

Abstract
Although the principal pharmacological targets of local anaesthetics (LAs) are voltage-gated Na+ channels, other targets have also been suggested. Here we examined the effects of LAs on the N-methyl-D-aspartate (NMDA) receptor, a receptor involved in the process of nociception.
LAs (bupivacaine, lidocaine, procaine, and tetracaine) reversibly and concentration-dependently inhibited recombinant ε1/ζ1 and ε2/ζ1 NMDA receptors expressed in Xenopus oocytes (IC50s for bupivacaine, lidocaine, procaine, and tetracaine were 1032.0, 1174.1, 642.1 and 653.8 μM at the ε1/ζ1 receptor; and 1090.8, 1821.3, 683.0 and 662.5 μM respectively (at the ε2/ζ1 receptor). Bupivacaine and procaine were non-competitive antagonists; bupivacaine possesses non-competitive and competitive actions when interacting with glycine, whereas procaine has only non-competitive action.
Mutation of asparagine residue at position 598 (Asp598) in the ζ1 subunit, a residue associated with the blockade site for Mg2+ and ketamine, to glutamine or arginine reduced the sensitivity to procaine but not to bupivacaine. Thus, procaine may interact with sites of action that are closely related to those of Mg2+ and ketamine blockade.
These results suggest that LAs inhibit the NMDA receptor by various mechanisms.
Keywords: Local anaesthetic, NMDA receptor, subunit, site-directed mutagenesis, Xenopus oocyte, electrophysiology
Introduction
Local anaesthetics (LA)s exert their regional anaesthetic effects and anti-arrhythmic properties by acting on voltage-gated Na+ channels (Berde & Strichartz, 1999). However, LAs modulate a wide range of other ion channels and receptors in the central nervous system (Fan et al., 1995; Hara et al., 1995; Sugimoto et al., 2000). Although most general anaesthetics primarily act at the GABAA receptor, gaseous anaesthetics such as nitrous oxide and xenon mainly act by inhibiting NMDA receptors (Franks et al., 1998; Jevtovic-Todorovic et al., 1998). The NMDA receptor has an ion channel and is important in fast excitatory neurotransmission. Five NMDA receptor subunits have been identified and divided into two families (Mori & Mishina, 1995). The mouse NMDA receptor consists of the ζ and ε subunit families, which are homologous to NMDAR1 and NMDAR2 in the rat. There are four members in the ε subfamily (ε1–4), whereas the ζ subfamily has only one member (ζ1) and a splice variant.
Surprisingly, there is little information about the interaction of LAs and NMDA receptors (Lu et al., 1996; Nishizawa et al., 2002). LA-mediated modulation of NMDA receptors could contribute to regional anaesthesia since the NMDA receptor is involved in mediating nociception and plasticity in the spinal cord and peripheral nervous systems (Dingledine et al., 1999).
Therefore, we examined the effects of LAs (lidocaine, bupivacaine, procaine, and tetracaine) on heteromeric NMDA receptors (ε1/ζ1, ε2/ζ1) expressed in Xenopus oocytes using two-electrode voltage clamp. Site-directed mutagenesis of the ζ1 subunit in the NMDA receptor was undertaken to explore the sites of action of LAs.
Methods
Site-directed mutagenesis of the ζ1 NMDA receptor subunit
Site-directed mutagenesis of the mouse ζ1 subunit (that is, replacement of the conserved asparagine (N) residue with glutamine (Q) or arginine (R) at 598 in the channel lining segment M2 of the ζ1 subunit) was performed, using the QuikChange™ Site-Directed Mutagenesis Kit (Strategene, La Jolla, CA, U.S.A.) as per the manufacturer's protocol. The primers, 5′-GGGGCGTCCTGCTCAGGTCTGGCATTGGGG-3′, 5′-CCCCAATGCCAGACCTGAGCAGGACGCCCC-3∞ (N598R) and 5′-GGGGCGTCCTGCTCCAGTCTGGCATTGGGG-3′, 5′-CCCCAATGCCAGACTGGAGCAGGACGCCCC-3′ (N598Q) were designed to incorporate the base sequence for R or Q instead of N at position 598 of the ζ1 subunit (mismatched base pairs are underlined). All the DNA sequences created by PCR were verified using an automatic sequencer.
Preparation of messenger RNAs for the NMDA receptor subunits
Mouse cDNAs encoding ε1, ε2, and mutant or wild type ζ1 were subcloned into the pSP35T transcription vector. To facilitate stable mRNA expression in oocytes, the multiple cloning sites of vectors were flanked by β-globulin from Xenopus laevis. Plasmid cDNAs were purified using the Qiagen plasmid kit (Qiagen, Chatworth, CA, U.S.A.) and resuspended in sterile water. We confirmed the identity of cloned DNA using restriction digests. Each cDNA template was linearized by restriction enzymes (Wako Chemical, Osaka, Japan) (NotI for ζ1, ζ1–N598Q and ζ1–N598R, EcoRI for ε1 and ε2). Capped mRNAs were synthesized using an SP6 RNA message machine kit (Ambion, Austin, TX, U.S.A.), using the protocol recommended by the manufacturer. Each mRNA stock was stored in RNAse-free water at −80°C until it is used.
Oocyte expression
In a study protocol approved by the Animal Research Committee of the Osaka University Medical School, female frogs (Xenopus laevis) were anaesthetized with 1% tricaine (3-aminobenzoic acid ethyl ester) and placed on ice under sterile conditions. Thereafter oocytes were harvested through a 5-mm laparotomy incision. The frogs were returned to the main tank following 2 days of post-surgical recovery in isolation. Manual defolliculation with forceps was followed by treating with 1.5 mg ml−1 collagenase type 1A (Sigma, St. Louis, MO, U.S.A.) in Ca2+-free ND96 (in mM): NaCl 96, KCl 2, HEPES 5, MgCl2 1, for 30 min at room temperature. Healthy oocytes at stages 4 and 5 were selected and thoroughly rinsed with ND96. Subunit mRNAs (0.1 mg ml−1) were mixed in a 1 : 1 molar ratio and injected (50–100 nl) into oocytes using a Nanoject injector (Drummond Scientific, Broomall, PA, U.S.A.). Prior to electrophysiological experiments, oocytes were incubated for 24–48 h at 20°C in ND96 solution containing 1.8 mM CaCl2 and 2.5 mM sodium pyruvate.
Electrophysiology and drug application
Oocytes were continuously perfused with Ba2+ Ringer's solution (mM): NaCl 115, KCl 2.5, BaCl2 1.8, HEPES 10; pH 7.4, at a rate of 10 ml min−1. Ba2+ Ringer's solution was used to minimize the effects of secondary activated Ca2+-dependent Cl− currents. Polyethylene (PE) tubing was used in the perfusion system and the absorption of LA in PE tubing was negligible (data not shown). Each oocyte was impaled with two 2–5 MΩ glass electrodes filled with 3 M KCl and voltage clamped at −80 mV using a two-electrode voltage amplifier (Nihon Kohden, Tokyo, Japan). Drugs and Ringer's solution were applied to the cell alternatively by using three-way stopcocks. Drug-containing solutions were perfused for at least 20 s to determine the peak current. Applications of NMDA/glycine were separated by a few minute intervals; at high concentrations (>100 μM for NMDA and >10 μM for glycine), 5 to 10 min intervals were allowed for the resolution of receptor desensitization. To test for cumulative desensitization, the same low concentration of agonist (NMDA 10 μM/glycine 1 μM) was applied after every application. The currents were digitally recorded with AxoScope software (Axon Instruments, Burlingame, CA, U.S.A.), running on an IBM personal computer. All electrophysiological experiments were performed at the room temperature.
Data analysis
Peak amplitudes of currents elicited by agonist and co-agonist were evaluated directly from the data recorded by AxoScope. To draw concentration–response curves for agonist-induced currents and inhibition curves for LAs, peak amplitudes were normalized and plotted, and the data fits to the following equations using Origin software (Microcal Software, Northampton, MA, U.S.A.).
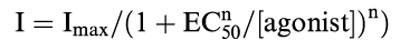 |
where I is the peak current at a given concentration of agonist and Imax is the maximum current. EC50 denotes the concentration of agonist eliciting a half-maximum response and n the Hill slope.
For the inhibition studies with LAs, the data fitted to the following equation:
Where I is the reduced current normalized with control data at a given concentration of LA and IC50 denotes the concentration of LAs that produce half maximal currents. All data are expressed as mean±s.e.mean. Standard errors on the EC50 and IC50 are estimated from the nonlinear fitting routine. Statistical analysis was performed using the unpaired Student's t-test or one-way analysis of variance (ANOVA), followed by Dunnett's post hoc test, if appropriate. P<0.05 was considered significant.
Chemicals
Drugs (bupivacaine, lidocaine, procaine, tetracaine, glycine, NMDA and MK-801) were purchased from Sigma (St. Louis, MO, U.S.A.). All drugs were directly dissolved in frog Ringer's solution on the day of the experiment. The pH was re-adjusted to pH 7.4 with 1N HCl or NaOH after each LA was dissolved.
Results
The effects of LAs on the recombinant NMDA receptors
In the adult mouse, the ε1, ε2 and ζ1 subunits are widely distributed in the brain, whereas the ε3 subunit is expressed predominantly in the cerebellum and the ε4 subunit is expressed only in the thalamus and brainstem (Mori & Mishina, 1995; Tolle et al., 1993). Therefore, we chose to examine ε1/ζ1 and ε2/ζ1. Expression of functional NMDA receptors was confirmed by the presence of an NMDA/glycine-induced current that was blocked by 10−4 M Mg2+ and 10−5 M MK-801 (data not shown). The slow perfusion rate limited the temporal resolution of the data; hence, we could only measure the peak current after fast desensitization. To confirm minimal effects of slow desensitization, consistent responses to NMDA 10 μM/glycine 1 μM between recordings were measured.
LAs are categorized into two groups: amides (bupivacaine and lidocaine) and esters (procaine and tetracaine), depending on the linkage between the hydrophilic and hydrophobic domains. Both types of LA inhibited NMDA/glycine-induced currents of both the ε1/ζ1 and ε2/ζ1 subunit of NMDA receptors in a concentration-dependent manner (Figure 1). The application of LA alone produced no measurable currents (data not shown). Table 1 summarizes the IC50 values and Hill slopes for LA-induced inhibition. Both receptors showed similar sensitivity to the LAs, with the ester-type LAs inhibiting currents more potently than does the amide-type LAs (IC50s: bupivacaine or lidocaine vs procaine and tetracaine P<0.05, ANOVA, Dunnett's test).
Figure 1.
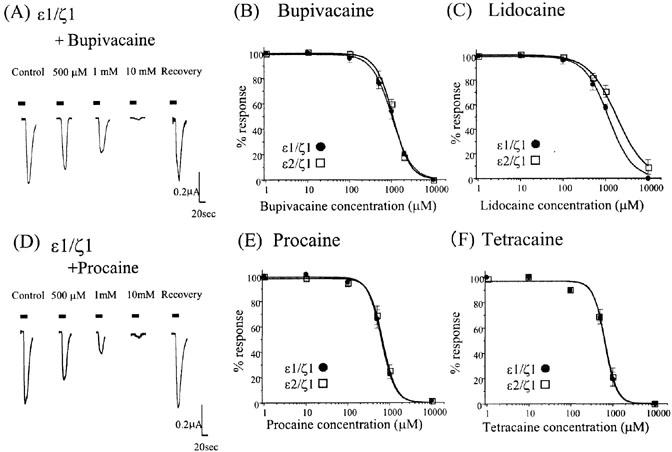
Effect of local anaesthetics on NMDA/glycine-induced currents. (A,D) The current traces for the NMDA 10 μM/glycine 1 μM responses with and without bupivacaine (A) or procaine (D) (500 μM–10 mM). Both LAs reversibly and concentration-dependently inhibited NMDA/glycine-induced currents of the ε1/ζ1 subunit NMDA receptors. Each horizontal bar indicates a period of drug application (>20 s). (B,C,E,F) Concentration-inhibition curves of the four local anaesthetics (B, bupivacaine; C, lidocaine; E, procaine; F, tetracaine) for NMDA/glycine-induced currents of the ε1/ζ1 and ε2/ζ1 sub-unit NMDA receptors. Each datum point shows the average for four to seven oocytes and is expressed as the mean±s.e.mean.
Table 1.
The IC50s and Hill slope values of local anaesthetics on the recombinant NMDA receptors
Inhibitory effects of bupivacaine and procaine as a function of NMDA and glycine concentration
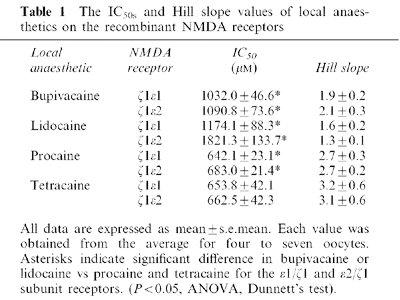
Subsequently we examined whether inhibition by bupivacaine and procaine was dependent on the concentration of NMDA and glycine. While bupivacaine reduced the maximum response, it did not significantly change the potency of NMDA (EC50 NMDA alone: 6.5±2.0 μM; NMDA+bupivacaine: 10.5±2.5 μM; P>0.05, t-test). Similarly, procaine reduced the maximum NMDA response without affecting its potency (EC50 NMDA alone: 6.5±2.0 μM; NMDA+procaine: 7.1±2.2 μM; P>0.05, t-test).
Glycine is an obligatory co-agonist at NMDA receptors, acting at a different receptor site to NMDA. We therefore tested whether bupivacaine and procaine could modulate the action of glycine on these receptors. Procaine reduced the maximum response of glycine without changing the potency (EC50: 0.9±0.03 μM without, and 0.9±0.04 μM with 500 μM procaine: P>0.05, t-test; Figure 2D, Table 2). In contrast, bupivacaine significantly reduced both the maximum response and potency of glycine (from 0.9±0.03 to 1.9±0.1 μM: P<0.05, t-test; Figure 2B, Table 2). These results suggest that bupivacaine and procaine inhibit NMDA receptors in different ways.
Figure 2.
The effects of bupivacaine and procaine on the concentration–response curves for NMDA and glycine at the ε1/ζ1 receptor. (A–C) The effects of 2 mM bupivacaine and 500 μM procaine on the concentration–response curves of NMDA with 10 μM glycine. Bupivacaine and procaine both reduced the maximum NMDA-induced responses. Bupivacaine slightly but not significantly shifted the concentration–response curve of NMDA to the right: the EC50 values changed from 6.5±2.0 μM to 10.5±2.5 μM (P>0.05, t-test). Procaine did not significantly affect the EC50 values of NMDA (7.1±2.2 μM in the presence of procaine, P>0.05, t-test). (B,D) The effects of 2 mM bupivacaine and 500 μM procaine on the concentration–response curves of glycine with 100 μM NMDA. As shown by the NMDA concentration–response curves, both LAs significantly reduced the maximal current of glycine. EC50 values in the glycine concentration–response curve were significantly increased by bupivacaine from 0.9±0.03 μM to 1.9± 0.1 μM (P<0.05, t-test) while there was no significant change with procaine (0.9±0.03 μM without procaine and 0.9±0.04 μM with procaine: P>0.05, t-test). All the responses were normalized to the peak current amplitude induced by NMDA 1000 μM/glycine 10 μM (A, C) or NMDA 100 μM/glycine 100 μM (B, D). Each datum point shows the average for five to seven oocytes, and is expressed as the mean±s.e.mean.
Table 2.
Inhibitory effects of bupivacaine and procaine on the concentration response curves of NMDA and glycine
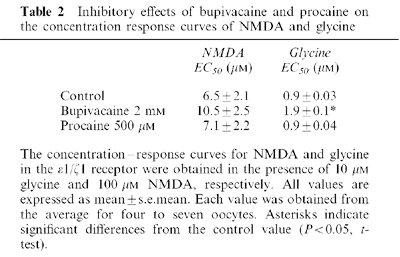
Inhibitory effects of bupivacaine and procaine at different potentials on the NMDA/glycine-induced currents
The effects of LAs on the NMDA/glycine-induced currents at the ε1/ζ1 subunit receptor were examined at various membrane potentials (Figure 3). The effects of 1 mM bupivacaine and 1 mM procaine were not significantly different for membrane potentials ranging from −80 to −10 mV (P>0.05, ANOVA). Thus, the inhibitory effects of these LAs show no voltage dependencies.
Figure 3.
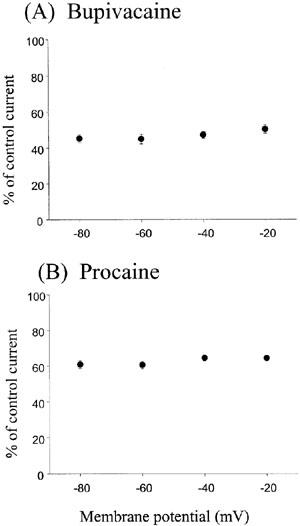
The effects of bupivacaine and procaine on NMDA/glycine-induced currents in the ε1/ζ1 NMDA receptor at different membrane potentials. The percentages of the control currents induced by NMDA 10 μM/glycine 1 μM NMDA at holding potentials ranging from −80 to −10 mV were plotted in the presence of 1 mM bupivacaine (A) and 1 mM procaine (B). No significant difference (P>0.05, ANOVA) was observed at four different potentials, showing no voltage dependency for both local anaesthetics. Each datum point shows the average for five oocytes and is expressed as the mean±s.e.mean.
Effect of point mutation on inhibition by LAs
A conserved asparagine (Asp598) in the M2 channel-lining segment of the ζ1 subunit is associated with Mg2+ blockade site (Mori et al., 1992), as well as the site for non-competitive antagonists PCP, ketamine, and MK-801 (Yamakura et al., 1993). To determine whether LAs mediate their inhibitory activity through the same site, two NMDA receptors containing point mutations at that site (ζ1-N598Q or ζ1-N598R) were examined. The ε1/ζ1–N598Q and ε1/ζ1–N598R receptors showed wild type NMDA/glycine dose-responses and sensitivity to blockade by ZnCl2. However, the mutant receptors showed drastic decreases in sensitivity to Mg2+ and ketamine blockade, as previously reported (Mori et al., 1992; Yamakura et al., 1993; data not shown).
The mutant receptors demonstrated reduced sensitivity to procaine without showing changes in bupivacaine sensitivity (Figure 4A–D). These results indicate that the Mg2+-insensitive single mutation of the ζ1 subunit (ζ1–N598Q or ζ1–N598R) can only affect the modulation of procaine, but not the bupivacaine, in NMDA receptor function.
Figure 4.
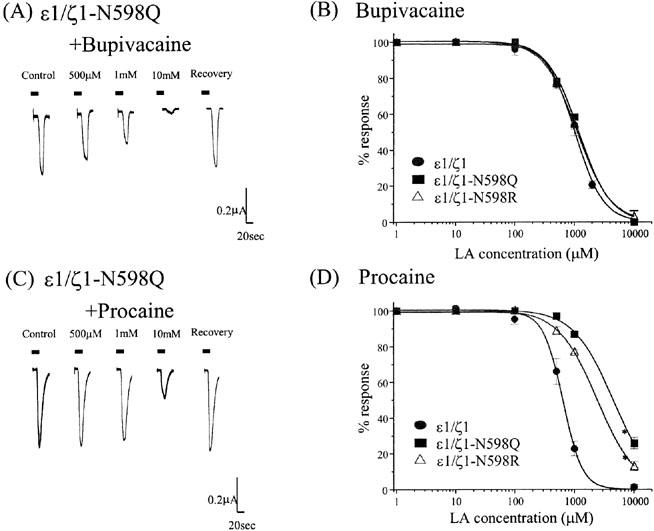
Effect of point mutation (ζ1–N598Q subunit) in the NMDA receptors on inhibitory actions of LAs. (A,C) The traces of the NMDA 10 μM/glycine 1 μM current responses with and without bupivacaine (A) or procaine (C) (500 μM–10 mM). Although procaine reversibly and concentration-dependently inhibited the NMDA/glycine-induced currents in the ε1/ζ1–N598Q subunit NMDA receptor, the mutated ζ1 subunit significantly reduced the sensitivity to procaine in comparison to that in the wild type ε1/ζ1 subunit (refer to Figure 1D). Each horizontal bar indicates a period of drug application. (B) The mutation of the ε1/ζ1–N598Q or ε1/ζ1–N598R subunit did not alter the effect of bupivacaine. The IC50 values (mean±s.e.mean) of bupivacaine on the mutated receptor were 1177.3±82.4 μM (ε1/ζ1–N598Q) and 1110.4±18.2 μM (ε1/ζ1–N598R). Each datum point shows the average for five to seven oocytes. (D) The mutation of the N598Q or N598R in the ζ1 subunit significantly reduced the sensitivity to procaine in the wild type receptor co-expressed with the ε1 subunit. The IC50 values (mean±s.e.mean.) of procaine on the mutated receptor were 4437.8±228.5 μM (ε1/ζ1–N598Q) and 2411.7±70.2 μM (ε1/ζ1–N598R) (P<0.05 vs control, t-test). Each datum point shows the average for five to seven oocytes.
Discussion
The NMDA receptor, one of the major receptor channels for rapid excitatory neurotransmission in the central nervous system, can be modulated by a number of important drugs, including general anaesthetics (Mori & Mishina, 1995; Dingledine et al., 1999; Yamakura & Shimoji, 1999). Gaseous anaesthetics, nitrous oxide and xenon mainly inhibit the NMDA receptors at clinical concentrations, suggesting their involvement in the mechanism of action of these anaesthetics (Franks et al., 1998; Jevtovic-Todorovic et al., 1998).
This study exhibits some new implications for the interaction between LAs and NMDA receptors. All LAs inhibited the function of NMDA receptors. The ester-type LAs were more potent than the amide-types, although there were no significant differences in the sensitivity of the two recombinant receptors. Since the inhibitory actions of procaine, an ester-type LA could not be cancelled by higher concentrations of NMDA and glycine, without affecting the EC50 values, there is a strong possibility that procaine acts at an allosteric site different from the sites for NMDA and glycine in the NMDA receptor. However bupivacaine, an amide-type LA may act both competitively and non-competitively at least at the glycine site while acting non-competitively at the NMDA site. It appears that bupivacaine may act at two different sites in the NMDA receptor; one is at the site related to glycine and the other one is at a different site other than glycine and NMDA.
To define the molecular basis of LA-mediated inhibition, we tested two mutant ζ1 subunits, N598Q and N598R, which endowed the insensitivities to Mg2+ as well as to typical non-competitive inhibitors (PCP, ketamine, and MK-801), but otherwise has a normal function (Yamakura et al., 1993). Although these mutant receptors demonstrated a similar sensitivity to bupivacaine as did wild type receptors, the sensitivity to procaine block was partially reduced. This suggests that the site-of-action for procaine might be in close proximity, but is not identical to, that for Mg2+ and ketamine blockade. This disparate site of action is consistent with the observed difference in Hill values for ketamine and Mg2+ blockade vs LAs in wild type receptors. The Hill slope values for all the tested LAs in wild type receptors were greater than 1.0. This feature is qualitatively different from the blockade by Mg2+ and Ketamine, with Hill slopes of less than 1.0 (Liu et al., 2001b).
While the Hill slopes for ε1/ζ1, ε1/ζ1–N598Q, and ε1/ζ1–N598R receptors in the presence of bupivacaine were very similar, the Hill slope value for procaine declined from 2.7 (ε1/ζ1) to 1.3 (ε1/ζ1–N598Q, ε1/ζ1–N598R). Unless the binding affinity of procaine can be changed, the cooperativity arises entirely from the conformational change (Colquhoun, 1998). The fall of the Hill slope in the mutation may indicate a reduction of the ability to change conformation which can relate to the action of procaine, not of bupivacaine and NMDA/glycine. However, if the binding affinity of procaine can be changed by the mutation, i.e., the second binding becomes weaker in the case of a two binding reaction, the Hill slope is reduced. This may be referred to as cooperativity of binding to distinguish it from the cooperativity that arises from the conformational change. The mutation of the asparagine residue in the M2 region may induce those conformational changes in the NMDA receptor and result in the reduction of cooperativity to procaine and the impairment of its action. Furthermore, mutagenesis studies of the channel pore region and binding domain in the ζ1 subunit will clarify the site and mechanism of action of procaine and other LAs.
NMDA receptors in the spinal cord play an important role in synaptic plasticity, such as central sensitization (wind up), after nociceptive stimuli (Tolle et al., 1993; Woolf & Salter, 2000). Soon after direct spinal injection, LAs in the spinal cord can reach concentrations of 10−4 M–10−3 M (Converse et al., 1954; Bromage et al., 1963; Cohen, 1968; Post & Freedman, 1984). In this in vitro study, the inhibitory effects of bupivacaine, lidocaine, procaine, and tetracaine on NMDA receptors were tested and it was found that sufficient inhibition can be achieved with the above mentioned concentrations. Thus, in addition to the effects on voltage-gated sodium, potassium, and calcium channels (Komai & McDowell, 2001; Liu et al., 2001a), the inhibition of NMDA receptors by LAs in the spinal cord could be important in preventing development of pain sensitization and play an important role in the treatment of pain by performing spinal and epidural anaesthesia with these agents.
Another relevant clinical insight arising from this study concerns the mechanism of LA-induced convulsions. Previous studies have suggested that LAs may induce convulsions both by suppression of inhibitory synapses such as GABAA receptor-mediated transmission (Hara et al., 1995; Sugimoto et al., 2000) and by facilitation of excitatory synapses, where NMDA receptors are involved (Kasaba et al., 1998; Ushijima et al., 1998). Although there is no data to indicate what brain concentrations of LAs produce convulsions, these pharmacological findings imply the involvement of NMDA receptors in LA-induced convulsions (Berde & Strichartz, 1999). By contrast, our findings demonstrate that LAs do not potentiate, but rather inhibit NMDA receptors. Thus our study does not support the involvement of LAs in excitatory synaptic responses, which were mediated by NMDA receptors, in the mechanism of LA-induced convulsions.
In summary, the interactions between LAs and recombinant NMDA receptors were investigated. That LAs concentration-dependently inhibit the NMDA receptor was demonstrated. Site-directed mutagenesis of the ζ1 subunit revealed that different mechanisms were involved for inhibitions by bupivacaine and procaine, and the site-of-action of procaine may be closely related to the blocking site for Mg2+ and ketamine. This is the first study to show the molecular site-of-action of LAs on the NMDA receptor using a recombinant receptor and site-directed mutagenesis.
Acknowledgments
This work was supported in part by a grant-in-aid for Scientific Research from the Ministry of Education, Science, and Culture of Japan. The authors thank Dr Mishina for providing the mouse NMDA receptor subunit clones and Drs Raymond Price, Nobuya Matsuoka and George Kcracke for editing our manuscript.
Abbreviations
- cDNA
complementary DNA
- GABAA
gamma-aminobutyric acid type A
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid
- LA
local anaesthetic
- MK-801
dizocilpine
- NMDA
N-methyl-D-aspartate
- PCP
phencyclidine
- PCR
polymerase chain reaction
- Tricaine
3-aminobenzoic acid ethyl ester
References
- BERDE C.B., STRICHARTZ G.R.Local anesthetics Anesthesia 1999Philadelphia: Churchill Livingstone; 491–522.ed. Miller, R.D. [Google Scholar]
- BROMAGE P.R., YOYAL A.D., BINNEY J.C. Local anesthetic drugs: Penetration from the spinal extradural space into the neuraxis. Science. 1963;140:392–393. doi: 10.1126/science.140.3565.392. [DOI] [PubMed] [Google Scholar]
- COHEN E.N. Distribution of local anesthetic agents in the neuraxis of the dog. Anesthesiology. 1968;29:1002–1005. doi: 10.1097/00000542-196809000-00028. [DOI] [PubMed] [Google Scholar]
- COLQUHOUN D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONVERSE J.G., LANDMESSER C.M., HARMED M.H. The concentration of procaine hydrochloride in the cerebrospinal fluid during spinal anesthesia, and the influence of epinephrine in prolonging the sensory anesthetic effect. Anesthesiology. 1954;15:1–10. doi: 10.1097/00000542-195401000-00001. [DOI] [PubMed] [Google Scholar]
- DINGLEDINE R., BORGES K., BOWIE D., TRAYNELIS S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- FAN P., OZ M., ZHANG L., WEIGHT F.F. Effect of cocaine on the 5-HT3 receptor-mediated ion current in xenopus oocytes. Brain Res. 1995;673:181–184. doi: 10.1016/0006-8993(94)01316-a. [DOI] [PubMed] [Google Scholar]
- FRANKS N.P., DICKINSON R., DESOUSA S.L.M., HALL A.C., LIEB W.R. How does xenon produce anaesthesia. Nature. 1998;396:324. doi: 10.1038/24525. [DOI] [PubMed] [Google Scholar]
- HARA M., KAI Y., IKEMOTO Y. Local anesthetics reduce the inhibitory neurotransmitter-induced current in dissociated hippocampal neurons of the rat. Eur. J. Pharmacol. 1995;283:83–89. doi: 10.1016/0014-2999(95)00293-t. [DOI] [PubMed] [Google Scholar]
- JEVTOVIC-TODOROVIC V., TODOROVIC S.M., MENNERICK S., POWELL S., DIKRANIAN K., BENSHOFF N., ZORUMSKI C.F., OLNEY J.W. Nitrous oxide (laughing gas) is an NMDA antagonist, neuroprotectant and neurotoxin. Nat. Med. 1998;4:460–463. doi: 10.1038/nm0498-460. [DOI] [PubMed] [Google Scholar]
- KASABA T., SHIRAISHI S., TANIGUCHI M., TAKASAKI M. Bupivacaine-induced convulsion is suppressed by MK-801. Reg. Anesth. Pain Med. 1998;23:71–76. doi: 10.1016/s1098-7339(98)90113-4. [DOI] [PubMed] [Google Scholar]
- KOMAI H., MCDOWELL T.S. Local anesthetic of voltage-activated potassium currents in rat dorsal root ganglion neuron. Anesthesiology. 2001;94:1089–1095. doi: 10.1097/00000542-200106000-00025. [DOI] [PubMed] [Google Scholar]
- LIU B.G., ZHUANG X.L., LI S.T., XU G.H., BRULL G.H., ZHANG J.M. Effects of bupivacaine and Ropivacaine on high-voltage activated calcium currents of the dorsal horn neurons in newborn rats. Anesthesiology. 2001a;95:139–143. doi: 10.1097/00000542-200107000-00024. [DOI] [PubMed] [Google Scholar]
- LIU H.T., HOLLMANN M.W., LIU W.H., HOENEMANN C.W., DURIEUX M.E. Modulation of NMDA receptor function by ketamine and magnesium: Part I. Anesth. Analg. 2001b;92:1173–1181. doi: 10.1097/00000539-200105000-00019. [DOI] [PubMed] [Google Scholar]
- LU W.Y., BIEGER D. Inhibition of nicotinic cholinoreceptor mediated current in vagal motor neurons by local anesthetics. Can. J. Physiol. Pharmacol. 1996;74:1265–1269. [PubMed] [Google Scholar]
- MORI H., MASAKI H., YAMAKURA T., MISHINA M. Identification by mutagenesis of a Mg2+-block site of the NMDA receptor channel. Nature. 1992;358:673–675. doi: 10.1038/358673a0. [DOI] [PubMed] [Google Scholar]
- MORI H., MISHINA M. Structure and function of the NMDA receptor channel. Neuropharmacol. 1995;34:1219–1237. [Google Scholar]
- NISHIZAWA N., SHIRASAKI T., NAKAO S., MATSUDA H., SHINGU K. The inhibition of the N-Methyl-D-Aspartate receptor channel by local anesthetics in mouse CA1 pyramidal neurons. Anesth. Analg. 2002;94:325–330. doi: 10.1097/00000539-200202000-00017. [DOI] [PubMed] [Google Scholar]
- POST C., FREEDMAN J. A new method for studying the distribution of drugs in spinal cord after intrathecal injection. Acta. Pharmacol. Toxicol. (Copenh) 1984;54:253–257. doi: 10.1111/j.1600-0773.1984.tb01926.x. [DOI] [PubMed] [Google Scholar]
- SUGIMOTO M., UCHIDA I., FUKAMI S., TAKENOSHITA M., MASHIMO T., YOSHIYA I. The α and γ subunit-dependent effects of local anesthetics on recombinant GABAA receptors. Eur. J. Pharmacol. 2000;401:329–337. doi: 10.1016/s0014-2999(00)00463-5. [DOI] [PubMed] [Google Scholar]
- TOLLE T.R., BERTHELE A., ZIEGLGANSBERGER W., SEEBURG P.H., WISDEN W. The differential expression of 16 NMDA and non-NMDA receptor subunits in the rat spinal cord and in periaqueductal gray. J. Neurosci. 1993;13:5009–5028. doi: 10.1523/JNEUROSCI.13-12-05009.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- USHIJIMA I., KOBAYASHI T., SUETSUGI M., WATANABE K., YAMADA M., YAMAGUCHI K. Cocaine: evidence for NMDA-, beta-carboline- and dopaminergic-mediated seizures in mice. Brain Res. 1998;797:347–350. doi: 10.1016/s0006-8993(98)00434-x. [DOI] [PubMed] [Google Scholar]
- YAMAKURA T., MORI H., MASAKI H., SHIMOJI K., MISHINA M. Different sensitivities of NMDA receptor channel subtypes to non-competitive antagonists. NeuroReport. 1993;4:687–690. doi: 10.1097/00001756-199306000-00021. [DOI] [PubMed] [Google Scholar]
- YAMAKURA T., SHIMOJI K. Subunit- and site-specific pharmacology of the NMDA receptor channel. Prog. Neurobiol. 1999;59:279–298. doi: 10.1016/s0301-0082(99)00007-6. [DOI] [PubMed] [Google Scholar]
- WOOLF C.J., SALTER M.W. Neuronal plasticity: Increasing the gain in pain. Science. 2000;288:1765–1768. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]