

Abstract
Thrombin, a mitogen for human cultured airway smooth muscle (HASM), has many actions that have been attributed to activation of protease-activated receptor (PARs). However, the role of PARs in the proliferative action has not been clearly identified. Moreover, thrombin elicits cytokine production in a number of cell types, but these effects have not been characterized in human ASM.
Thrombin (0.03–3 U ml−1)-stimulated increases in the levels of the pro-inflammatory and fibrogenic cytokine, granulocyte-macrophage colony-stimulating factor (GM-CSF) were observed over the same concentration range observed for thrombin-stimulated mitogenesis.
Inhibition of thrombin proteolytic activity, with either D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone (PPACK)- or hirudin-treated thrombin (0.3 U ml−1) or in the presence of the thrombin serine protease-selective inhibitor, SDZ 217-766 (0.15 μM), reduced the thrombin-stimulated GM-CSF levels by 91±3, 65±12 and 83±9% (n=8, P<0.05), respectively. PPACK treatment, hirudin and SDZ 217-766 inhibited thrombin-stimulated increase in cell number by 70±8, 63±11 and 69±8%, respectively.
PAR-selective peptides SFLLRN (PAR1; 10 μM), SLIGKV (PAR2; 10 μM), GYPGQV (PAR4; 100 μM) or the combination of SFLLRN and GYPGQV elicited mitogenic responses of only 15% of that to thrombin and surprisingly, had no effect on GM-CSF levels (n=8). Nevertheless, inhibition of thrombin responses by pertussis toxin (50 ng ml−1) suggests that the PAR-independent actions also involve a G-protein-coupled receptor.
PAR1 receptor expression was evident by immunohistochemistry and these receptors were coupled to increases in intracellular calcium, but not to the phosphorylation of ERK or the increases in cyclin D1 protein levels that are essential for cell proliferation. Cross-desensitization of intracellular calcium increases by thrombin and the PAR1-selective peptide provides evidence that the PAR1 receptor responds to both ligands.
The failure of PAR-selective peptides to mimic thrombin responses together with the inhibition of thrombin responses by serine protease inhibitors suggest the involvement of novel proteolytic receptor targets for thrombin-induced mitogenesis and cytokine production.
Keywords: Thrombin, smooth muscle, airway, protease-activated receptor, granulocyte-macrophage colony-stimulating factor, growth, proliferation
Introduction
Thrombin is a serine protease generated from prothrombin as a result from the activation of the coagulation cascade (Grand et al., 1996). The role of thrombin was initially thought to be restricted to regulating haemostasis and thrombosis. However, this protease is now known to elicit a vast array of cellular responses: being chemotactic for inflammatory cells such as macrophages, monocytes and neutrophils; stimulating cell rounding; initiating bone cell resorption and neuronal proliferation (Grand et al., 1996). Thrombin is also implicated in vascular remodelling diseases such as atherosclerosis through pro-aggregatory actions and by directly stimulating smooth muscle proliferation (Tapparelli et al., 1993).
Smooth muscle cell hyperplasia contributes to the airway wall remodelling (AWR) that is associated with asthma (Ebina et al., 1993; Stewart et al., 1993). Our studies and those of several other groups have established that thrombin is a mitogen for human airway smooth muscle (HASM) (Panettieri et al., 1995; Tomlinson et al., 1994), but the receptor-dependence of this action has not been delineated. Elevated levels of thrombin are found in the bronchoalveolar lavage fluid (BALF) of asthmatics (Gabazza et al., 1999). Thus, thrombin has the potential to contribute to acute and chronic airway inflammation and remodelling, which are important pathological characteristics of asthma.
Eosinophil-derived mediators contribute to bronchospasm, acute and chronic inflammation and AWR. In vitro and in vivo studies show that granulocyte-macrophage colony-stimulating factor (GM-CSF) promotes the differentiation, proliferation, function, migration and survival of eosinophils (Lopez et al., 1986; Park et al., 1998). BALF GM-CSF levels in asthmatics are twice as high as those detected in healthy volunteers (Mattoli et al., 1991). In the context of AWR, GM-CSF can also stimulate collagen deposition by mesenchymal cells (Plenz et al., 1999). In epithelial cells and keratinocytes, serine proteases such as house dust mite allergens and tryptase have been shown to stimulate GM-CSF production (Sun et al., 2001; Vliagoftis et al., 2001; Wakita et al., 1997). Thus, we have explored the possible stimulatory effects of thrombin on GM-CSF production by HASM.
In smooth muscle cells, thrombin elevates intracellular calcium and activates the mitogen-activated protein kinase family (MAPK), particularly extracellular signal-regulated kinases 1 and 2 (ERK 1/2). Activation of ERK is followed by increases in cyclin D1 protein levels that are important in the mitogenic signalling required for entry of cells into the S phase of the cell cycle (Ravenhall et al., 2000; Stewart, 2001). A number of other signalling mechanisms have been implicated in thrombin-mediated mitogenesis, including PI3-kinase and p70S6k (Stewart, 2001). However, there is little known about the signalling pathways by which serine-proteases such as thrombin elicit cytokine production by mesenchymal cells.
Many of the responses to thrombin have been attributed to its ability to activate a specific type of G-protein-coupled receptor, the protease-activated receptor (PAR). Nevertheless, there is evidence that fibroblast, monocyte and macrophage mitogenic responses are independent of thrombin proteolytic activity (Bar-Shavit et al., 1983; 1990). There are four genetically distinct subtypes of the PAR family (Macfarlane et al., 2001), of which thrombin predominantly activates PAR1 through proteolytic cleavage of the N-terminal domain of the receptor. The nascent N-terminus functions as a ‘tethered ligand' that activates the receptor. Thrombin is less active on PAR3 and PAR4 and does not appear to activate PAR2 (Macfarlane et al., 2001). The two sites important for thrombin activity are its catalytic site and the anion binding site (Grand et al., 1996). The anion binding site co-ordinates the binding of thrombin to PARs or to specific, negatively charged substrates, such as fibrinogen. In human lung fibroblasts (Bar-Shavit et al., 1986), vascular smooth muscle cells (Herbert et al., 1992) and umbilical vein endothelial cells (Herbert et al., 1994), mitogenic responses to thrombin require serine-protease activity. However, the extent to which these thrombin responses are mimicked by PAR-selective peptides remains controversial, since substantial differences in cellular responses to thrombin and PAR-selective peptides have been observed. For example, bovine coronary artery smooth muscle proliferation induced by thrombin is not mimicked by PAR1-selective peptide (SFLLRN) (Bretschneider et al., 1997).
The main purpose of this study was to compare the effects of thrombin and PAR-selective peptides on functional responses of HASM, because definition of the importance of PARs in these responses is essential to any attempts to block the inflammation and remodelling actions of thrombin.
Methods
Cell culture
HASM cell cultures were generated from bronchi (0.5–2 cm diameter) obtained from lung resected from heart–lung transplant recipients. ASM was micro-dissected (with the aid of a binocular operating microscope) and enzymatically digested with elastase (0.5 mg ml−1) and collagenase (1 mg ml−1) (Worthington Biochemical Corporation, Lakewood, NJ, U.S.A.) to generate cell suspensions that were then cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% w v−1 foetal calf serum (FCS), as previously described (Stewart et al., 1995). Cells were passaged weekly at a 1 : 4 split ratio by a 10 min exposure to 0.5% w v−1 trypsin (CSL, Victoria, Australia) (in phosphate buffered saline (PBS, Oxoid, Hampshire, U.K.) containing 1 mM EDTA) generating a cell density of 1×104 cells per cm2. Cell passage numbers 4–14 were used for experiments; a period in which there is no relationship between cell passage number and responsiveness to growth factors or inhibitors and the expression of smooth muscle α-actin is maintained (Stewart et al., 1995).
HASM cells were plated into 6-well plates and allowed to grow to 70–80% monolayer confluency in complete DMEM. Subsequently, the cells were incubated with serum-free DMEM for 24 h to arrest growth. Drug treatments were added 30 min before stimulation for 48 h with thrombin in the presence of the growth supplement Monomed A (1% v v−1, containing insulin, tranferrin and selenium, CSL). When the incubation period was complete, cell supernatants were removed and stored at −20°C until required for GM-CSF determination, whilst the cells were harvested as described below to determine cell number.
Cell number determination
After incubation with thrombin for 48 h, the cells were washed twice with PBS and then dislodged with 300 μL of 0.5% w v−1 trypsin (CSL) for 5–10 min. PBS (500 μL) containing 2% FCS, was then added to neutralize the activity of trypsin and the cells were collected into 1.5 ml plastic tubes and centrifuged (760×g, 5 min at 4°C). Supernatants were discarded and pellets were resuspended in 200 μL of PBS containing 2% FCS and the cells counted using a haemocytometer. Trypan blue exclusion was used to identify non-viable cells.
Measurement of GM-CSF levels
Levels of GM-CSF were quantified using enzyme-linked immunosorbent assays (ELISA). ELISA plates were coated with rat anti-human GM-CSF monoclonal coating antibody (1 μg ml−1, Endogen, MA, U.S.A.); in 0.1 M sodium carbonate buffer, pH 9.4–9.8) and then left to incubate overnight at room temperature. The plate was washed three times with buffer (PBS containing 0.1% Tween-20, PBS-T) before incubation with a blocking buffer (PBS containing 4% FCS) for 1 h. Biotin-labelled, rat anti-human GM-CSF antibody (0.25 μg ml−1, Endogen) diluted in PBS was incubated with samples or human recombinant GM-CSF standards (ranging from 0–1000 pg ml−1, Endogen) for 2 h at room temperature before extensive washing. The signal was generated during a 30 min incubation with poly HRP-streptavidin solution (Endogen) and the substrate, 3,3′,5,5′-Tetramethylbenzidine (BD PharMingen, San Diego, CA, U.S.A.) according to methods supplied by the manufacturer (Endogen) and absorbance was determined at 450 nm (Victor 1420 Multilabel Counter, Wallac).
Preparation of PPACK-treated thrombin
The preparation of D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone (PPACK)-treated thrombin was based on a modification of a previously described method (Greco et al., 1990). Briefly, thrombin was incubated with 10-fold molar excess of PPACK (Biomol, PA, U.S.A.) for 1 h at room temperature. Excess unbound PPACK (3 ml) was dialyzed against 0.38 M NaCl, 0.36 mM NaPO4, pH 7.4 for 48 h with four changes of fresh buffer (250 ml). As a control, thrombin was incubated with PBS and dialyzed under the same conditions (treatment condition labelled as sham-thrombin). To determine whether all of the thrombin had reacted with PPACK, the preparation was tested for its fibrinogen clotting activity and in a fluorogenic thrombin serine protease activity assay (see below).
Measurement of protease activity using fibrinogen clotting assay and fluorogenic assay
The level of thrombin protease inhibition produced by PPACK, SDZ-217-766 (gift from Sandoz) and hirudin (Sigma) was determined by measuring effects on both fibrinogen clotting activity and fluorogenic activity.
Fibrinogen clotting assay
In a 96-well plate, 20 μL of thrombin (0.3 U ml−1 final concentration) treated with PPACK, SDZ-217-766 (0.15 or 1.5 μM), or hirudin (0.3 U ml−1) was added to 180 μL fibrinogen (4 mg ml−1, Sigma) in PBS. Changes in opacity (resulting from thrombin catalytic activity) at 560 nm were measured after a 5 min incubation using a microplate reader (Victor 1420 Multilable Counter, Wallac), and absorbance was compared against a thrombin (0.003–3 U ml−1) concentration-response curve.
Fluorogenic assay
140 μL PBS was added to 96-well plate. 20 μL of PPACK treated-thrombin (0.3 U ml−1), SDZ-217-766 treated-thrombin (0.3 U ml−1) or hirudin treated-thrombin (0.3 U ml−1) was added to the appropriate wells. A further 20 μL PBS was added to all wells. To start the reaction, 20 μL of the fluorogenic thrombin substrate III (0.1 mM, Benzoyl-Phe-Val-Arg-AMC, diluted in 100% dimethylsulphoxide, DMSO, Calbiochem, San Diego, U.S.A.) was added (total volume of 200 μL). The plate was incubated at 37°C in 5% CO2 for 48 h. Hydrolysis of the substrate by thrombin was monitored following the change in absorbance (355–460 nm) using a microplate reader (Victor 1420 Multilable Counter, Wallac). We confirmed that PPACK, SDZ 217-766 and hirudin caused more than 96% inhibition of the protease activity of thrombin (data not shown).
Immunoblot analysis
Cells were grown in 6-well plates under the same conditions described for the cell counting. Thrombin or one of PAR1-selective peptides was added to appropriate wells for 6 h (ERK1/2) or 20 h (Cyclin D1). These incubation times have been shown in our laboratory to be appropriate for the measurement of ERK phosphorylation and cyclin D1 in relation to cell-cycle progression (Ravenhall et al., 2000; Stewart et al., 1999). Following the incubation, the cells were washed twice with ice-cold PBS and lysed for 20 min on ice with 300 μL lysis buffer (containing 100 mM NaCl, 10 mM Tris-HCL, pH 7.5; 2 mM EDTA, 0.5% w v−1 deoxycholate, 1% v v−1 triton-X 100, 1 mM phenylmethylsulphonyl fluoride, 10 mM MgCl2, 100 IU ml−1 aprotinin, 100 μM orthovanadate). The cells were scraped, transferred to 1.5 ml plastic tubes, centrifuged at 760×g for 5 min, and the supernatant collected and stored at −20°C. Aliquots (7 μL) were taken from the supernatants and protein content was determined using Bio-Rad protein assay (BioRad, New South Wales, Australia). The samples were then denatured by boiling for 4 min in a ratio of 1 : 1 with loading buffer (62.5 nM Tris-HCl, pH 6.8, 20% glycerol, 2% sodium dodecylsulphate, SDS). Samples (60 μg protein per lane) were separated electrophoretically on a 12% SDS-polyacrylamide gel and proteins were transferred onto nitrocellulose membranes (Hi-Bond C, Amersham, Cardiff, U.K.) for Western blotting. Membranes were blocked with 5% (w v−1) skim milk in TBS-T (cyclin D1) or PBS (ERK1/2) for 1 h at room temperature, and then incubated overnight at 4°C with ERK1/2 antibody (rabbit polyclonal IgG, 1 : 1000, New England Biolabs Inc., Beverly, MA, U.S.A.) or cyclin D1 antibody (rabbit polyclonal antibody IgG, 1 : 1000, Upstate Biotechnology, Lake Placid, NY, U.S.A.). The membranes were then incubated for 1 h at room temperature with the secondary antibody (horseradish peroxide (HRP) conjugated anti-rabbit IgG, 1 : 1000 dilution, Silenus laboratories, Victoria, Australia; 1 : 2000 dilution, New England Biolabs Inc., respectively) and visualized with an enhanced chemiluminescence kit (Amersham). Scanning and quantification of the relative levels of phosphorylated ERK1/2 and cyclin D1 was performed using the Kodak image station 440CF (Perkin Elmer Wallac) equipped with Kodak ID image analysis software (Eastman Kodak company, Rochester, NY, U.S.A.).
Measurement of intracellular Ca2+ levels
Glass coverslips were sterilized in 70% (v v−1) ethanol and then rinsed with DMEM before being placed on the bottom of the 6-well culture plates. The cells were seeded as described in the cell culture method section. Once confluent, the medium was replaced with DMEM containing 1 μM of the calcium-sensitive dye, FURA-2 acetoxy methyl ester (FURA-2AM, Molecular Probes, Eugene, U.S.A.) as described in our previous studies (Stewart et al., 1994; Tomlinson et al., 1995). Briefly, the cells were left to incubate at room temperature in a shaker apparatus (60 strokes min−1) for 40 min. The medium was replaced with Tyrode buffer (pH 7.4, Sigma) containing 0.25% (w v−1) BSA (Sigma) and incubated for 15 min at 37°C. The measurement of intracellular calcium was carried out using an F-2000 fluorescence spectrophotometer (Hitachi, Japan) equipped with filters, magnetic stirring, and heating (37°C). The emission wavelength was set at 510 nm and the excitation wavelengths alternated at 0.5 s intervals between 340 and 380 nm. Basal and agonist-stimulated elevations in intracellular calcium were assessed from the fluorescence ratio (340 and 380 nm) and have been expressed as a percentage of the response to the initial agonist.
The compounds added to the cells were prepared and warmed to 37°C. Drug additions were made approximately 50 s after placement of the slide in the cuvette. For desensitization experiments, the agonists were added sequentially (without any separate washes between treatments) when calcium levels returned to baseline or to a new steady state level. Endothelin-1 (100 nM) was added at the end of the protocol to ascertain responsiveness to a ligand with no relationship to PAR or thrombin mechanisms.
Immunohistochemistry
Immunohistochemistry was carried out using methods based on a modification of those previously described (Stewart et al., 1997). Cells were cultured into 8-well glass chamber slides and allowed to grow to confluence. After washing in PBS, the cells were fixed in ice-cold acetone for 2 min, cells were incubated for 20 min at room temperature with 1.5% normal horse serum (NHS, using a Vectastain Elite ABC kit) to prevent non-specific secondary antibody binding, then incubated with the primary mouse monoclonal antibody for human PAR1 antibody (ATAP2, 5 μg ml−1; obtained from initial investigation of optimal PAR1 antibody dilution) or isotype control, mouse IgG1 (Dako, Copenhagen, Denmark); overnight at room temperature. After two 5 min washes in PBS, the cells were exposed to a horse Biotinylated anti-mouse IgG secondary antibody (diluted in 1.5% v v−1 NHS) for 45 min, at room temperature. Endogenous peroxidase activity was blocked by immersion in methanol containing 0.12% (v v−1) hydrogen peroxide for 10 min. Following washing in PBS, cells were incubated for 45 min at room temperature with the avidin-biotin complex (Vectastain Elite ABC kit; 0.5% final; diluted in 1.5% NHS). Immunoreactivity was visualized by addition of the metal enhanced diaminobenzidine (Immunopure® Metal Enhanced DAB substrate kit). All slides were counter-stained with haemotoxylin, and observed by light microscopy. The cellular composition of the HASM cultures were also concurrently examined using primary mouse monoclonal antibodies to smooth muscle α-actin (Dako; 1 : 25 dilution in PBS containing 0.25% BSA), or smooth muscle myosin (1 : 1000, Dako) as in previous studies (Stewart et al., 1997).
Drugs
The PAR peptides (SFLLRN, SLIGKV and GYPGQV) were synthesized with carboxy-terminal amidation and purified to ⩾95% via HPLC by Auspep (Victoria, Australia). Stock solutions of SDZ 217-766 and thrombin fluorogenic substrate III were prepared in 100% v v−1 DMSO. The highest concentration of vehicle DMSO (0.01%), was well below the threshold concentration for influencing HASM DNA synthesis (0.3%) (Fernandes et al., 2000). Stock solutions of thrombin (Sigma) were prepared in 0.25% BSA in PBS. All other drugs were prepared in distilled water. Intermediate dilutions of stock solutions were made in 0.25% BSA-containing DMEM.
Statistical analysis of results
Supernatants collected for GM-CSF assay were assayed in duplicate. All results are expressed as mean±standard error of mean (mean±s.e.mean); n represents the number of different cell cultures obtained from different donors. Differences were determined by one-way analysis of variance (ANOVA) with repeated measures, followed by post-hoc Bonferroni test, where appropriate. Data for levels of GM-CSF are expressed as fmol per 106 cells. Cell number was expressed as fold increment over basal. Graphpad Prism for Windows (version 3) was used for all of the statistical analyses. Differences were considered to be significant when the probability was less than 0.05 (P<0.05).
Results
Thrombin concentration-dependently stimulated increases in the levels of GM-CSF
Incubation of quiescent ASM with increasing concentrations of thrombin (0.003–3 U ml−1) for 48 h caused a marked increase in the levels of GM-CSF (Figure 1a) over the basal level of GM-CSF that was close to or below the detection limit of the assay (<1 fmol). The concentration of thrombin (0.3 U ml−1) that produced maximum levels of GM-CSF and increases in cell number (Figure 1b) was used throughout the study.
Figure 1.
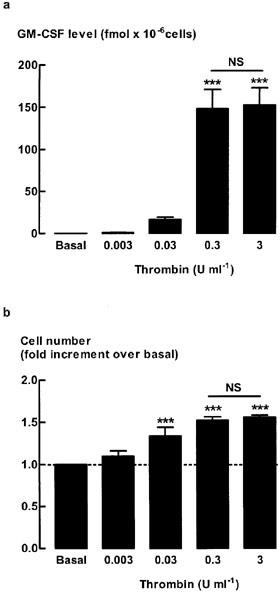
Effect of increasing concentrations of thrombin on airway smooth muscle (a) GM-CSF levels and (b) cell number. Quiescent cells were stimulated for 48 h with increasing concentrations of thrombin (0.003–3 U ml−1). Data represent the mean and s.e.mean of the level determined from four different cell cultures. GM-CSF level are expressed as fmol per 106 cells and the basal levels of GM-CSF were below the detection limit of the assay (<1 fmol). Cell number data are expressed as fold increment over the number of unstimulated cells (basal). ***P<0.001 compared with basal. NS, not significant 0.3 U ml−1 compared with 3 U ml−1. The average number of unstimulated cells at the end of the 48 h incubation was 1.64±0.3×105 cells per well.
Effect of thrombin proteolytic activity on increases in GM-CSF levels and cell proliferation
To examine whether proteolytic activity of thrombin was required for thrombin-stimulated increases in GM-CSF levels and cell number, three types of thrombin inhibitors were used: PPACK; SDZ 217-766; and hirudin (Tapparelli et al., 1993). Inhibition of thrombin protease activity by pretreatment with PPACK reduced the level of GM-CSF to close to the detection limit of the assay, whereas mitogenesis was reduced by approximately 70% (Figure 2). Pre-incubation with SDZ 217-766 (0.15 μM), which inhibited thrombin (0.3 U ml−1)-stimulated increases in DNA synthesis (Fernandes & Stewart, unpublished observations), significantly reduced thrombin-stimulated increases in GM-CSF levels by 73±9%, and mitogenesis by 66±8% (Figure 3). SDZ 217-766 alone had no effect on unstimulated cell number or GM-CSF levels (data not shown). There was no further inhibition of either the GM-CSF level or cell number following a 10 fold higher concentration of SDZ 217-766 (1.5 μM, data not shown). Increases in both GM-CSF levels and cell number were inhibited by hirudin (65±12% and 63±11% inhibition, respectively, Figure 3).
Figure 2.
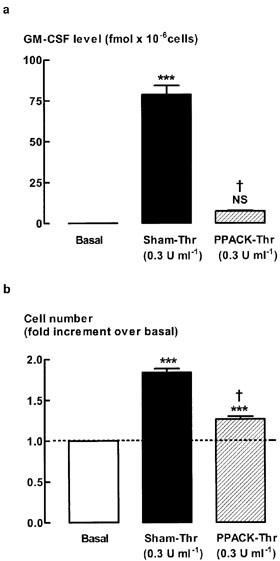
Effect of PPACK-treated thrombin on (a) GM-CSF levels and (b) cell number. Quiescent human ASM cells were stimulated for 48 h with either thrombin (0.3 U ml−1) or PPACK-treated thrombin (0.3 U ml−1). ***P<0.001, NS=not significant compared with basal, †P<0.001 compared with Sham-Thr, n=8. The average number of unstimulated cells (basal) at the end of the 48 h stimulation was 2.1±0.2×105 cells per well.
Figure 3.
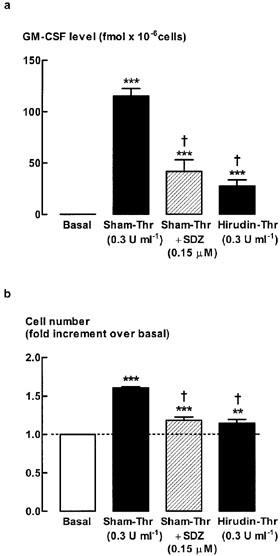
Effect of SDZ 217-766 or hirudin on (a) GM-CSF levels and (b) cell number. Quiescent cells were stimulated for 48 h with thrombin (0.3 U ml−1) in the presence or absence of the thrombin protease inhibitor, SDZ 217-766 (0.15 μM, was added 30 min prior to stimulation with thrombin). Thrombin was incubated for 30 min with hirudin (1 : 1) separately prior to being added to the cells. Sham-treated thrombin (Sham-Thr) was subjected to the same protocol except that hirudin was omitted from the incubation. ***P<0.001, **P<0.01 compared with basal, n=6-8. †P<0.05, compared with sham-Thr. The average number of unstimulated cells (basal) at the end of the 48 h incubation was 1.7±0.3×105 cells per well.
Effect of PAR-selective peptides on GM-CSF level
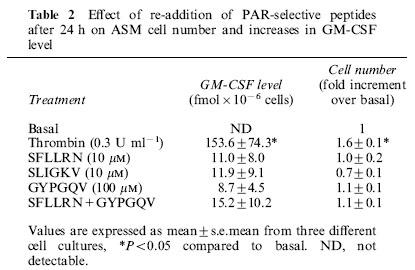
To determine whether the thrombin-stimulated increase in GM-CSF level was mediated via PARs in human ASM, peptides selective for PAR1 (SFLLRN), PAR2 (SLIGKV) or PAR4 (GYPGQV) were incubated with ASM for 48 h, after which time the level of GM-CSF was determined (Figure 4a). The concentrations of PAR ligands are based on those previously determined to be maximally effective in several different cell types (Hollenberg, 1999). None of the PAR-selective peptides alone mimicked the effect of thrombin-stimulated increases in GM-CSF level. Given that PAR4 is also activated by thrombin (Hollenberg, 1999), it was possible that both PAR1 and PAR4 could be required to increase GM-CSF levels. Incubation of ASM with peptides selective for PAR1 and PAR4 in combination did not significantly (P>0.05) increase GM-CSF levels (Figure 4a). Higher concentrations of each of the PAR-selective peptides were also examined (Table 1), but no significant responses were observed. To account for the possible degradation of the PAR-selective peptides over the 48 h incubation period, PAR-selective peptides were also re-added after 24 h (Table 2). The level of GM-CSF remained indistinguishable from the detection limit of the assay (<1 fmol).
Figure 4.
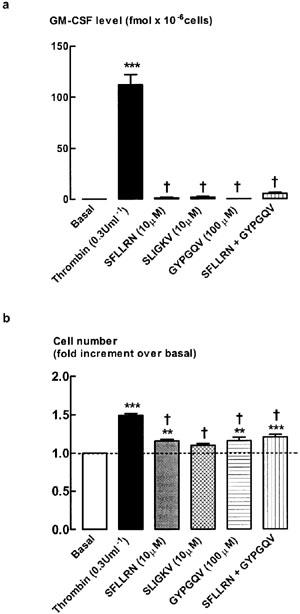
Effect of PAR-selective peptides on (a) GM-CSF levels and (b) cell number. Quiescent cells were stimulated for 48 h with either thrombin (0.3 U ml−1) or one of the PAR-selective peptides (SFLLRN, PAR1, 10 μM; SLIGKV, PAR2, 10 μM; GYPGQV, PAR4, 100 μM). **P<0.01 and ***P<0.001 compared with basal, n=8. †P<0.05, compared with Thr. The average number of unstimulated cells (basal) at the end of the 48 h incubation was 2.0±0.3×105 cells per well.
Table 1.
Effect of higher concentrations of PAR-selective peptides on ASM cell number and increases in GM-CSF level
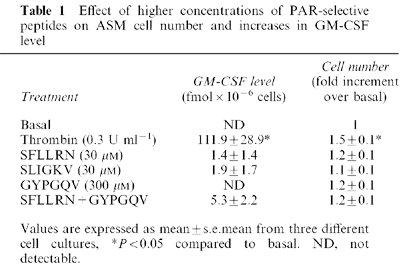
Table 2.
Effect of re-addition of PAR-selective peptides after 24 h on ASM cell number and increases in GM-CSF level
Effect of PAR-selective peptides on cell number
PAR1- and PAR4-selective peptides alone or in combination caused a modest increase in cell number (Figure 4b). However, these responses were significantly less than those to thrombin. No increase in cell number was observed following stimulation with the PAR2-selective peptide. Furthermore, the effects of the PAR2-selective peptide on increases in cell number following the addition of both PAR1- and PAR4-selective peptides were neither synergistic nor additive.
Effect of pertussis toxin on thrombin-stimulated increases in GM-CSF level and cell number
To examine the potential role of G proteins on thrombin-stimulated increase in GM-CSF and cell number, cells were stimulated with thrombin in the presence or absence of the Giα inhibitor, pertussis toxin (Figure 5). Thrombin-stimulated increases in GM-CSF level were attenuated in the presence of pertussis toxin (54±12% inhibition), whereas cell number increases were completely inhibited (Figure 5). To ascertain whether the effects of pertussis toxin were selective for the thrombin responses, its effects on basic Fibroblast Growth Factor (bFGF) were investigated. bFGF (300 pM)-stimulated increases in cell number were not significantly (P>0.05) altered by pertussis toxin pretreatment (bFGF 1.85±0.14 increase in cell number; bFGF+pertussis toxin 1.64±0.11; n=8). Similarly, bFGF-induced increases in GM-CSF levels were also unaffected (P>0.05) by pertussis toxin pre-treatment (bFGF 52.7±13.3 fmol×10−6 cells; bFGF+pertussis toxin, 38.4±9.1, n=8).
Figure 5.
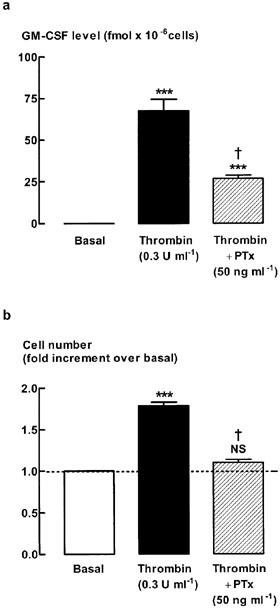
Effect of pertussis toxin (PTx) on (a) GM-CSF level and (b) cell number. Quiescent cells were stimulated for 48 h with thrombin (0.3 U ml−1) in the presence or absence of the Giα inhibitor, pertussis toxin (50 ng ml−1). Pertussis toxin was added 30 min prior to stimulation with thrombin. ***P<0.001, NS=not significant compared with basal, n=8. †P<0.05 compared with Thr. The number of cells (basal) at the end of the 48 h incubation in the absence of mitogen was 1.5±0.3×105 cells per well.
ERK and cyclin D1 protein expression
We examined the effects of thrombin and PAR1 on two important signalling proteins involved in cell proliferation, ERK1/2 phosphorylation and cyclin D1 levels (Figure 6). Previous studies have established that although ERK1/2 phosphorylation is increased up to 20 h after addition of mitogen, the addition of the ERK inhibitor, PD98059 has no effect when added 8 h or more after mitogen, indicating that during the first 8 h of exposure to mitogen ERK1/2 activity is essential to cell cycle progression (Fernandes et al., 1999; Fernandes & Stewart, unpublished observations). We therefore chose the 6 h time point for evaluation of ERK1/2 phosphorylation in the present study. Previous time-course studies of the increases in expression of cyclin D1 indicate that protein expression is increased at 20 h and is accompanied by phosphorylation of retinoblastoma protein (Stewart et al., 1997), indicating that this is a suitable time point to evaluate cyclin D1 protein levels. As expected, both ERK1/2 phosphorylation and cyclin D1 protein levels significantly increased following a 6 or 20 h stimulation with thrombin (0.3 U ml−1), respectively, as compared to basal levels. In contrast, addition of PAR1-selective peptide for 6 or 20 h was without effect on levels of phosphorylated ERK1/2 or cyclin D1 protein.
Figure 6.
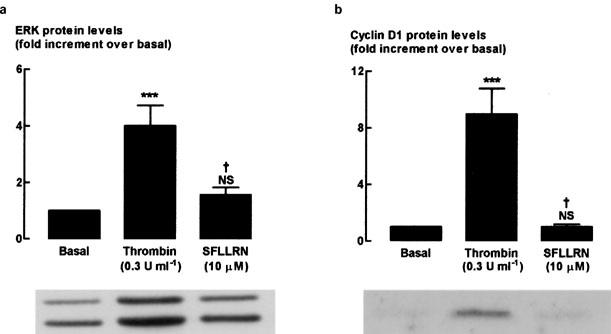
Western blot analysis showing the effect of PAR1-selective peptide on (a) phosphorylated ERK1/2 and (b) cyclin D1 protein expression. Cells were stimulated for 6 or 20 h respectively with 0.3 U ml−1 thrombin or 10 μM PAR1-selective peptide, SFLLRN. Grouped data represent results obtained from five different cultures. †P<0.05, compared with Thr. ***P<0.001, NS=not significant compared with unstimulated cells (basal).
PAR1 receptor expression by HASM
To verify the presence of PAR1 receptors on HASM, immunohistochemical localization of PAR-1 was carried out (Figure 7) and functional effects of the PAR1 on intracellular Ca2+ levels were measured (Figure 8). Specific immunoreactivity for PAR1 was observed in HASM (Figure 7). PAR1 expression was predominantly cytoplasmic and perinuclear. As expected, thrombin (0.3 U ml−1) induced a significant increase in intracellular calcium level with an increment in fluorescence ratio (340 and 380 nm) of approximately 0.5 units. Calcium levels were maximal within 10 s and declined to basal or steady state levels within 1 min. Stimulation with PAR1-selective peptide (10 μM) also increased calcium levels (Figure 8), confirming that the peptide was biologically active. To confirm that PAR1 was present on HASM, desensitisation studies were carried out. Re-exposure to the same agonist, SFLLRN or thrombin, resulted in a reduction in the increase in calcium level (Figure 8). Stimulation with PAR1-selective peptide following pre-exposure to thrombin attenuated the increase in the level of calcium by 70±19 and 100% desensitization was observed when the order of ligand addition was reversed. Endothelin-1 was added at the end of all desensitization experiments to ensure that the desensitization responses were related to thrombin and PARs and not due to depletion of the releasable calcium pool. Endothelin-1-stimulated increases in intracellular calcium levels were intact.
Figure 7.
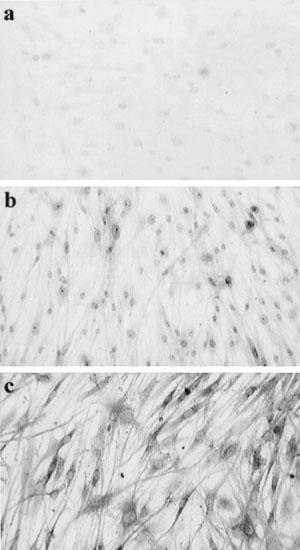
Localization of PAR1 immunoreactivity. (a) Isotype control, (b) smooth muscle α-actin, (c) PAR1.
Figure 8.
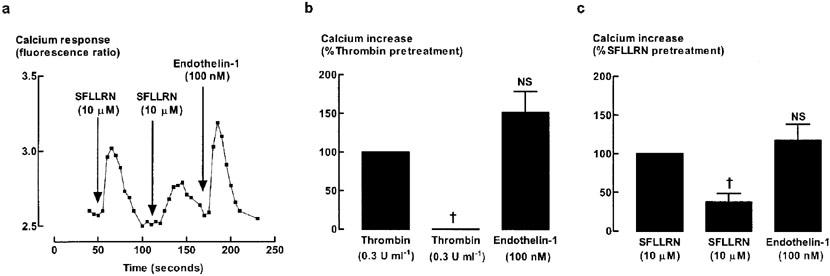
Desensitisation of PAR1-stimulated increase in intracellular Ca2+ levels. Cells were exposed to the indicated agonists at intervals of 1 min without intervening washes. (a) A trace representative of four different cell culture preparations. Grouped data (n=4) showing desensitization of (b) thrombin- or (c) SFLLRN-induced increase in calcium levels. NS, not significant, †P<0.05, compared with original response.
Discussion
The two major findings in this study were that thrombin: (i) increased levels of GM-CSF over the same concentration range that stimulated proliferation; (ii) effects on GM-CSF levels and mitogenesis appear to be independent of known PARs. Our data suggest the involvement of novel proteolytic receptor target(s) for thrombin-induced cytokine production and mitogenesis.
Thrombin-stimulated increases in GM-CSF levels were completely dependent on proteolytic activity, consistent with a PAR-mediated mechanism. However, none of the PAR-selective peptides that were examined in this study increased GM-CSF levels. The trivial mitogenic responses induced by PAR-selective peptides by comparison with those to thrombin also suggest a role for non-PAR mechanisms in the mitogenic actions of thrombin. The lack of activity of peptides cannot be ascribed to metabolism, as neither re-addition after 24 h incubation, nor a 3 fold increase in concentration increased either the GM-CSF or mitogenic responses. The PAR2 peptide has been shown to increase [3H]-thymidine uptake at the same concentration used in the present study (Chambers et al., 2001). PAR1 receptors were expressed in HASM as evidenced by immunohistochemical localization and by intracellular Ca2+ responses. These receptors were functionally active and showed cross-desensitization with thrombin. However, they were not linked to the persistent activation of ERK and increases in cyclin D1 levels that are known to be essential for the HASM proliferative response (Ravenhall et al., 2000). In rat cultured microglial cells, the PAR1-selective peptide also failed to mimic thrombin-stimulated increases in NF-κB activation (Ryu et al., 2000), a transcription factor that is essential for regulation of GM-CSF expression (Cakouros et al., 2001).
Although PAR2 is also present in HASM (Knight et al., 2001; Schmidlin et al., 2001), PAR2-selective peptides failed to increase cell proliferation (present study) and evoked only modest increases in thymidine incorporation (Chambers et al., 2001). Thus, thrombin-induced cell proliferation is not likely to be mediated by PAR2 activation. Our studies do not directly exclude a role for PAR3, as PAR3 may function as a cofactor for PAR4 (Macfarlane et al., 2001). Nevertheless, as PAR3 is thought to function as a co-factor for the activation of PAR4 and the PAR4 peptide was inactive, it seems unlikely that the effects of thrombin could be mediated by PAR3, which in any case is not a high affinity substrate for thrombin (Ayala et al., 2001). Clearly, PAR-selective antagonists would be useful in studying the roles and interactions of these PARs. However, there are still problems with the selectivity and potency of current PAR antagonists (Macfarlane et al., 2001). Current substituted peptide compounds have been shown to inhibit PAR1-selective peptide stimulation, but have not been shown to be effective against thrombin stimulation (Macfarlane et al., 2001).
The difference between thrombin and PAR peptide responses may be explained by the low efficiency of presentation or binding of the selective peptides as compared to the natural tethered ligand produced from thrombin cleavage (Chambers et al., 2001). Thus, a relatively high concentration of the free PAR-selective peptide required to cause receptor activation may be necessary to compensate for the lack of covalent tethering in the vicinity of the active site of the PAR. However, the contrast between the PAR peptides and thrombin as ligands is unlikely to explain our findings, as these peptide concentrations were sufficient to stimulate increases in intracellular calcium. Moreover, concentrations as high as 30 μM (SFLLRN and SLIGKV) or 300 μM (GYPQV) failed to elicit significant cytokine or mitogenic responses, whereas one-third these concentrations elevated intracellular Ca2+ and showed cross-desensitization with thrombin. The use of concentrations higher than those in the current study would be inappropriate, since there would be a high likelihood of unspecific action. In addition, this would take the concentration range well outside that which has been required in other bioassays for the peptides to mimic the actions of thrombin or other serine proteases (e.g. Hauck et al., 1999).
Our observations suggest that there are additional ‘receptors' that mediate thrombin-induced mitogenesis and GM-CSF production. The bradykinin (BK) receptor can also be activated by proteases such as trypsin in CHO, HEK 293 and MDCK cells, suggesting that the BK receptor may belong to a new group of serine protease-activated receptors (Hecquet et al., 2000). However, BK lacks mitogenic actions in HASM (Ammit et al., 1999) making BK receptor activation by thrombin an untenable explanation of the current findings. The GPIbα subunit of GPIb-IX-V, which has a well-characterized role in von Willebrand factor-mediated platelet adhesion, also has affinity for thrombin and may initiate transmembrane signalling itself, or may serve as a cofactor for PAR1 activation by thrombin or other surface proteins (de candia et al., 2001). GPIbα is highly expressed on megakaryocytes, platelets and endothelial cells (Wu et al., 1997), but its expression on HASM is unknown.
Serine proteases such as trypsin, tryptase, and the house dust mite protease allergens, Der p3 and Der p 9 increase GM-CSF levels in human keratinocytes and epithelial cells, suggesting a PAR2-mediated mechanism of action (Sun et al., 2001; Vliagoftis et al., 2001; Wakita et al., 1997). Although the mitogenic actions of thrombin in HASM are well established, this is the first study to show that thrombin is a potent stimulator of increases in GM-CSF levels. Thrombin-stimulated increases in GM-CSF levels were markedly reduced by pertussis toxin, suggesting that Giα proteins are implicated in the non-PAR signalling. As GM-CSF has both pro-inflammatory and fibrogenic actions that can contribute to AWR, HASM production of GM-CSF in response to growth factors, in addition to established stimuli, such as TNFα and IL-1α, may be important in perpetuating inflammation and remodelling in asthma.
The PAR-independence of thrombin-stimulated increase in GM-CSF level and HASM mitogenesis is in accordance with a recent study showing that the tryptase-stimulated increase in HASM proliferation was mediated via non-PAR mechanisms (Brown et al., 2002). The thrombin-induced mitogenesis is partly independent of its enzymatic activity, as thrombin-stimulated increases in cell number were not fully inhibited by concentrations of hirudin, PPACK or SDZ 217-766 that completely prevented the action of thrombin on fibrinogen and on a thrombin fluorogenic substrate. Nevertheless, the residual effect of thrombin in the absence of proteolytic activity is modest (∼30%). Mannose receptors may mediate the non-proteolytic action of tryptase in stimulating mitogenesis as tryptase contains several glycosylated residues on its surface (Brown et al., 2002). In addition, various mannosyl-rich glycoproteins are mitogenic for bovine ASM (Lew et al., 1994). Thrombin also has a site that may be glycosylated (Grand et al., 1996).
Our findings indicate that attempts to therapeutically target the contribution of thrombin to chronic inflammation and tissue remodelling should consider inhibition of serine protease activity as a primary strategy, since there appear to be multiple defined and undefined protease activity-dependent ‘receptors' for thrombin.
Acknowledgments
The authors thank Ms Trudi Harris for her expertise in cell culturing and the late Dr Carlo Tapparelli (Sandoz, Basle) for a gift of SDZ 217-766. This project was supported by the National Health and Medical Research Council of Australia. We also thank A/Prof John Wilson and Dr Trevor Williams (Respiratory Medicine) and the Anatomical pathology Department, Alfred Hospital for provision of human airway specimens; Dr Tom Cocks for gift of ATAP2 and Ms Tiffany Bamford (Department of Pharmacology) for assistance with the immunohistochemistry.
Abbreviations
- ASM
airway smooth muscle
- ANOVA
analysis of variance
- DMEM
Dulbecco's modified Eagle's medium
- ERK1/2
extracellular signal-regulated kinase 1/2
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- GPCR
G-protein-coupled receptors
- PARs
protease-activated receptors
- PBS
phosphate-buffered saline
- PPACK
D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone
- PTx
pertussis toxin
References
- AMMIT A.J., KANE S.A., PANETTIERI R.A., JR Activation of K-p21ras and N-p21ras, but not H-p21ras, is necessary for mitogen-induced human airway smooth-muscle proliferation. Am. J. Respir. Cell Mol. Biol. 1999;21:719–727. doi: 10.1165/ajrcmb.21.6.3731. [DOI] [PubMed] [Google Scholar]
- AYALA Y.M., CANTWELL A.M., ROSE T., BUSH L.A., AROSIO D., DI CERA E. Molecular mapping of thrombin-receptor interactions. Proteins. 2001;45:107–116. doi: 10.1002/prot.1130. [DOI] [PubMed] [Google Scholar]
- BAR-SHAVIT R., BENEZRA M., ELDOR A., HY-AM E., FENTON J.W., 2ND, WILNER G.D., VLODAVSKY I. Thrombin immobilized to extracellular matrix is a potent mitogen for vascular smooth muscle cells: nonenzymatic mode of action. Cell Regul. 1990;1:453–463. doi: 10.1091/mbc.1.6.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAR-SHAVIT R., KAHN A., WILNER G.D., FENTON J.W., 2ND Monocyte chemotaxis: stimulation by specific exosite region in thrombin. Science. 1983;220:728–731. doi: 10.1126/science.6836310. [DOI] [PubMed] [Google Scholar]
- BAR-SHAVIT R., KAHN A.J., MANN K.G., WILNER G.D. Identification of a thrombin sequence with growth factor activity on macrophages. Proc. Natl. Acad. Sci. U.S.A. 1986;83:976–980. doi: 10.1073/pnas.83.4.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRETSCHNEIDER E., WITTPOTH M., WEBER A.A., GLUSA E., SCHROR K. Thrombin but not thrombin receptor activating peptide is mitogenic for coronary artery smooth muscle cells. Thromb. Res. 1997;87:493–497. doi: 10.1016/s0049-3848(97)00164-3. [DOI] [PubMed] [Google Scholar]
- BROWN J.K., JONES C.A., ROONEY L.A., CAUGHEY G.H., HALL I.P. Tryptase's potent mitogenic effects in human airway smooth muscle cells are via nonproteolytic actions. Am. J. Physiol. 2002;282:L197–L206. doi: 10.1152/ajplung.2002.282.2.L197. [DOI] [PubMed] [Google Scholar]
- CAKOUROS D., COCKERILL P.N., BERT A.G., MITAL R., ROBERTS D.C., SHANNON M.F. A NF-kB/Sp1 region is essential for chromatin remodelling and correct transcription of a human granulocyte colony-stimulating factor transgene. J. Immunol. 2001;167:302–310. doi: 10.4049/jimmunol.167.1.302. [DOI] [PubMed] [Google Scholar]
- CHAMBERS L.S., BLACK J.L., PORONNIK P., JOHNSON P.R. Functional effects of protease-activated receptor-2 stimulation on human airway smooth muscle. Am. J. Physiol. 2001;281:L1369–L1378. doi: 10.1152/ajplung.2001.281.6.L1369. [DOI] [PubMed] [Google Scholar]
- DE CANDIA E., HALL S.W., RUTELLA S., LANDOLFI R., ANDREWS R.K., DE CRISTOFARO R. Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J. Biol. Chem. 2001;276:4692–4698. doi: 10.1074/jbc.M008160200. [DOI] [PubMed] [Google Scholar]
- EBINA M., TAKAHASHI T., CHIBA T., MOTOMIYA M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma: a 3-D morphometric study. Am. Rev. Respir. Dis. 1993;148:720–726. doi: 10.1164/ajrccm/148.3.720. [DOI] [PubMed] [Google Scholar]
- FERNANDES D., GUIDA E., KOUTSOUBOS V., HARRIS T., VADIVELOO P., WILSON J.W., STEWART A.G. Glucocorticoids inhibit proliferation, cyclin D1 expression, and retinoblastoma protein phosphorylation, but not activity of the extracellular-regulated kinases in human cultured airway smooth muscle. Am. J. Respir. Cell Mol. Biol. 1999;21:77–88. doi: 10.1165/ajrcmb.21.1.3396. [DOI] [PubMed] [Google Scholar]
- FERNANDES D., VLAHOS R., STEWART A.G. Thrombin-stimulated DNA synthesis in human cultured airway smooth muscle occurs independently of products of cyclo-oxygenase or 5-lipoxygenase. Pulm. Pharmacol. Ther. 2000;13:241–248. doi: 10.1006/pupt.2000.0251. [DOI] [PubMed] [Google Scholar]
- GABAZZA E.C., TAGUCHI O., TAMAKI S., TAKEYA H., KOBAYASHI H., YASUI H., KOBAYASHI T., HATAJI O., URANO H., ZHOU H., SUZUKI K., ADACHI Y. Thrombin in the airways of asthmatic patients. Lung. 1999;177:253–262. doi: 10.1007/pl00007645. [DOI] [PubMed] [Google Scholar]
- GRAND R.J., TURNELL A.S., GRABHAM P.W. Cellular consequences of thrombin-receptor activation. Biochem. J. 1996;313:353–368. doi: 10.1042/bj3130353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRECO N.J., TENNER T.E., JR, TANDON N.N., JAMIESON G.A. PPACK-thrombin inhibits thrombin-induced platelet aggregation and cytoplasmic acidification but does not inhibit platelet shape change. Blood. 1990;75:1989–1990. [PubMed] [Google Scholar]
- HAUCK R.W., SCHULZ C., SCHOMIG A., HOFFMAN R.K., PANETTIERI R.A., JR alpha-Thrombin stimulates contraction of human bronchial rings by activation of protease-activated receptors. Am. J. Physiol. 1999;277:L22–L29. doi: 10.1152/ajplung.1999.277.1.L22. [DOI] [PubMed] [Google Scholar]
- HECQUET C., TAN F., MARCIC B.M., ERDOS E.G. Human bradykinin B(2) receptor is activated by kallikrein and other seine proteases. Mol. Pharmacol. 2000;58:828–836. doi: 10.1124/mol.58.4.828. [DOI] [PubMed] [Google Scholar]
- HERBERT J.M., DUPUY E., LAPLACE M.C., ZINI J.M., BAR SHAVIT R., TOBELEM G. Thrombin induces endothelial cell growth via both a proteolytic and a non-proteolytic pathway. Biochem. J. 1994;303:227–231. doi: 10.1042/bj3030227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERBERT J.M., LAMARCHE I., DOL F. Induction of vascular smooth muscle cell growth by selective activation of the thrombin receptor. Effect of heparin. FEBS Lett. 1992;301:155–158. doi: 10.1016/0014-5793(92)81237-g. [DOI] [PubMed] [Google Scholar]
- HOLLENBERG M.D. Protease-activated receptors: PAR4 and counting: how long is the course. Trends Pharmacol. Sci. 1999;20:271–273. doi: 10.1016/s0165-6147(99)01333-4. [DOI] [PubMed] [Google Scholar]
- KNIGHT D.A., LIM S., SCAFFIDI A.K., ROCHE N., CHUNG K.F., STEWART G.A., THOMPSON P.J. Protease-activated receptors in human airways: upregulation of PAR-2 in respiratory epithelium from patients with asthma. J. Allergy Clin. Immunol. 2001;108:797–803. doi: 10.1067/mai.2001.119025. [DOI] [PubMed] [Google Scholar]
- LEW D.B., SONGU-MIZE E., PONTOW S.E., STAHL P.D., RATTAZZI M.C. A mannose receptor mediates mannosyl-rich glycoprotein-induced mitogenesis in bovine airway smooth muscle cells. J. Clin. Invest. 1994;94:1855–1863. doi: 10.1172/JCI117535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOPEZ A.F., WILLIAMSON D.J., GAMBLE J.R., BEGLEY C.G., HARLAN J.M., KLEBANOFF S.J., WALTERSDORPH A., WONG G., CLARK S.C., VADAS M.A. Recombinant human granulocyte-macrophage colony-stimulating factor stimulates in vitro mature human neutrophil and eosinophil function, surface receptor expression, and survival. J. Clin. Invest. 1986;78:1220–1228. doi: 10.1172/JCI112705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACFARLANE S.R., SEATTER M.J., KANKE T., HUNTER G.D., PLEVIN R. Proteinase-activated receptors. Pharmacol. Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- MATTOLI S., MATTOSO V.L., SOLOPERTO M., ALLEGRA L., FASOLI A. Cellular and biochemical characteristics of bronchoalveolar lavage fluid in symptomatic nonallergic asthma. J. Allergy Clin. Immunol. 1991;87:794–802. doi: 10.1016/0091-6749(91)90125-8. [DOI] [PubMed] [Google Scholar]
- PANETTIERI R.A., JR, HALL I.P., MAKI C.S., MURRAY R.K. alpha-Thrombin increases cytosolic calcium and induces human airway smooth muscle cell proliferation. Am. J. Respir. Cell Mol. Biol. 1995;13:205–216. doi: 10.1165/ajrcmb.13.2.7626288. [DOI] [PubMed] [Google Scholar]
- PARK C.S., CHOI Y.S., KI S.Y., MOON S.H., JEONG S.W., UH S.T., KIM Y.H. Granulocyte-macrophage colony-stimulating factor is the main cytokine enhancing survival of eosinophils in asthmatic airways. Eur. Respir. J. 1998;12:872–878. doi: 10.1183/09031936.98.12040872. [DOI] [PubMed] [Google Scholar]
- PLENZ G., REICHENBERG S., KOENIG C., RAUTERBERG J., DENG M.C., BABA H.A., ROBENEK H. Granulocyte-macrophage colony-stimulating factor (GM-CSF) modulates the expression of type VIII collagen mRNA in vascular smooth muscle cells and both are codistributed during atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1999;19:1658–1668. doi: 10.1161/01.atv.19.7.1658. [DOI] [PubMed] [Google Scholar]
- RAVENHALL C.E., GUIDA E., HARRIS T., KOUTSOUBOS V., STEWART A.G. The importance of ERK activity in the regulation of cyclin D1 levels and DNA synthesis in human cultured airway smooth muscle. Br. J. Pharmacol. 2000;131:17–28. doi: 10.1038/sj.bjp.0703454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RYU J., PYO H., JOU I., JOE E. Thrombin induces NO release from cultured rat microglia via protein kinase C, mitogen-activated protein kinase, and NF-kappa B. J. Biol. Chem. 2000;275:29955–29959. doi: 10.1074/jbc.M001220200. [DOI] [PubMed] [Google Scholar]
- SCHMIDLIN F., AMADESI S., VIDIL R., TREVISANI M., MARTINET N., CAUGHEY G., TOGNETTO M., CAVALLESCO G., MAPP C., GEPPETTI P., BUNNETT N.W. Expression and function of proteinase-activated receptor 2 in human bronchial smooth muscle. Am. J. Respir. Crit. Care Med. 2001;164:1276–1281. doi: 10.1164/ajrccm.164.7.2101157. [DOI] [PubMed] [Google Scholar]
- STEWART A.G. Airway wall remodelling and hyperresponsiveness: modelling remodelling in vitro and in vivo. Pulm. Pharmacol. Ther. 2001;14:255–265. doi: 10.1006/pupt.2001.0290. [DOI] [PubMed] [Google Scholar]
- STEWART A.G., GRIGORIADIS G., HARRIS T. Mitogenic actions of endothelin-1 and epidermal growth factor in cultured airway smooth muscle. Clin. Exp. Pharmacol. Physiol. 1994;21:277–285. doi: 10.1111/j.1440-1681.1994.tb02513.x. [DOI] [PubMed] [Google Scholar]
- STEWART A.G., HARRIS T., FERNANDES D.J., SCHACHTE L.C., KALAFATIS V., GILLZAN K.M., RAVENHALL C.E., TOMLINSON P.R.T., WILSON J. β2-adrenergic agonists and cAMP arrest human cultured airway smooth muscle cells in G1 phase of the cell cycle: role of mitogen-activated protein kinase, cyclin D1 and p27Kip1. Molecular Pharmacology. 1999;56:1079–1086. doi: 10.1124/mol.56.5.1079. [DOI] [PubMed] [Google Scholar]
- STEWART A.G., TOMLINSON P.R., WILSON J. Airway wall remodelling in asthma: a novel target for the development of anti-asthma drugs. Trends Pharmacol. Sci. 1993;14:275–279. doi: 10.1016/0165-6147(93)90130-c. [DOI] [PubMed] [Google Scholar]
- STEWART A.G., TOMLINSON P.R., WILSON J.W. Regulation of airway wall remodelling: prospects for the development of novel anti-asthma drugs. Adv. Pharmacol. 1995;33:209–253. doi: 10.1016/s1054-3589(08)60670-5. [DOI] [PubMed] [Google Scholar]
- STEWART A.G., TOMLINSON P.R., WILSON J.W. Beta 2-adrenoceptor agonist-mediated inhibition of human airway smooth muscle cell proliferation: importance of the duration of beta 2-adrenoceptor stimulation. Br. J. Pharmacol. 1997;121:361–368. doi: 10.1038/sj.bjp.0701128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUN G., STACEY M.A., SCHMIDT M., MORI L., MATTOLI S. Interaction of mite allergens Der p3 and Der p9 with protease-activated receptor-2 expressed by lung epithelial cells. J. Immunol. 2001;167:1014–1021. doi: 10.4049/jimmunol.167.2.1014. [DOI] [PubMed] [Google Scholar]
- TAPPARELLI C., METTERNICH R., EHRHARDT C., COOK N.S. Synthetic low-molecular weight thrombin inhibitors: molecular design and pharmacological profile. Trends Pharmacol. Sci. 1993;14:366–376. doi: 10.1016/0165-6147(93)90095-2. [DOI] [PubMed] [Google Scholar]
- TOMLINSON P.R., WILSON J.W., STEWART A.G. Inhibition by salbutamol of the proliferation of human airway smooth muscle cells grown in culture. Br. J. Pharmacol. 1994;111:641–647. doi: 10.1111/j.1476-5381.1994.tb14784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOMLINSON P.R., WILSON J.W., STEWART A.G. Salbutamol inhibits the proliferation of human airway smooth muscle cells grown in culture: relationship to elevated cAMP levels. Biochem. Pharmacol. 1995;49:1809–1819. doi: 10.1016/0006-2952(94)00532-q. [DOI] [PubMed] [Google Scholar]
- VLIAGOFTIS H., BEFUS A.D., HOLLENBERG M.D., MOQBEL R. Airway epithelial cells release eosinophil survival-promoting factors (GM-CSF) after stimulation of proteinase-activated receptor 2. J. Allergy Clin. Immunol. 2001;107:679–685. doi: 10.1067/mai.2001.114245. [DOI] [PubMed] [Google Scholar]
- WAKITA H., FURUKAWA F., TAKIGAWA M. Thrombin and trypsin induce granulocyte-macrophage colony-stimulating factor and interleukin-6 gene expression in cultured normal human keratinocytes. Proc. Assoc. Am. Physicians. 1997;109:190–207. [PubMed] [Google Scholar]
- WU G., ESSEX D.W., MELONI F.J., TAKAFUTA T., FUJIMURA K., KONKLE B.A., SHAPIRO S.S. Human endothelial cells in culture and in vivo express on their surface all four components of the glycoprotein Ib/IX/V complex. Blood. 1997;90:2660–2669. [PubMed] [Google Scholar]