with proteins of the exocytotic apparatus that could provide a molecular basis for the influence of depolarization on G-protein coupled receptor signalling.
We do not believe that receptor desensitization restricted group III mGluR activation because the L-AP4 response did not desensitize within 15 min of exposure and because the MSOP-mediated increase in EPSC amplitudes during stimulation trains revealed similar inhibition levels at peak and tail periods (P(B) was 0.33 in the two-way ANOVA).
A number of studies suggest that group III mGluRs may have significant tonic activity levels possibly restricting the amount of attainable activity-dependent activation. Tonic group III mGluR activation via ambient neurotransmitter is likely involved in cases where competitive antagonists increased the amplitude of single evoked EPSCs. Such observations were made at baroreceptor terminals in the nucleus tractus solitarius (Pamidimukkala & Hay, 2001) and in glutamatergic terminals onto magnocellular neurones in the hypothalamic supraoptic nucleus (Oliet et al., 2001; Schrader & Tasker, 1997). In mGluR4 knock-out mice, PPF and PTP at the PF to PC synapse were reduced rather than increased relative to wild-type mice (Pekhletski et al., 1996) prompting the authors to propose a tonic inhibitory function for mGluR4a receptors, with a higher release probability for the first EPSC in mutant animals and subsequently reduced facilitation of the following test EPSC. It was not demonstrated whether the proposed tonic activity was intrinsic or caused by steady-state ligand-mediated activation. Slightly different conditions might prevail at this synapse in mice and rats because Pekhletski et al. (1996) also report that MAP4 (500 μM) was applied to rat cerebellar slices, in an attempt to reproduce the observations made in mutant mice, and no effect on PPF (latencies 50–500 ms) or PTP (7 pulses, 40 Hz, testpulse latency 300 ms) was observed. In our recordings, no tonic activation of group III mGluRs via extracellular glutamate was observed as the competitive antagonist MSOP had no effect on single EPSCs. We cannot rule out the possibility that group III mGluRs might have intrinsic activity, insensitive to MSOP, from our experiments.
In summary, we observed potent, efficacious inhibition of synaptic strength at PF to PC synapses following activation of presynaptic group III mGluRs via exogenous L-AP4 which contrasts with the moderate inhibition achieved via synaptically released endogenous agonist. This was unexpected because mGluR4a is reported to be localized close to release sites and to possess high glutamate affinity. However, our observations are in line with findings made by other laboratories in other areas of the brain where group III mGluRs are less prominently expressed. Whilst we cannot exclude the possibility that endogenous agonists other than glutamate may be required for full receptor activation (e.g. aspartate or L-SOP: Hampson et al., 1999; Klunk et al., 1991; Yuzaki et al., 1996), our observations suggest the existence of additional, as yet uncharacterized mechanisms regulating the activity-dependent activation of presynaptic group III mGluRs.
Acknowledgments
We thank Dr Vincent Mutel for sharing his insight into mGluRs with us and Dr Gerd Trube for statistical advice and comments on the manuscript.
Abbreviations
- aCSF
artificial cerebrospinal fluid
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate
- EC50
50% maximum effective concentration
- EPSC
excitatory postsynaptic current
- fEPSP
field excitatory postsynaptic potential
- GABA
γ-aminobutyric acid
- Irms
‘root-mean-square' current noise
- KB
antagonist equilibrium dissociation constant
- MAP4
(S)-2-amino-2-methyl-4-phosphonobutanoic acid
- mEPSC
miniature excitatory postsynaptic current
- mGluR
metabotropic glutamate receptor
- NMDA
N-methyl-D-aspartic acid
- PC
Purkinje cell
- PF
parallel fibre
- PPF
paired-pulse facilitation
- PTP
post-tetanic potentiation
References
- ANWYL R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res. Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- BARBOUR B., KELLER B.U., LLANO I., MARTY A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–1343. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- BATCHELOR A.M., GARTHWAITE J. Novel synaptic potentials in cerebellar Purkinje cells: probable mediation by metabotropic glutamate receptors. Neuropharmacology. 1993;32:11–20. doi: 10.1016/0028-3908(93)90124-l. [DOI] [PubMed] [Google Scholar]
- BERTHELE A., PLATZER S., LAURIE D.J., WEIS S., SOMMER B., ZIEGLGANSBERGER W., CONRAD B., TOLLE T.R. Expression of metabotropic glutamate receptor subtype mRNA (mGluR1-8) in human cerebellum. NeuroReport. 1999;10:3861–3867. doi: 10.1097/00001756-199912160-00026. [DOI] [PubMed] [Google Scholar]
- BRADLEY S.R., STANDAERT D.G., RHODES K.J., REES H.D., TESTA C.M., LEVEY A.I., CONN P.J. Immunohistochemical localization of subtype 4a metabotropic glutamate receptors in the rat and mouse basal ganglia. J. Comp. Neurol. 1999;407:33–46. [PubMed] [Google Scholar]
- BRASNJO G., OTIS T.S. Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron. 2001;31:607–616. doi: 10.1016/s0896-6273(01)00377-4. [DOI] [PubMed] [Google Scholar]
- BRODY D.L., YUE D.T. Relief of G-protein inhibition of calcium channels and short-term synaptic facilitation in cultured hippocampal neurons. J. Neurosci. 2000;20:889–898. doi: 10.1523/JNEUROSCI.20-03-00889.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN C.Y., LING E.H., HOROWITZ J.M., BONHAM A.C. Synaptic transmission in nucleus tractus solitarius is depressed by Group II and III but not Group I presynaptic metabotropic glutamate receptors in rats. J. Physiol. 2002;538:773–786. doi: 10.1113/jphysiol.2001.012948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONQUET F., BASHIR Z.I., DAVIES C.H., DANIEL H., FERRAGUTI F., BORDI F., FRANZ-BACON K., REGGIANI A., MATARESE V., CONDE F., COLLINGRIDGE G.L., CREPEL L. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- CORTI C., ALDEGHERI L., SOMOGYI P., FERRAGUTI F. Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience. 2002;110:403–420. doi: 10.1016/s0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- CORTI C., RESTITUITO S., RIMLAND J.M., BRABET I., CORSI M., PIN J.P., FERRAGUTI F. Cloning and characterization of alternative mRNA forms for the rat metabotropic glutamate receptors mGluR7 and mGluR8. Eur. J. Neurosci. 1998;10:3629–3641. doi: 10.1046/j.1460-9568.1998.00371.x. [DOI] [PubMed] [Google Scholar]
- DANIEL H., CREPEL F. Control of Ca2+ influx by cannabinoid and metabotropic glutamate receptors in rat cerebellar cortex requires K+ channels. Journal of Physiology. 2001;537:793–800. doi: 10.1111/j.1469-7793.2001.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVARE M.A., AVDONIN V., HALL D.D., PEDEN E.M., BURETTE A., WEINBERG R.J., HORNE M.C., HOSHI T., HELL J.W. A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- DE BLASI A., CONN P.J., PIN J.P., NICOLETTI F. Molecular determinants of metabotropic glutamate receptor signaling. Trends Pharmacol. Sci. 2001;22:114–120. doi: 10.1016/s0165-6147(00)01635-7. [DOI] [PubMed] [Google Scholar]
- DEBANNE D., GUERINEAU N.C., GAHWILER B.H., THOMPSON S.M. Action-potential propagation gated by an axonal I(A)-like K+ conductance in hippocampus [published erratum appears in Nature 1997 Dec 4;390(6659):536] Nature. 1997;389:286–289. doi: 10.1038/38502. [DOI] [PubMed] [Google Scholar]
- DITTMAN J.S., REGEHR W.G. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J. Neurosci. 1996;16:1623–1633. doi: 10.1523/JNEUROSCI.16-05-01623.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUBE G.R., MARSHALL K.C. Activity-dependent activation of presynaptic metabotropic glutamate receptors in locus coeruleus. J. Neurophysiol. 2000;83:1141–1149. doi: 10.1152/jn.2000.83.3.1141. [DOI] [PubMed] [Google Scholar]
- DUVOISIN R.M., ZHANG C., RAMONELL K. A novel metabotropic glutamate receptor expressed in the retina and olfactory bulb. J. Neurosci. 1995;15:3075–3083. doi: 10.1523/JNEUROSCI.15-04-03075.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMPSON D.R., HUANG X.-P., PEKHLETSKI R., PELTEKOVA V., HORNBY G., THOMSEN C., THOGERSEN H. Probing the ligand-binding domain of the mGluR4 subtype of metabotropic glutamate receptor. J. Biol. Chem. 1999;274:33488–33495. doi: 10.1074/jbc.274.47.33488. [DOI] [PubMed] [Google Scholar]
- HARTELL N.A. Strong activation of parallel fibers produces localized calcium transients and a form of LTD that spreads to distant synapses. Neuron. 1996;16:601–610. doi: 10.1016/s0896-6273(00)80079-3. [DOI] [PubMed] [Google Scholar]
- IACOVELLI L., BRUNO V., SALVATORE L., MELCHIORRI D., GRADINI R., CARICASOLE A., BARLETTA E., DEBLASI A., NICOLETTI F. J. Neurochem. 2002. pp. 216–223. [DOI] [PubMed]
- ILOUZ N., BRANSKI L., PARNIS J., PARNAS H., LINIAL M. Depolarization affects the binding properties of muscarinic acetylcholine receptors and their interaction with proteins of the exocytic apparatus. J. Biol. Chem. 1999;274:29519–29528. doi: 10.1074/jbc.274.41.29519. [DOI] [PubMed] [Google Scholar]
- KEW J.N., DUCARRE J.M., PFLIMLIN M.C., MUTEL V., KEMP J.A. Activity-dependent presynaptic autoinhibition by group II metabotropic glutamate receptors at the perforant path inputs to the dentate gyrus and CA1. Neuropharmacology. 2001;40:20–27. doi: 10.1016/s0028-3908(00)00118-0. [DOI] [PubMed] [Google Scholar]
- KEW J.N., PFLIMLIN M.C., KEMP J.A., MUTEL V. Differential regulation of synaptic transmission by mGlu2 and mGlu3 at the perforant path inputs to the dentate gyrus and CA1 revealed in mGlu2 -/- mice. Neuropharmacology. 2002;43:215–221. doi: 10.1016/s0028-3908(02)00084-9. [DOI] [PubMed] [Google Scholar]
- KINOSHITA A., OHISHI H., NOMURA S., SHIGEMOTO R., NAKANISHI S., MIZUNO N. Presynaptic localization of a metabotropic glutamate receptor, mGluR4a, in the cerebellar cortex: a light and electron microscope study in the rat. Neurosci. Lett. 1996;207:199–202. doi: 10.1016/0304-3940(96)12519-2. [DOI] [PubMed] [Google Scholar]
- KLUNK W.E., MCCLURE R.J., PETTEGREW J.W. L-phosphoserine, a metabolite elevated in Alzheimer's disease, interacts with specific L-glutamate receptor subtypes. J. Neurochem. 1991;56:1997–2003. doi: 10.1111/j.1471-4159.1991.tb03458.x. [DOI] [PubMed] [Google Scholar]
- LITSCHIG S., GASPARINI F., RUEEGG D., STOEHR N., FLOR P.J., VRANESIC I., PREZEAU L., PIN J.P., THOMSEN C., KUHN R. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol. Pharmacol. 1999;55:453–461. [PubMed] [Google Scholar]
- LLINAS R.R., WALTON K.D. Cerebellum. New York, Oxford: Oxford University Press; 1998. [Google Scholar]
- LUJAN R., ROBERTS J.D., SHIGEMOTO R., OHISHI H., SOMOGYI P. Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J. Chem. Neuroanat. 1997;13:219–241. doi: 10.1016/s0891-0618(97)00051-3. [DOI] [PubMed] [Google Scholar]
- MATEOS J.M., AZKUE J., SARRIA R., KUHN R., GRANDES P., KNOPFEL T. Localization of the mGlu4a metabotropic glutamate receptor in rat cerebellar cortex. Histochem. Cell Biol. 1998;109:135–139. doi: 10.1007/s004180050211. [DOI] [PubMed] [Google Scholar]
- MATEOS J.M., ELEZGARAI I., BENITEZ R., OSORIO A., BILBAO A., AZKUE J.J., KUHN R., KNOPFEL T., GRANDES P. Clustering of the group III metabotropic glutamate receptor 4a at parallel fiber synaptic terminals in the rat cerebellar molecular layer. Neurosci. Res. 1999;35:71–74. doi: 10.1016/s0168-0102(99)00066-8. [DOI] [PubMed] [Google Scholar]
- MITCHELL S.J., SILVER R.A. Glutamate spillover suppresses inhibition by activating presynaptic mGluRs. Nature. 2000;404:498–502. doi: 10.1038/35006649. [DOI] [PubMed] [Google Scholar]
- MONASTYRSKAIA K., LUNDSTROM K., PLAHL D., ACUNA G., SCHWEITZER C., MALHERBE P., MUTEL V. Effect of the umami peptides on the ligand binding and function of rat mGlu4a receptor might implicate this receptor in the monosodium glutamate taste transduction. Br. J. Pharmacol. 1999;128:1027–1034. doi: 10.1038/sj.bjp.0702885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEALE S.A., GARTHWAITE J.M., BATCHELOR A. Metabotropic glutamate receptor subtypes modulating neurotransmission at parallel fibre-Purkinje cell synapses in rat cerebellum. Neuropharmacology. 2001;41:42–49. doi: 10.1016/s0028-3908(01)00046-6. [DOI] [PubMed] [Google Scholar]
- OHISHI H., AKAZAWA C., SHIGEMOTO R., NAKANISHI S., MIZUNO N. Distributions of the mRNAs for L-2-amino-4-phosphonobutyrate-sensitive metabotropic glutamate receptors, mGluR4 and mGluR7, in the rat brain. J. Comp. Neurol. 1995;360:555–570. doi: 10.1002/cne.903600402. [DOI] [PubMed] [Google Scholar]
- O'LEARY D.M., CASSIDY E.M., O'CONNOR J.J. Group II and III metabotropic glutamate receptors modulate paired pulse depression in the rat dentate gyrus in vitro. Eur. J. Pharmacol. 1997;340:35–44. doi: 10.1016/s0014-2999(97)01405-2. [DOI] [PubMed] [Google Scholar]
- OLIET S.H.R., PIET R., POULAIN D.A. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- PAMIDIMUKKALA J., HAY M. Frequency dependence of endocytosis in aortic baroreceptor neurons and role of group III mGluRs. Am. J. Physiol. 2001;281:H387–H395. doi: 10.1152/ajpheart.2001.281.1.H387. [DOI] [PubMed] [Google Scholar]
- PEKHLETSKI R., GERLAI R., OVERSTREET L.S., HUANG X.P., AGOPYAN N., SLATER N.T., ABRAMOW-NEWERLY W., RODER J.C., HAMPSON D.R. Impaired cerebellar synaptic plasticity and motor performance in mice lacking the mGluR4 subtype of metabotropic glutamate receptor. J. Neurosci. 1996;16:6364–6373. doi: 10.1523/JNEUROSCI.16-20-06364.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERROY J., RICHARD S., NARGEOT J., BOCKAERT J., FAGNI L. Permissive effect of voltage on mGlu 7 receptor subtype signaling in neurons. Journal of Biological Chemistry. 2002;277:1223–1228. doi: 10.1074/jbc.M109141200. [DOI] [PubMed] [Google Scholar]
- PIN J.P., DE COLLE C., BESSIS A.S., ACHER F. New perspectives for the development of selective metabotropic glutamate receptor ligands. Eur. J. Pharmacol. 1999;375:277–294. doi: 10.1016/s0014-2999(99)00258-7. [DOI] [PubMed] [Google Scholar]
- SALIN P.A., MALENKA R.C., NICOLL R.A. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- SCANZIANI M., SALIN P.A., VOGT K.E., MALENKA R.C., NICOLL R.A. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- SCHOEPP D.D. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J. Pharmacol. Exp. Ther. 2001;299:12–20. [PubMed] [Google Scholar]
- SCHOEPP D.D., JANE D.E., MONN J.A. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- SCHRADER L.A., TASKER J.G. Presynaptic modulation by metabotropic glutamate receptors of excitatory and inhibitory synaptic inputs to hypothalamic magnocellular neurons. J. Neurophysiol. 1997;77:527–536. doi: 10.1152/jn.1997.77.2.527. [DOI] [PubMed] [Google Scholar]
- SEMYANOV A., KULLMANN D.M. Modulation of GABAergic signaling among interneurons by metabotropic glutamate receptors. Neuron. 2000;25:663–672. doi: 10.1016/s0896-6273(00)81068-5. [DOI] [PubMed] [Google Scholar]
- SHIGEMOTO R., KINOSHITA A., WADA E., NOMURA S., OHISHI H., TAKADA M., FLOR P.J., NEKI A., ABE T., NAKANISHI S., MIZUNO N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J. Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEFANI A., SPADONI F., BERNARDI G. Group III metabotropic glutamate receptor agonists modulate high voltage-activated Ca2+ currents in pyramidal neurons of the adult rat. Exp. Brain Res. 1998;119:237–244. doi: 10.1007/s002210050337. [DOI] [PubMed] [Google Scholar]
- TABATA T., AIBA A., KANO M. Extracellular calcium controls the dynamic range of neuronal metabotropic glutamate receptor responses. Mol. Cell Neurosci. 2002;20:56–68. doi: 10.1006/mcne.2002.1118. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI M., KOVALCHUK Y., ATTWELL D. Pre- and postsynaptic determinants of EPSC waveform at cerebellar climbing fiber and parallel fiber to Purkinje cell synapses. J. Neurosci. 1995;15:5693–5702. doi: 10.1523/JNEUROSCI.15-08-05693.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS N.K., JANE D.E., TSE H.W., WATKINS J.C. alpha-Methyl derivatives of serine-O-phosphate as novel, selective competitive metabotropic glutamate receptor antagonists. Neuropharmacology. 1996;35:637–642. doi: 10.1016/0028-3908(96)84635-1. [DOI] [PubMed] [Google Scholar]
- THOMSEN C., PEKHLETSKI R., HALDEMAN B., GILBERT T.A., O'HARA P., HAMPSON D.R. Cloning and characterization of a metabotropic glutamate receptor, mGluR4b. Neuropharmacology. 1997;36:21–30. doi: 10.1016/s0028-3908(96)00153-0. [DOI] [PubMed] [Google Scholar]
- VON GERSDORFF H., SCHNEGGENBURGER R., WEIS S., NEHER E. Presynaptic depression at a calyx synapse: the small contribution of metabotropic glutamate receptors. J. Neurosci. 1997;17:8137–8146. doi: 10.1523/JNEUROSCI.17-21-08137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YUZAKI M., FORREST D., CURRAN T., CONNOR J.A. Selective activation of calcium permeability by aspartate in Purkinje cells. Science. 1996;273:1112–1114. doi: 10.1126/science.273.5278.1112. [DOI] [PubMed] [Google Scholar]