

Abstract
Our aims were to characterize activation of the transcription factor, nuclear factor kappa-B(NF-κB), during myocardial ischaemia-reperfusion and to assess its functional role in the evolution of acute ischaemia-reperfusion injury in intact myocardium in vivo.
Under pentobarbitone anaesthesia, rabbits underwent sham operation, 30 min left coronary artery occlusion followed by 0, 10 or 180 min reperfusion. Saline or NF-κB inhibitor diethyldithiocarbamic acid (DDTC, 50, 100 or 200 mg kg−1) was given intravenously 5 min prior to reperfusion.
Electromobility shift assay revealed that 30 min ischaemia alone did not activate NF-κB compared to time-matched sham-operated controls (85±13% vs 100±28%, respectively). However, ischaemia plus 10 min reperfusion markedly increased activation of NF-κB (295±77%). DDTC 50 mg kg−1 did not inhibit NF-κB activation (278±67%) but at the higher doses complete inhibition was observed (54±20%, 31±16%, respectively).
Infarct to risk ratio was determined by triphenyltetrazolium chloride staining after 30 min ischaemia and 180 min reperfusion. DDTC 50 or 100 mg kg−1 significantly reduced infarct size compared to the saline-treated control group (34.9±5.2%, 37.1±5.9%, vs 51.3±3.6%, P<0.05, respectively), whereas there was no protection with 200 mg kg−1 (45.6±5.3%).
We conclude that ischaemia alone does not activate NF-κB, but post-ischaemic reperfusion robustly activates NF-κB in the myocardium. DDTC limited irreversible injury at low doses, but this effect appears to be dissociated from inhibition of NF-κB. Thus, activation of NF-κB during reperfusion does not appear to play a role in the evolution of myocardial infarction during the early phase of reperfusion.
Keywords: Ischaemia-reperfusion, infarct size, transcription factor, nuclear factor kappa-B, in vivo myocardium, diethyldithiocarbamic acid, antioxidant, pro-oxidant, oxidative stress
Introduction
Myocardial ischaemia develops when coronary blood supply to myocardium is reduced, in either absolute terms or relative to tissue demand. A pivotal feature of ischaemia is that oxygen supply to the mitochondria is inadequate to support oxidative phosphorylation. Uncoupling of oxidative phosphorylation rapidly produces profound biochemical and morphological changes within the myocardium, the severity of which are ultimately determined by the degree and duration of oxygen deprivation. In the absence of reperfusion (the re-admission of oxygen and metabolic substrates) irreversible tissue injury will occur resulting in myocardial infarction. Although reperfusion is essential for the salvage of myocardium, the process of reperfusion is associated with profound disturbances of cellular metabolism, notably oxidant stress, that may predispose to further tissue injury during the early phase of reperfusion (Yellon & Baxter, 2000).
Nuclear factor kappa-B (NF-κB) is a ubiquitously expressed redox-sensitive transcription factor that can respond rapidly to a variety of stimuli including cytokines, endotoxin and oxidative stress. The numerous gene products that are regulated by NF-κB co-ordinate cell homeostasis following stress and ultimately control the balance between cell survival and cell death (Perkins, 1997). This dual function of NF-κB is finely regulated by a mechanism that involves ligand binding to a cell surface receptor and multiple phosphorylation steps through a protein kinase cascade (Zechner et al., 1998; Valen et al., 2001). Phosphorylation and ubiquitination of the inhibitor protein, IκB, in the cytosol releases the sequestered NF-κB. NF-κB then translocates into the nucleus followed by phosphorylation and binds to the promoter region of the target gene. Genes regulated by this transcription factor include inducible nitric oxide synthase (iNOS), cyclooxygenase II (COX-2), manganese-conjugated superoxide dismutase (MnSOD), antiapoptotic proteins such as Bcl2, inhibitor of apoptosis factors (IAFs), A1, and members of the cytokine family (e.g. TNFα), interleukins (e.g IL-1 and IL-6), cell adhesion molecules (ICAM, VCAM, selectins), FAS ligand and other transcription factors (p53).
There is evidence that activation of NF-κB occurs rapidly in response to ischaemia in many tissues including kidney cells, brain, liver and myocardium. In isolated rat heart, Li et al. (1999) reported a maximal 2 fold increase of NF-κB DNA binding activity following brief ischaemia. When reperfusion was instituted following 15 min ischaemia, activity was increased 4 fold, suggesting that ischaemia activates NF-κB and that this activation is further augmented by reperfusion (Li et al., 1999). Activation of NF-κB following ischaemia-reperfusion periods has led to the suggestion that this transcription factor plays a decisive role in the pathological response of tissues to ischaemia-reperfusion. Sawa et al. (1997) demonstrated that transfection of rat myocardium with a ‘decoy'-oligonucleotide against the NF-κB binding site led to improved left ventricular recovery when the hearts were subjected to ischaemia-reperfusion. Recently, inhibition of activation of NF-κB by peptide inhibitors PR11 and PR39 has been reported to limit infarct size in rat heart (Bao et al., 2001). Zingarelli et al. (2002a) have shown that the sesquiterpene lactone parthenolide, a supposedly specific inhibitor of IKK and NF-κB, reduced reperfusion-induced damage in rat hearts. However, this apparently deleterious role of NF-κB in ischaemia-reperfusion must be viewed alongside other studies implicating NF-κB activation in the cardioprotective effects of both early and delayed forms of ischaemic preconditioning (Maulik et al., 1999; Morgan et al., 1999; Xuan et al., 1999).
The aims of the present studies were to examine whether NF-κB is activated during the severe ischaemia-reperfusion conditions that lead to myocardial infarction and if so, whether this activation plays any role in the evolution of tissue injury. To examine the hypothesis that NF-κB is activated, and plays a role in myocardial infarction we undertook studies in a well-characterized in vivo rabbit preparation, closely modelling acute myocardial infarction in humans. We sought to identify the pattern of activation of NF-κB in this preparation and then evaluated the contribution of NF-κB to the development of irreversible tissue injury by using the inhibitor compound, diethyldithiocarbamic acid (Orrenius et al., 1996).
Methods
Animals
Male New-Zealand White rabbits (2.0–3.0 kg, Charles River, Bicester, U.K.) were used in these studies. All procedures were in accordance with UK Home Office guidelines on the Animals (Scientific Procedures) Act 1986 (The Stationery Office, London, U.K.). The animals were housed in individual cages and had free access to a standard pelleted diet and water.
Materials
Diethyldithiocarbamic acid (DDTC), triphenyltetrazolium chloride (TTC) and Evans blue were obtained from Sigma (Gillingham, Dorset, U.K.). NF-κB consensus oligonucleotide and T4 polynucleotide kinase were from Promega (Southampton, U.K.), Poly-dIdC was from Amersham (Buckinghamshire, U.K.) and Cocktail EDTA-free protease inhibitor tablets were from Roche (Welwyn Garden City, U.K.). Other chemicals and reagents were of analytical quality, supplied by Merck/BDH (Poole, U.K.).
Surgical procedure
Rabbits were premedicated with ‘Hypnorm' 0.1 ml kg−1 i.m. (Janssen Pharmaceuticals, Wantage, U.K., containing fentanyl citrate (315 μg ml−1) and fluanisone (10 mg ml−1)) and anaesthetized with sodium pentobarbitone 30 mg kg−1 i.v. through the left marginal ear vein. Tracheotomy was performed, the animals were intubated and artificially ventilated with a small animal respirator (Harvard Apparatus, Edenbridge, U.K.) using room air supplemented with O2. Tidal volume was adjusted to maintain arterial pH between 7.4 and 7.5. The right common carotid artery was cannulated with a rigid polyethylene catheter connected to a pressure transducer (Lectromed, Welwyn Garden City, U.K.) for continuous monitoring of blood pressure and heart rate. The right marginal ear vein was cannulated for administration of heparin, saline or DDTC. Thereafter, Hypnorm, 0.1 ml kg−1 i.m., and pentobarbitone sodium, 5 to 10 mg kg−1 i.v., were administered as required to maintain surgical anaesthesia. Rectal temperature was maintained within physiological range (38–39.5°C) by a heating pad. A medium sternotomy was performed and the pericardium was incised to expose the heart. A 3-0 silk suture was passed around the major anterolateral branch of the left coronary artery and a polypropylene tube was pulled over the ends of the suture. Regional ischaemia was induced by pulling the tube tight against the myocardium and clamping it in a fixed position. Ischaemia was confirmed by regional hypokinesis and cyanosis. After 30 min of ischaemia the clamp was released and reperfusion was confirmed by evident reactive hyperaemia.
Determination of myocardial infarction
After 180 min reperfusion 300 U kg−1 i.v. heparin was administered. The animals were killed by an overdose of pentobarbitone, the heart was excised and attached to a Langendorff perfusion apparatus by the aortic root and perfused retrogradely with saline to remove blood. The coronary artery was ligated and 0.25% Evans blue dissolved in saline was infused through the aorta to stain the non-ischaemic area. The heart was frozen for 1.5 h at −20°C, cut into 2 mm slices and the slices were incubated in phosphate buffer (pH=7.45) containing 1% TTC for 10–15 min. The volumes of infarcted tissue and area at risk were determined by computerized planimetry (Summa Sketch II, Summa Graphics, Newport, CT, U.S.A.). Infarct size was expressed as the percentage of the ischaemic risk volume.
Experimental protocols
All experimental protocols are illustrated in Figure 1.
Figure 1.
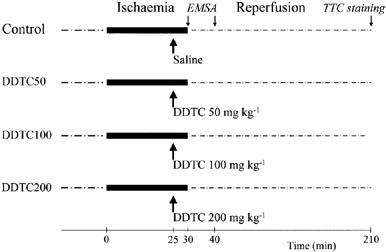
The experimental protocol outlining the procedures that were applied in anaesthetised rabbits.
Assessment of the effect of ischaemia-reperfusion on NF-κB activation
Under Hypnorm and pentobarbitone anaesthesia as described above, rabbits were subjected to sham operation with no myocardial ischaemia-reperfusion (n=4), to 30 min ischaemia alone (n=3) or to 30 min ischaemia plus 10 min reperfusion (n=5). DDTC was given as an intravenous bolus 5 min prior to reperfusion at doses of 50 (n=3), 100 (n=3) or 200 (n=3) mg kg−1. At the end of reperfusion the animals were euthanized by anaesthetic overdose, the heart was rapidly excised, rinsed in saline, the ischaemic risk zone and non-ischaemic zone were separated and frozen in liquid nitrogen.
Assessment of the effect of NF-κB inhibition on myocardial infarction
In these experiments hearts were subjected to 30 min coronary artery occlusion followed by 180 min reperfusion. Saline (n=11) or DDTC 50 (n=6), 100 (n=7) or 200 (n=7) mg kg−1 was given as a slow i.v. injection (2.5 ml over 2–3 min), beginning 5 min prior to reperfusion. At the end of experiments infarct size was determined by TTC staining as described above.
Protein extraction
Proteins from frozen tissue samples were extracted by Schreiber's method (Schreiber et al., 1989). Frozen tissue samples from the ischaemic risk zone of the left ventricle were placed in 4 ml ice cold phosphate buffered saline (PBS) solution, homogenized with a IKA-Ultra-Turrax homogenizer (Staufen, Germany) and centrifuged for 5 min at 3500 r.p.m., 4°C to remove blood elements. Supernatant was discarded and the pellet was washed again with 1 ml cold PBS. PBS was replaced with 0.5 ml ice cold hypotonic buffer containing (mM): HEPES 10 (pH=7.9), KCl 10, EDTA 0.1 (pH=8.0), EGTA 0.1 (pH=8.0), DTT 1, PMSF 0.5 and various protease inhibitors (Cocktail Inhibitor EDTA free, Roche). The extract was incubated for 15 min then 50 μl 10% Nonidet P40 was added, the sample was vortexed, incubated for 5 min and centrifuged at 14 000 r.p.m., 4°C for 30 s. The supernatant was stored as cytosolic protein fraction at −80°C for further studies. The pellet was resuspended in 350 μl ice cold high salt buffer containing (mM) HEPES 20 (pH=7.9), NaCl 400, EDTA 1 (pH=8.0), EGTA 1 (pH=8.0), DTT 1, PMSF 1 and protease inhibitors. The samples were incubated on a rocking platform on ice for 30 min and centrifuged at 14 000 r.p.m., 4°C for 15 min. The pellet was stored as nuclear protein fraction at −80°C for further studies. Total protein content was determined using Bio-Rad reagent according to the manufacturer's protocol (Bio-Rad Laboratories, Munich, Germany).
Electromobility shift assay
Nuclear proteins (15 μg) were incubated for 25 min at room temperature with labelled oligonucleotide in a binding reaction containing 0.2 μg μl−1 polydIdC, 4% glycerol (mM) Tris 10 (pH=7.5), NaCl 100, EDTA 1, MgCl2 1, DTT 5, 10 μg BSA. NF-κB oligonucleotide (5′-AGT TGA GGG GAC TTT CCC AGG C-3′ ) was 5′ end-labelled as dsDNA with [γ-32P]ATP (ICN Biochemicals, Germany) using T4 polynucleotide kinase. 5% nondenaturing gel was pre-run for 2–3 h at room temperature in 0.25×Tris-Borate-EDTA buffer at 125 V. Supershift assay was performed on samples incubated for an additional 10 min with anti-p65 antibody (Santa Cruz, U.S.A.). For competition assay cold, unlabelled oligonucleotide was given in excess (40×) prior to adding labelled oligonucleotide. The samples were loaded onto the gel and run for 2.5 h. Gel was dried, packed in cling film and exposed to Kodak film at −80°C. The films were scanned and band densities were compared using NIH Image software (http://rsb.info.nih.gov/nih-image).
Statistical analysis
Data are expressed as means±s.e.mean. Infarct size and risk zone size data were compared by one-way ANOVA with Fisher's protected least significant difference test (Statview computerized program). Haemodynamic data were analysed by repeated measures ANOVA. Differences between mean values were considered significant when P<0.05.
Results
A total of 55 rabbits were used for these studies. Twenty-one rabbits were used to examine the responses of NF-κB to ischaemia-reperfusion and the effects of DDTC on this activation. Thirty-four rabbits were used to examine the effects of NF-κB inhibition on acute myocardial infarction of which three were excluded (one was excluded due to failure of reperfusing the coronary artery and two were excluded due to arrhythmias during reperfusion). Thus, we report data from 31 successfully completed infarct experiments.
Activation of NF-κB by ischaemia-reperfusion
We hypothesized that 30 min ischaemia followed by a short reperfusion period would rapidly translocate and activate NF-κB. Activation of NF-κB was measured by EMSA in frozen tissue samples taken from the risk area of the heart. A representative EMSA blot is illustrated in Figure 2A and the corresponding densitometric analysis is illustrated in Figure 2B. A very low basal level of DNA binding activity was observed in myocardium from sham-operated animals (lane 1 & Figure 2B). In myocardium subjected to 30 min ischaemia, NF-κB was not activated above control levels (lane 2 & Figure 2B). However, NF-κB activation was significantly increased in myocardium subjected to 30 min ischaemia and 10 min reperfusion (3 fold), indicating a rapid and reperfusion-dependent activation process (lane 3 & Figure 2B). DDTC 50 mg kg−1 immediately prior to reperfusion did not abolish this activation (lane 4 & Figure 2B). However, DDTC 100 mg kg−1 or 200 mg kg−1 completely prevented activation of NF-κB by reperfusion (lane 5 and 6 & Figure 2B). Supershift with anti-p65 antibody and competition assays with cold oligonucleotide in excess confirmed the specificity of NF-κB (lane 7 and 8).
Figure 2.
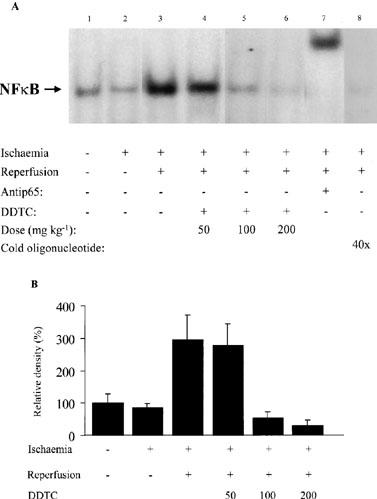
Electromobility shift assay to measure activation of NF-κB. Nuclear proteins were extracted as described in Methods. (A) Representative EMSA blot including supershift assay and competition assay. Tissue samples were taken from sham-operated rabbits, from rabbits that were subjected to ischaemia, ischaemia-reperfusion, and from rabbits that were given DDTC 5 min prior to reperfusion (50, 100 and 200 mg kg−1). Incubation of the nuclear proteins with anti-p65 antibody resulted in a supershift in the mobility of the transcription factor. The anti-p65 antibody and cold oligonucleotide in excess specified the transcription factor as NF-κB. (B) densitometric measurement of bands representing NF-κB. Each bar is expressed as the mean±s.e.mean of at least three individual experiments.
Effect of DDTC on myocardial infarct size
Ischaemic risk zone and absolute infarct size data are presented in Table 1. Ischaemic risk zone size is an important determinant of infarct size. Risk volumes were comparable among the treatment groups. Infarct size normalized as a percentage of the ischaemic risk zone, thereby taking account of small variations in risk zone volume, showed a modest but statistically significant reduction following treatment with DDTC 50 or 100 mg kg−1 (34.9±5.2% and 37.1±5.9%, respectively vs 51.5±3.8% in the control group, P<0.05, Figure 3). However, DDTC at the dose of 200 mg kg−1 did not significantly reduce the infarct size (45.6±5.3%, Figure 3).
Table 1.
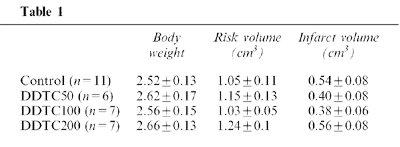
Figure 3.
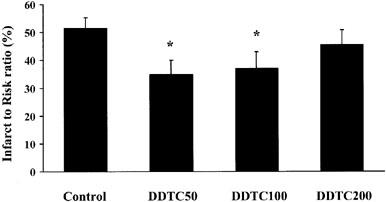
The effect of DDTC on myocardial infarction assessed by TTC staining. Infarct size is expressed as the percentage of the area at risk. Saline or DDTC (50, 100 and 200 mg kg−1) was given as a single slow bolus 5 min prior to reperfusion. *P<0.05 vs saline-treated control group (one way ANOVA followed by Fischer's post hoc test).
Haemodynamic effect of DDTC during myocardial ischaemia-reperfusion
The haemodynamic effects of DDTC given intravenously as a single bolus injection are summarized in Figure 4. DDTC 50 mg kg−1 had little effect on blood pressure (Panel A and B). At the higher doses, dose-dependent decreases in systolic and mean arterial blood pressures were observed (Panel A and B). These reductions were evident within a few minutes of the administration of DDTC. DDTC 200 mg kg−1 induced a significant hypotension that was sustained throughout the reperfusion period. Heart rate did not differ significantly between the treatment groups (Panel C).
Figure 4.
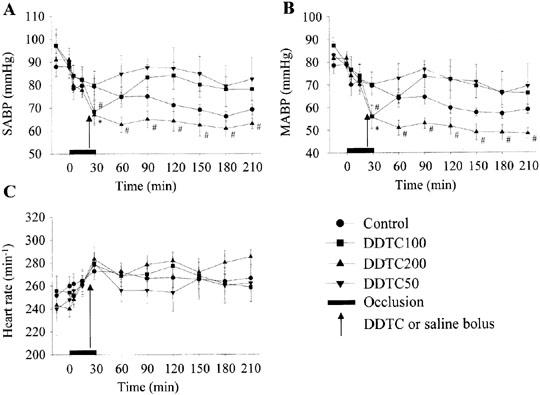
The haemodynamic effects of coronary artery occlusion followed by reperfusion, and administration of DDTC 5 min prior to reperfusion. DDTC was given intravenously as a single slow bolus as indicated by the arrow (50, 100 and 200 mg kg−1). *P<0.05, #P<0.01 vs before DDTC treatment (Repeated measures-ANOVA). (A) Changes in systolic arterial blood pressure. (B) Changes in mean arterial blood pressure. (C) Changes in heart rate.
Discussion
The results of this study show that NF-κB is rapidly activated by reperfusion as early as 10 min after ischaemia in the in vivo myocardium and that the DNA binding activity of the transcription factor is completely inhibited by administration of DDTC at the time of reperfusion. DDTC had a dose-dependent effect on myocardial infarct size but this was clearly dissociated from inhibition of NF-κB activation. Thus, these results suggest that although NF-κB is activated during acute myocardial infarction, activation plays no detectable deleterious role in the early evolution of acute myocardial infarction.
Characterization of activation of NF κB induced by ischaemia-reperfusion
Several previous studies have reported the activation of NF-κB by myocardial ischaemia-reperfusion in different species. It has been shown that short, sublethal cycles of ischaemia and reperfusion (ischaemic preconditioning) activate NF-κB in rabbit hearts in vivo (Morgan et al., 1999; Xuan et al., 1999) and in perfused rat hearts in vitro (Li et al., 1999; Maulik et al., 1999). However, Li et al. (1999) have also demonstrated that a prolonged, lethal period of ischaemia activates NF-κB in perfused rat hearts and this activation was further increased by reperfusion. Chandrasekar et al. (2001) have shown that there was a 5–6 fold increase in NF-κB activation after 45 min ischaemia followed by 15 min or 30 min reperfusion, after which the activation gradually decreased. This is similar to the finding we report here that severe ischaemia alone was insufficient to activate NF-κB but that reperfusion was necessary for activation that occurs rapidly after the onset of reperfusion. There is recent evidence from Hiasa et al. (2001) that NF-κB activation occurs in two distinct phases during reperfusion following a prolonged ischaemic period. The first phase is immediately after reperfusion and the second phase occurs several hours later probably due to circulating de novo synthesized cytokines and interleukins. A similar pattern of activation of NF-κB has recently been reported by Zingarelli et al. (2002b).
Inhibition of NF-κB activation during myocardial ischaemia-reperfusion
Patterns of gene expression initiated by NF-κB activation are complex. Some gene products that are regulated by NF-κB are expressed in support of cell survival, principally by preventing apoptosis (MnSOD, Bcl-2). Other proteins include cytokines such as the pro- and anti-apoptotic cytokine TNFα, and cell adhesion molecules, frequently characterized as mediators of a ‘deleterious inflammatory' cascade in reperfused myocardium. Adhesion molecules are expressed on vascular endothelial cells, polymorphonuclear leukocytes (neutrophils) and cardiac myocytes (Frangogiannis et al., 1998; Kacimi et al., 1998). Various cell adhesion molecules such as ICAM and P-selectin appear rapidly (within 1 h of reperfusion) on the cell membrane (Frangogiannis et al., 1998; Sun et al., 2001). This will initiate an immune response including the activation of endothelial cells and attached neutrophils. Although the neutrophil activation in response to ischaemia-reperfusion may be associated with the generation of reactive oxygen species, the contribution of neutrophil recruitment and activation during reperfusion as an injurious mechanism contributing to the extent of infarction has not been proven. Indeed, neutrophil recruitment may be a necessary and desirable early step in the response to injury (Baxter, 2002).
Our findings that NF-κB inhibition did not modify reperfusion injury are at odds with other reports supporting a contributory role of NF-κB in the development of myocardial ischaemia-reperfusion injury. Transfection of a decoy oligonucleotide before ischaemia improved post-reperfusion myocardial function in isolated rat hearts (Sawa et al., 1997; Sakaguchi et al., 2001). PR39 and PR11 are novel non-specific inhibitors of NF-κB that have been shown to reduce infarct size in rats when injected directly into the ischaemic zone at the onset of reperfusion (Bao et al., 2001). The protection was attributed to reduced neutrophil infiltration and decreased myeloperoxidase activity in the infarcted myocardium. In the latter study the protection due to suppression of NF-κB was manifested at the later phase of reperfusion eg. 24 h later. In our study the hearts were reperfused for 3 h. Although it is possible that NF-κB-regulated mechanisms such as inflammation do not manifest during the early phase of reperfusion, the close association between infarct size determined after 3–5 h reperfusion and that observed 24–48 h later has been confirmed and validated (Fishbein et al., 1981). Thus, duration of reperfusion is unlikely to account for differences between the studies. It is conceivable that NF-κB-mediated ‘inflammatory' processes are more relevant to the initiation of infarct healing and remodelling several days or weeks following acute injury. Indeed, iNOS knock-out mice were more susceptable to lethal ischaemia-reperfusion induced injury and the damage was increased due to suppressed activation of NF-κB and enhanced apoptosis (Zingarelli et al., 2002b). This study supports our findings that activation of NF-κB by reperfusion after myocardial ischaemia is not fundamentally linked to deleterious mechanisms.
DDTC and mechanism of protection
DDTC has been widely used as an inhibitor of NF-κB but its action is not specific and the mechanism(s) by which NF-κB inhibition by DDTC is achieved are not clear. DDTC has antioxidant as well as pro-oxidant properties (Orrenius et al., 1996). NF-κB activation occurs in response to the generation of oxidative radicals by an unidentified mechanism. It is thought that antioxidants inhibit NF-κB by eliminating the oxidative stimulus. However, these antioxidants may achieve their NF-κB inhibitory effects by other mechanisms. For example, it has recently been shown that the oxyradical scavenger N-acetylcysteine inhibits NF-κB by preventing the degradation of IκB in the cytosol (Pajonk et al., 2002). DDTC also blocks NF-κB activation by mechanisms other than as an antioxidant. DDTC is a cation chelator and by this action may actually become a prooxidant agent. By chelating Cu2+, DDTC creates a complex that will oxidize the essential thiol group of NF-κB, thereby inhibiting its DNA binding activity (Orrenius et al., 1996). Nevertheless, the inhibitory effect of DDTC does not rule out the possibility that as an antioxidant containing two thiol groups it can reduce reperfusion injury or at least delay cell necrosis. It has also been shown that DDTC inhibits caspases (Orrenius et al., 1996) and this may also contribute to the protective effect since caspase inhibitors given during reperfusion have been shown to limit infarct size (Mocanu et al., 2000).
DDTC given intravenously dropped both the systolic and mean arterial blood pressure and this effect was dose dependent. The decrease in these parameters by DDTC was sustained throughout the reperfusion period at the dose of 200 mg kg−1. The possibility that the highest dose of DDTC did not confer protection due to sustained low blood pressure seems unlikely. It has been demonstrated previously that in pentobarbitone anaesthetized rabbits the development of infarction does not depend on the haemodynamic parameters significantly (Ytrehus et al., 1994). Unloading of the left ventricle during ischaemia and reperfusion has been shown not to influence infarct size in isolated rabbit myocardium (van winkle et al., 1990).
Although we did not specifically investigate the mechanism behind the modest protection afforded by DDTC at 50 or 100 mg kg−1, it is possible that it was due to its antioxidant and not its NF-κB inhibitory effect. It has been shown previously that a variety of antioxidants given at reperfusion can reduce infarct size but this effect may be highly unreliable. For example, mercaptopropionylglycine (MPG) infused for the first 4 h of reperfusion markedly reduced the infarction in canine hearts 48 h later (Horwitz et al., 1994). In contrast, Venturini's group could not confirm the protective effect of the antioxidant MPG in a similar canine model of infarction (Venturini et al., 1998). Miki et al. (1999) have shown that MPG given during reperfusion preserved viability of ischaemic rabbit hearts for up to 3 h when it was assessed by tetrazolium staining. However, no protection was detectable three days later when infarct size was assessed by histology (Miki et al., 1999). In relation to the present study, protection with DDTC was not observed at the highest dose examined (200 mg kg−1) and the reason for this difference between low-dose and high-dose treatment is not known at present. Such discrepancies indicate that antioxidants may not be reliable agents for consideration as adjunctive treatments during reperfusion.
Conclusion
Our results suggest that NF-κB activated by reperfusion after myocardial ischaemia does not mediate injury associated with reperfusion during the first 3 h. Although the non-specific NF-κB inhibitor DDTC limited infarct size at low dose, this effect was clearly independent of NF-κB inhibition. However, we cannot exclude the possibility for a role in later reperfusion events, including those that are critical to the initiation of scar formation and longer-term ventricular remodelling. Indeed, since NF-κB is a key regulator of the balance between cell death and survival, the place of therapies directed against this protein should be weighed very carefully.
Acknowledgments
The authors gratefully acknowledge the support of a British Heart Foundation project grant PG/2000027.
Abbreviations
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- COX-2
cyclooxygenase II
- DDTC
diethyldithiocarbamic acid
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- EMSA
electromobility shift assay
- HEPES
N-[2-hydroxyethyl]piperazine-N′-[2-ethansulphonic acid]
- HR
heart rate
- IAF
inhibitor of apoptosis factor
- ICAM
intercellular cell adhesion molecule
- IκB
inhibitor of κ-B
- IL-1
interleukin-1
- IL-6
interleukin-6
- iNOS
inducible nitric oxide synthase
- KCl
potassium chloride, NF-κB, nuclear factor kappa-B
- MABP
mean arterial blood pressure
- MgCl2
magnesium chloride
- MnSOD
manganese-conjugated superoxide dismutase
- MPG
mercaptopropionyl glycine
- NaCl
sodium chloride
- PBS
phosphate-buffered saline
- PMSF
phenylmethylsulphonyl fluoride
- polydIdC
polydeoxyinosinic deoxycytidylic acid
- SABP
systolic arterial blood pressure
- TNFα
tumor necrosis factor alpha
- TTC
triphenyltetrazolium chloride
- VCAM
vascular cell adhesion molecule
References
- BAO J., SATO K., LI M., GAO Y., ABID R., AIRD W., SIMONS M., POST M.J. PR-39 and PR-11 peptides inhibit ischaemia-reperfusion injury by blocking proteasome-mediated IκBα degradation. Am. J. Physiol. Heart Circ. Physiol. 2001;281:H2612–H2618. doi: 10.1152/ajpheart.2001.281.6.H2612. [DOI] [PubMed] [Google Scholar]
- BAXTER G.F. The neutrophil as a mediator of myocardial ischemia-reperfusion injury: time to move on. Basic Res. Cardiol. 2002;97:268–275. doi: 10.1007/s00395-002-0366-7. [DOI] [PubMed] [Google Scholar]
- CHANDRASEKAR B., SMITH J.B., FREEMAN G.L. Ischemia-reperfusion of rat myocardium activates nuclear factor-κB and induces neutrophil infiltration via lipopolysaccharide-induced CXC chemokine. Circulation. 2001;103:2296–2302. doi: 10.1161/01.cir.103.18.2296. [DOI] [PubMed] [Google Scholar]
- FISHBEIN M.C., MEERBAUM S., RIT J., LANDO U., KANMATSUSE K., MERCIER J.C., CORDAY E., GANZ W. Early phase acute myocardial infarct size quantification: validation of the triphenyl tetrazolium chloride tissue enzyme staining technique. Am. Heart J. 1981;101:593–600. doi: 10.1016/0002-8703(81)90226-x. [DOI] [PubMed] [Google Scholar]
- FRANGOGIANNIS N.G., YOUKER K.A., ROSSEN R.D., GWECHENBERGER M., LIDSEY M.H., MENDOZA L.H., MICHAEL L.H., BALLANTYNE C.M., SMITH C.W., ENTMAN M.L. Cytokines and the microcirculation in ischemia and reperfusion. J. Mol. Cell. Cardiol. 1998;30:2567–2576. doi: 10.1006/jmcc.1998.0829. [DOI] [PubMed] [Google Scholar]
- HIASA G., HAMADA M., IKEDA S., HIWADA K. Ischemic preconditioning and lipopolysaccharide attenuate nuclear factor-kappaB activation and gene expression of inflammatory cytokines in the ischemia-reperfused rat heart. Jpn. Circ. J. 2001;65:984–990. doi: 10.1253/jcj.65.984. [DOI] [PubMed] [Google Scholar]
- HORWITZ L.D., FENNESSEY P.V., SHIKES R.H., KONG Y. Marked reduction in myocardial infarct size due to prolonged infusion of an antioxidant during reperfusion. Circulation. 1994;89:1792–1801. doi: 10.1161/01.cir.89.4.1792. [DOI] [PubMed] [Google Scholar]
- KACIMI R., KARLINER J.S., KOUDSSI F., LONG C.S. Expression and regulation of adhesion molecules in cardiac cells by cytokines. Response to acute hypoxia. Circ. Res. 1998;82:576–586. doi: 10.1161/01.res.82.5.576. [DOI] [PubMed] [Google Scholar]
- LI C., BOWDER W., KAO R.L. Early activation of transcription factor NF-κB during ischaemia in perfused rat heart. Am. J. Physiol. Heart Circ. Physiol. 1999;276:H543–H552. doi: 10.1152/ajpheart.1999.276.2.H543. [DOI] [PubMed] [Google Scholar]
- MAULIK N., ENGELMAN R.M., ROUSOU J.A., FLACK J.E., DEATON D., DAS D.K. Ischemic preconditioning reduces apoptosis by upregulating antideath gene Bcl2. Circulation. 1999;100 suppl II:369–375. doi: 10.1161/01.cir.100.suppl_2.ii-369. [DOI] [PubMed] [Google Scholar]
- MIKI T., COHEN M.V., DOWNEY J.M. Failure of N-2-mercaptopropionyl glycine to reduce myocardial infarction after 3 days of reperfusion in rabbits. Basic Res. Cardiol. 1999;94:180–187. doi: 10.1007/s003950050141. [DOI] [PubMed] [Google Scholar]
- MOCANU M.M., BAXTER G.F., YELLON D.M. Caspase inhibition and limitation of myocardial infarct size: protection against lethal reperfusion injury. Br. J. Pharmacol. 2000;130:197–200. doi: 10.1038/sj.bjp.0703336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORGAN E.N., BOYLE E.M., YUN W., GRISCAVAGE-ENNIS J.M., FARR A.L., CANTY T.G., POHLMAN T.H., VERRIER E.D. An essential role for NF-κB in the cardioadaptive response to ischaemia. Ann. Thorac. Surg. 1999;68:377–382. doi: 10.1016/s0003-4975(99)00646-3. [DOI] [PubMed] [Google Scholar]
- ORRENIUS S., NOBEL C.S.I., VAN DEN DOBBELSTEEN D.J., BURKITT M.J., SLATER A.F.G. Dithiocarbamates and the redox regulation of cell death. Biochem. Soc. Transactions. 1996;24:1032–1038. doi: 10.1042/bst0241032. [DOI] [PubMed] [Google Scholar]
- PAJONK F., RIESS K., SOMMER A., McBRIDE W.H. N-acetyl-L-cysteine inhibits 26S proteasome function: implications for effects on NF-κB activation. Free Rad. Biol. & Med. 2002;32:536–543. doi: 10.1016/s0891-5849(02)00743-8. [DOI] [PubMed] [Google Scholar]
- PERKINS N.D. Achieving transcriptional specificity with NF-κB. Int. J. Biochem. Cell Biol. 1997;29:1433–1448. doi: 10.1016/s1357-2725(97)00088-5. [DOI] [PubMed] [Google Scholar]
- SAKAGUCHI T., SAWA Y., FUKUSHIMA N., NISHIMURA M., ICHIKAWA H., KANEDA Y., MATSUDA H. A novel strategy of decoy transfection against nuclear factor-κB in myocardial preservation. Ann. Thorac. Surg. 2001;71:624–630. doi: 10.1016/s0003-4975(00)01906-8. [DOI] [PubMed] [Google Scholar]
- SAWA Y., MORISHITA R., SUZUKI K., KAGISAKI K., KANEDA Y., MAEDA K., KADOBA K., MATSUDA H. A novel strategy for myocardial protection using in vivo transfection of cis element ‘decoy' against NFkB binding site. Circulation. 1997;96 suppl II:280–285. [PubMed] [Google Scholar]
- SCHREIBER E., MATTHIAS P., MULLER M.M., SCHAFFNER W. Rapid detection of octamer binding proteins with ‘mini-extracts', prepared from a small number of cells. Nucleic Acid Research. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUN B., FAN H., HONDA T., FUJIMAKI R., LAFOND-WALKER A., MASUI Y., LOWENSTEIN C.J., BECKER L.C. Activation of NF-κB and expression of ICAM-1 in ischaemic-reperfused canine myocardium. J. Mol. Cell. Cardiol. 2001;33:109–119. doi: 10.1006/jmcc.2000.1280. [DOI] [PubMed] [Google Scholar]
- VALEN G., YAN Z., HANSSON G.K. Nuclear factor kappa-B and the heart. J. Am. Coll. Cardiol. 2001;38:307–314. doi: 10.1016/s0735-1097(01)01377-8. [DOI] [PubMed] [Google Scholar]
- VAN WINKLE D.M., MATSUKI T., GAD N.M., JORDAN M.C., DOWNEY J.M. Left ventricular unloading during reperfusion does not limit myocardial infarct size. Circulation. 1990;81:1374–1379. doi: 10.1161/01.cir.81.4.1374. [DOI] [PubMed] [Google Scholar]
- VENTURINI C.M., FLICKINGER A.G., WOMACK C.R., SMITH M.E., MCMAHON E.G. The antioxidant, N-(2-mercaptopropionyl)-glycine (MPG), does not reduce myocardial infarct size in an acute canine model of myocardial ischaemia and reperfusion. J. Thromb. Thrombolysis. 1998;5:135–141. doi: 10.1023/A:1008830129106. [DOI] [PubMed] [Google Scholar]
- XUAN Y.T., TANG X.L., BANERJEE S., TAKANO H., LI R.C.X., HAN H., QIU Y., LI J.J., BOLLI R. Nuclear factor-κB plays an essential role in the late phase of ischaemic preconditioning in conscious rabbits. Circ. Res. 1999;84:1095–1109. doi: 10.1161/01.res.84.9.1095. [DOI] [PubMed] [Google Scholar]
- YELLON D.M., BAXTER G.F. Protecting the ischaemic and reperfused myocardium in acute myocardial infarction: distant dream or near reality. Heart. 2000;83:381–387. doi: 10.1136/heart.83.4.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YTREHUS K., LIU Y., TSUCHIDA A., MIURA T., LIU G.S., YANG X.M., HERBERT D., COHEN M.V., DOWNEY J.M. Rat and rabbit heart infarction: effects of anesthesia, perfusate, risk zone, and method of infarct sizing. Am. J. Physiol. 1994;267:H2383–H2390. doi: 10.1152/ajpheart.1994.267.6.H2383. [DOI] [PubMed] [Google Scholar]
- ZECHNER D., CRAIG R., HANFORD D.S., MCDONOUGH P.M., SABBADININ R.A., GLEMBOTSKI C.C. MKK6 activates myocardial cell NF-kappaB and inhibits apoptosis in a p38 mitogen-activated protein kinase-dependent manner. J. Biol. Chem. 1998;273:8232–8239. doi: 10.1074/jbc.273.14.8232. [DOI] [PubMed] [Google Scholar]
- ZINGARELLI B., HAKE P.W., DENENBERG A., WONG H.R. Sesquiterpene lactone parthenolide, an inhibitor of IkappaB kinase complex and nuclear factor-kappaB, exerts beneficial effects in myocardial reperfusion injury. Shock. 2002a;17:127–134. doi: 10.1097/00024382-200202000-00008. [DOI] [PubMed] [Google Scholar]
- ZINGARELLI B., HAKE P.W., YANG Z., OCONNOR M., DENENBERG A., WONG H.R. Absence of inducible nitric oxide synthase modulates early reperfusion-induced NF-κB and AP1 activation and enhances myocardial damage. FASEB J. 2002b;16:327–342. doi: 10.1096/fj.01-0533com. [DOI] [PubMed] [Google Scholar]