

Abstract
Cadmium is an extremely toxic metal commonly found in industrial workplaces, a food contaminant and a major component of cigarette smoke. Cadmium can severely damage several organs, including the brain. In this work, we have studied both the cadmium toxicity on rat cortical neurons in culture and the possible protective effect of serum.
Our results indicate that: (1) cadmium is taken up by the neurons in a dose and serum dependent way; (2) cadmium, at concentrations from 1 μM or 10 μM (depending on the absence or the presence of serum) up to 100 μM, decreases the metabolic capacity, which was evaluated by the XTT (tetrazolium salt) test; (3) cadmium induces apoptosis and LDH (lactate dehydrogenase) release in a dose dependent way; (4) in a serum-free medium, the cadmium-induced apoptosis is accompanied by caspase-3 activation; (5) both the caspase-3 activation and the cadmium-induced apoptosis are reversed by N-acethyl-Asp-Glu-Val-Asp-aldehyde (Ac-DEVD-CHO), a selective caspase-3 inhibitor, indicating that the caspase-3 pathway is involved in cadmium-induced apoptosis in cortical neurons; and (6) the cadmium concentrations which produce caspase-3 activation do not modify the intracellular ATP levels; however, higher cadmium concentrations lead to both intracellular ATP depletion and ATP release, but do not increase the caspase-3 activity, indicating that cadmium also produces cellular death by necrosis.
These results suggest that cadmium induces either apoptosis or necrosis in rat cortical neurons, depending on the cadmium concentration.
Keywords: Cortical neurons, cadmium toxicity, apoptosis, caspase-3, necrosis
Introduction
Cadmium is a potent cell poison, which causes different types of damage, including cell death or increase in cell proliferation. These toxic effects of cadmium are worrying because it is a highly toxic environmental pollutant released from the smelting and refining of metals and cigarette smoking. Its very long biological half-life (30 years) is responsible for the almost irreversible accumulation in human organs (Nordberg, 1984), thus causing several adverse effects. Food and cigarette smoke are the biggest sources of cadmium exposure for people in the general population (Waalkes et al., 1992). Oral exposure to cadmium may result in adverse effects on a number of tissues, including kidney (Sudo et al., 1996), liver (Koizumi et al., 1996), bone, thyroid gland, testes (Xu et al., 1996; Jones et al., 1997), the immune system, and the cardiovascular system. Cadmium also is believed to cause pulmonary emphysema. Due to its high blood–brain barrier permeability (Wong & Klaassen, 1982; Babitch, 1988; Sutoo, 1994; Choudhuri et al., 1996; Shukla et al., 1996), cadmium also affects the nervous system, and neurological disorders such as learning disabilities and hyperactivity in children may occur (Pihl & Parkes, 1977; Marlowe et al., 1985). Parkinsonism after acute exposure to cadmium has been described by Okuda et al. (1997), and neurochemical disturbances of the serotonergic system in the offspring during the lactational period have been shown in rats exposed to low levels of cadmium in the drinking water (Andersson et al., 1997). In neuronal cells, cadmium induces oxidative stress, which produces protein damage (Stadtman, 1992; Figueiredo-Pereira et al., 1998) and subsequently neurodegeneration (Coyle & Puttfarcken, 1993; Cohen & Werner, 1994; Williams, 1995; Okuda et al., 1997). Cadmium is known to enhance the production of free radicals in the brain (Shukla et al., 1987) and to interfere with the cellular defence mechanism against oxidation (Shukla et al., 1989). The toxic effects of cadmium are rather complex and still debated. Some authors have suggested that cadmium induces apoptosis in several cell-types (el Azzouzi et al., 1994; Habeebu et al., 1998; Ishido et al., 1999; Tsangaris & Tzortzatou-Stathopoulou, 1998; Kim et al., 2000; Risso de Faverney et al., 2001; Kondoh et al., 2002) and recent studies indicate that the cadmium-induced apoptosis can be inhibited by Zn and other caspase-3 inhibitors (Perry et al., 1997; Shimada et al., 1998; Galán et al., 2000). On the other hand, Yuan et al. (2000) have found that cadmium blocks the chromium-induced apoptosis in Chinese hamster ovary cells (CHO K1-BH4), via the inhibition of caspase-3. Thus, these results establish the question whether caspases are involved in apoptosis.
Cadmium may bind to intracellular proteins, including tubulin, troponin C and calmodulin (Akiyama et al., 1990), as well as human metallothionein (Masters et al., 1994). In addition, cadmium may bind to plasma transferring (de smet et al., 2001), serum albumin (Zhou et al., 1992; Goumakos et al., 1991) and glycoproteins (Zak et al., 1978). Beside that, cadmium is a potent inhibitor of voltage-dependent calcium channels, which have been suggested to be the primary route for the cadmium uptake by some secretor cells like pituitary GH4C1 cells (Hinkle et al., 1987), GH3 cells (Hinkle et al., 1992), rat pheochromocytoma PC12 cells (Hinkle & Osborne, 1994), rat melanotrophs (Shibuya & Douglas, 1992) and even mammalian neurons (Usai et al., 1999). Although cadmium toxicity is well proved, its cytotoxic effect is controversial, given that some authors have indicated that cadmium can kill cells after a prolonged exposure (Kaji et al., 1993), while others emphasize that it is a carcinogen in animals and humans (Jin & Ringertz, 1990; Koizumi & Li, 1992; Kazantzis et al., 1992). Since cadmium is a toxic agent for several cells, the aim of this work was to study the mechanism through which cadmium induces cell death on cortical neurons and the hypothetical protective role of serum, since cadmium may bind to the −SH groups of serum proteins.
Methods
Materials
Eagle's Minimum Essential Medium (EMEM) was obtained from Bio-Whittaker (Verviers, Belgium). Foetal calf serum (FCS) and horse serum (HS) were purchased from Sera-Lab (Sussex, U.K.). Cytosine arabinoside, poly-D-lysine, glutamine, penicillin G, streptomycin, rabbit polyclonal anti-GFAP and anti-rabbit IgG FITC antibodies were obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.). XTT test and Kinesis 50 test were purchased from Boehringer Mannheim (Mannheim, Germany), Annexin-V FLUOS test from Roche and ATP reagents from BioOrbit (Turku, Finland). Ac. DEVD-AMC [N-acethyl-Asp-Glu-Val-Asp-AMC (7-amino-4-methyl coumarin)] and Ac. DEVD-CHO were from Pharmingen. Other chemicals were reactive grade products from Merck (Darmstadt, Germany).
Cell culture
Cortical neurons were obtained from foetal rats at 19 days of gestation as described by Segal (1983) with minor modifications. Cell suspensions were plated at a density of 106 cells ml−1 in plastic Petri dishes coated with poly-D-lysine (10 mg ml−1) in EMEM supplemented with 0.6% glucose, 0.3 g l−1 glutamine, 100 μ ml−1 penicillin G, 100 μg ml−1 streptomycin, 10% (v v−1) FCS and 10% (v v−1) HS. Cells were incubated at 37°C in a humidified atmosphere containing 5%CO2/95% air. After 72 h of culture, the medium was changed and cells were incubated in fresh medium supplemented with cytosine arabinoside at a final concentration of 10 μM to prevent contaminating glial cell proliferation. Neurons were used on day 7. Cell viability was assessed by the trypan blue dye exclusion method and it was routinely higher than 95%. Both cell staining with cresyl violet to identify neurons and the specific anti-GFAP antibody to identify glial cells, were used to check the cell culture purity.
Assay of glial cell contamination
After 7 days of culture, cortical neurons were detached from the culture plates with 0.25 mM trypsin, which contained 1 mM EDT in Hank's balanced salt solution (HBSS). Once the cells were detached, 0.5 ml FCS was added in order to neutralise the trypsin action. The cell suspension was then centrifuged at 4000×g for 5 min. After the centrifugation, cells were fixed by 2% (w v) paraformaldehyde in PBS for 30 min and then washed with PBS. Subsequently, cells were incubated for 1 h with anti-GFAP antibody diluted 1 : 500 in PBS at room temperature. After a further wash with PBS, anti-rabbit IgG FITC conjugated was applied and incubated for 30 min as before. The secondary antibody was diluted 1 : 100 in PBS prior to addition. After a final wash with PBS, the glial cells were identified by flow cytometry analysis. Under these conditions the positive cells (glial contamination) were 9±3%.
Cadmium treatment
Neurons were incubated with cadmium chloride at concentrations between 100 nM and 100 μM in the absence or the presence of 5% FCS and 5% HS for several periods of time.
Cadmium determination
Neurons were exposed for 24 h at 37°C to different cadmium chloride concentrations in the absence or the presence of serum. After incubation, the medium was discarded and the cell layer was washed twice with PBS. The cell layer was exposed to 1 ml 0.1 M of acetate buffer (pH 5.5) containing 10 mM EDTA, for 72 h at 4°C. The extract was collected and the remaining cells were gently washed with 1 ml of the same buffer; the wash was combined with the extract. The extract was analysed for the content of cadmium by flameless atomic absorption spectrophotometry (Perkin Elmer 2280).
XTT test
This assay is based on the ability of metabolically active cells to reduce tetrazolium salt (XTT) to coloured formazan compounds; dead cells do not. Thus, this assay detects viable cells exclusively. The dye intensity can be read at 450 nm/630 nm wavelengths with a spectrophotometer. The intensity of the dye is proportional to the number of metabolically active cells. Neurons plated in a flat 96-well plate and exposed to different concentrations of cadmium chloride, were washed with PBS and treated with the reaction solution according to the Kit specifications. The absorbance of the samples was measured with an ELISA reader (wavelength 450 nm; reference wavelength 630 nm). Results are expressed as percentages with regard to the controls (untreated cells).
Assessment of cell morphology
Neuronal morphology was assessed by Phase Contrast Microscopy at 20× after various time intervals of incubation with cadmium. After the exposure to cadmium, neurons were washed twice with PBS. Cells were fixed by p-formaldehyde (4%; w v) for 30 min at room temperature. Finally the neurons were washed with PBS and observed under the light microscopy.
Propidium iodide staining
After the incubation with cadmium, the neurons were stained for 15 min with 0.005% of propidium iodide dissolved in PBS. Then, the cells were observed under fluorescence microscopy (Nikon Diaphot 300). Several fields were observed per condition.
Lactate dehydrogenase (LDH) release
An LDH assay was performed measuring the reduction of pyruvic acid to lactic acid according to the following reaction:
The NADH transformation in NAD was followed by monitoring the absorbance with a spectrophotometer at 340 nm. For the determination of LDH, the medium and the cell lysate were collected after 24 h of treatment. Neurons were lysed with 0.1 M Tris-HCl (pH 7.4) containing 0.1% Triton X-100. LDH activity was measured in both culture medium and cell lysate, following the addition of 1 mM pyruvate and 0.2 mM NADH in 0.1 M Tris-HCl (pH 7.4) in a volume of 1 ml. LDH release is given as percentage of LDH found in the culture medium with respect to the total LDH (the sum between LDH in the culture medium plus LDH inside the cells).
ATP content and ATP release
ATP was determined by chemiluminescence. This method is based on the measurement of ATP degradation by the luciferine/luciferase system according to the following reaction:
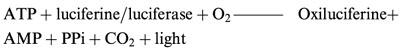 |
The light intensity is measured at 560 nm in a spectroluminometer (BioOrbit 1251). Culture medium was taken and ATP measured using a commercially available kit (ATP Biomass kit) following the kit instructions.
Cells, plated on ice, were washed twice with PBS and then lysed with 10 mM Tris-HCl (pH 7.5) containing 0.1% Triton X-100. The cell lysate was centrifuged at 4°C and 13,000 r.p.m. (Eppendorf centrifuge 5415 C) for 5 min. The supernatants were taken and placed on ice and the ATP immediately determined. The ATP inside the cells was considered as the intracellular ATP content. The released ATP was considered to be that found in the culture medium. The released ATP values are expressed as percentage of ATP in the culture medium over the total ATP content (ATP in the culture medium+ATP inside the cells).
Determination of neuronal apoptosis by flow cytometry
Apoptosis was assayed using a commercially available kit (Kinesis 50, BioRad), following the manufacturer's instructions. Neurons plated on Petri dishes and treated as indicated above were harvested using 0.02% EDTA. After centrifugation at 4000×g for 5 min, cell pellets were incubated with 0.25 ml PBS and 0.25 ml of the reactive solution, which includes propidium iodide (Kinesis 50) for 30 min in the dark at room temperature. Apoptotic neurons were quantified by flow cytometry (FACS-Scan Flow Cytometer, Becton-Dickinson).
Annexin test
This test is based on the property of the apoptotic cells to induce membrane changes as the translocation of the phosphatidylserine (PS) from the inner face of the plasma membrane to the outer leaflet. Under these conditions, Annexin-V may mark PS according to the specifications of the test. Cells were analysed under a confocal microscope.
Assay for caspase-3 activity
After various time intervals of incubation with cadmium, the cortical neurons were washed with PBS and then lysed with the following buffer (in mM): Tris-HCl 10 (pH 7.5), NaCl 130, NaH2PO4 10, Na2HP2O4 10, Triton X-100 0.1% and natriumpyrophosphate 10. The lysates were centrifuged at 5000×g for 5 min in an Eppendorf centrifuge 5415 C. The supernatant was assayed for caspase-3 activity without further purification. Cytosolic protein (at least 20 μg) was incubated with 20 μM of Ac. DEVD-AMC in 150 μl of incubation buffer (20 mM HEPES -pH 7.5-, 10% glycerol, 2 mM DTT). The release of AMC, after 2–4 h, was monitored using a spectrofluorimeter (Bio-Tek FL 600) (excitation at 380 nm, emission at 430–460 nm). Under these conditions the reaction was linear between 0–4 h, or even longer. In those experiments, in which caspase-3 activity was measured in the presence of the caspase-3 inhibitor (Ac. DEVD-CHO), this was added at a final concentration of 50 nM. Caspase-3 activity is expressed as fluorescent arbitrary units (FAU) μg−1 protein 2 h−1.
Protein determination
The protein concentration was determined by the Bradford assay (Bradford, 1976).
Data presentation
Data are expressed as mean±s.e.mean of three or four independent experiments, each one performed in duplicate with different batches of cells. Statistical comparisons were made using the Student's t-test.
Results
Cadmium uptake
As it is well demonstrated that cadmium is a contaminant cation that indices neurological disorders, the first purpose of this study was to check whether the cortical neurons in culture were able to uptake cadmium. Cadmium was taken up by the neurons in a way which was dependent on both the dose and the presence or absence of serum in the treatment medium (Figure 1). The rate of cadmium uptake was highly significant from a concentration of 1 μM, showing non-linear dose dependence. The entry of cadmium inside the cells could not be assessed at lower concentrations because of the low sensitivity of the method employed. After the treatment with 100 μM cadmium, the uptake in the absence of serum was six times higher than the uptake in a medium supplemented with serum (317±22 μg Cd2+ mg−1 protein and 50.0±3.1 μg Cd2+ mg−1 protein, respectively).
Figure 1.
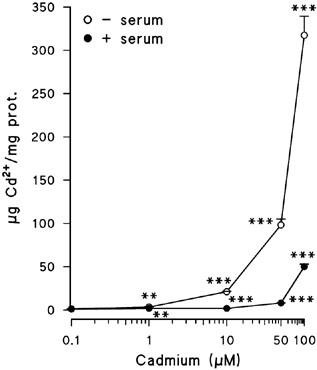
Cadmium uptake by cortical neurons treated for 24 h with the indicated cadmium concentrations, either in the presence or in the absence of serum. Results are expressed as μg Cd2+ mg−1 protein. Data are mean±s.e.mean of two separate experiments from cells of different cultures, each one performed in triplicate. **=P<0.01 and ***=P<0.001.
Cell metabolic capacity
The metabolic capacity was measured using the XTT test (Figure 2). The exposure of cortical neurons to 1 nM–100 μM cadmium in the absence of serum induced a decrease of their metabolic capacity from a cadmium concentration of 1 μM. In the presence of serum, the drop of metabolic activity was clear from a cadmium concentration of 10 μM. The cadmium decrease was always lower in a medium with serum than in its absence, but showed dose-dependence in both cases. All these results taken together indicate that serum protects cortical neurons from cadmium toxicity.
Figure 2.

Cortical neurons were incubated for 24 h with the indicated cadmium concentrations, either in the absence or in the presence of serum. Cell viability was checked by the XTT test. Results are expressed as percentages over control. Data are mean±s.e.mean of three separate experiments from cells of different cultures, each one performed in duplicate. ns=not significant. **=P<0.01 and ***=P<0.001.
Effect of cadmium treatment on neuronal morphology
Results from Figure 3 were obtained after the treatment of the cells with 10 μM of cadmium either in the presence or the absence of serum and at different treatment times. Cadmium modified the neuronal morphology after a 6 h-treatment in a serum-free medium (Figure 3B), whereas at 24 h it showed a great loss of neuronal integrity mainly evidenced by the almost complete disappearance of the axons (Figure 3C). However, when this treatment was performed in a medium supplemented with serum there were no morphological changes at 6 h (Figure 3E), while at 24 h they could be seen, but always to a lesser extent than in a serum-free medium (Figure 3F).
Figure 3.

Cadmium effect on morphology of cortical neurons. (A, B, C) treatment in the absence of serum; (D, E, F) in the presence of serum. (A and D) control neurons; (B and E) 6 h of treatment with 10 μM cadmium; (C and F) 24 h of treatment with 10 μM cadmium.
Analysis of cell-death induced by cadmium
Propidium iodide is a dye, which stains DNA in such a way that exclusion of propidium iodide is a signal of cellular integrity. The images from Figure 4 were obtained after propidium iodide staining in the absence of serum. They show a higher number of nuclei dying in neurons treated with 100 μM of cadmium than in control cells.
Figure 4.

Analysis of cellular death induced by cadmium by propidium iodide staining: (A) control neurons; (B) cortical neurons treated with 100 μM cadmium for 24 h in the absence of serum.
LDH release
Cadmium induced a dose-dependent LDH release to the extracellular medium (Figure 5). After 24 h of exposure in a serum-free medium, doses of 1 μM cadmium and higher induced LDH release. In a medium supplemented with serum, after 24 h of treatment, 10 μM cadmium and higher concentrations induced LDH release. LDH release was always higher in a medium deprived of serum than in a medium supplemented with serum.
Figure 5.

Effect of cadmium on LDH release. Cortical neurons were incubated with the indicated cadmium concentrations for 24 h, either in the absence or in the presence of serum. Results are expressed as percentages of LDH release after subtracting the control values. Data are mean±s.e.mean of three separate experiments from cells of different cultures, each one performed in triplicate. ns=not significant, **=P<0.01 and ***=P<0.001.
Thus, cadmium induces necrosis, this effect being dependent on (1) cadmium concentration and (2) the presence or the absence of serum in the treatment medium. Similarly to the results of metabolic ability, serum seems to protect cortical neurons from cadmium-induced necrosis.
Apoptosis induced by cadmium
Since lower concentrations of cadmium than 1 μM did not induce necrotic cell death, it was studied whether these concentrations could induce cell death by apoptosis. The results obtained (Figure 6) by flow cytometry indicated that the exposure to 100 nM and 1 μM of cadmium in the absence of serum caused apoptosis, but higher doses did not. On the other hand, in the presence of serum, cadmium induced apoptotic cell death at all the studied concentrations. Under those conditions in which cadmium induced apoptosis, in the absence of serum, the apoptotic event was always higher than in the presence of serum. It should be pointed out that serum deprivation itself induces apoptosis.
Figure 6.
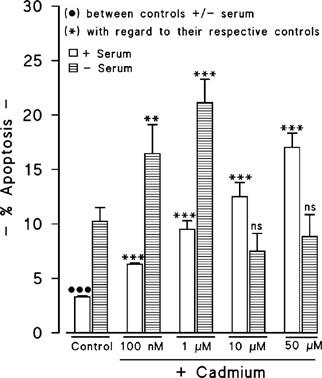
Quantification by flow cytometry of cadmium-induced apoptosis. Results are expressed as percentages of apoptosis with regard to the total cells. Data are mean±s.e.mean of three separate experiments from cells of different cultures, each one performed in duplicate. ns=not significant. **=P<0.01, *** or •••P<0.001.
Apoptosis measurement by microscopic analysis of apoptotic cortical neurons
In the early stages of apoptosis changes occur on the cell surface (Andree et al., 1990; Fadok et al., 1992). One of the membrane alterations is the translocation of phosphatidylserine (PS) from the inner face of the plasma membrane to the outer leaflet, by which PS becomes exposed at the external surface of the cells (Vermes et al., 1995). Annexin-V is a Ca2+-dependent phosphatidyl-binding protein, which shows a high affinity for PS. Thus, this protein may be used as a sensitive probe for membrane alterations and as a consequence, it may be suitable to detect apoptotic cells. But, since necrotic cells also expose PS according to the loss of the membrane integrity, apoptotic cells have to be differentiated from these necrotic cells. So, simultaneous DNA staining which is used for the dye exclusion test, allows the discrimination of necrotic cells from the Annexin-V positively-stained cell cluster. The images from Figure 7 show stained cells with both Annexin-V FLUOS and propidium iodide. One may observe both apoptotic cells (Figure 7C) and necrotic cells (Figure 7D) when neurons were treated with 10 μM cadmium for 8 h in a serum-free medium (Figure 7B). However, there were no PS positive cells in the control conditions (cells not treated with cadmium) (Figure 7A).
Figure 7.

Detection of the early phase of apoptosis induced by cadmium. (A) control neurons; (B) neurons treated with 1 μM of cadmium in a serum-free medium for 8 h; (C) apoptotic cells; (D) necrotic cells.
ATP content and ATP release
Necrotic neuronal death has been associated with an early loss of ATP. However, ATP is thought to be required for the apoptotic pathway (Ankarcrona et al., 1995; 1996; Bonfoco et al., 1995). In order to study the possible involvement of ATP concentrations in the cadmium-induced neuronal death, the intracellular ATP concentration was measured following the exposure of the cortical neurons to several cadmium concentrations for 24 h in the absence of serum (Figure 8). Cadmium concentrations of 0.1 μM and 1 μM did not alter the ATP levels with regard to the control, but higher doses induced almost a total depletion of the intracellular ATP levels. Additionally, 1 μM and higher doses of cadmium evoked ATP release to the extracellular medium in a dose-dependent way. This ATP release was inversely correlated with the intracellular ATP content.
Figure 8.
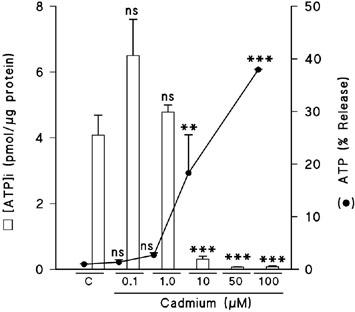
Effect of cadmium on ATP intracellular levels and ATP release. Cells were exposed for 8 h to the indicated cadmium concentrations in a serum-free medium. Results are expressed as percentages of ATP release or pmol [ATP]i μg−1 protein. Data are mean±s.e.mean of three experiments from cells of different cultures, each one performed in duplicate. ns=not significant, **=P<0.01, ***=P<0.001.
Caspase-3 activity
The caspase family of proteases plays a crucial role in apoptotic neuronal death. To identify the apoptotic mechanism involved in the cadmium-mediated neuronal death, the caspase-3-like activity in cortical neurons after the exposure to cadmium in the absence of serum for 24 h was investigated (Figure 9). A cadmium concentration of 1 μM, which had induced the highest apoptosis, increased caspase-3 activity, whereas higher cadmium concentrations (10 μM) did not affect or even inhibited caspase-3 activity with regard to the control.
Figure 9.

Effect of cadmium on caspase-3-like activity. Cortical neurons were treated with different cadmium concentrations in the absence of serum. Results are expressed as FAU μg−1 protein 2 h−1 incubation. Data are mean±s.e.mean of three separate experiments from cells of different cultures, each one performed in triplicate. ns=not significant, ***=P<0.001.
The addition of the caspase-3 inhibitor tetrapeptide Ac-DEVD-CHO (50 nM) reversed the cadmium-induced up-regulation of caspase-3 activity (Figure 10A). The inhibitor even decreased the caspase-3 activity found in the control. The caspase-3 inhibitor also decreased the 1 μM cadmium-induced apoptosis (Figure 10B), without affecting the apoptosis found in the control cells.
Figure 10.

Effect of caspase-3 inhibitor on the cadmium-induced modification of caspase-3 activity (A) and apoptosis (B). Cortical neurons were treated with 1 μM of cadmium in a serum-free medium for 24 h, in the absence or the presence of the caspase-3 inhibitor. Untreated cells were exposed to the inhibitor in order to discover the action of this inhibitor itself on the studied parameters. The insets in Figure 10B represent single results from flow cytometry analysis. Several of these were used to perform the statistical data indicated in bars. ns=not significant, **=P<0.01, *** or •••=P<0.001.
Discussion
Although the results presented in this work indicate that cadmium is taken up by the neurons after the exposure to 1 μM or higher doses of cadmium for 24 h, we think that lower doses may also be taken up by these cells, although we were unable to detect it because of the low sensitivity of the atomic spectrophotometric method used in the cadmium measurement.
The plotting of the uptake rate of cadmium at various concentrations was non-linear, suggesting that, in addition to a simple diffusion, other transport mechanisms might be involved. Thus, the cadmium entry could occur through the same pathways of Ca2+ influx as it has been already suggested by several authors in different cell types (Limaye & Shaikh, 1999; Souza et al., 1996; Quamme, 1992; Hinkle et al., 1987; 1992; Hinkle & Osborne, 1994; Kanthasamy et al., 1995; Usai et al., 1999). The cadmium uptake in cortical neurons was dependent on both the cadmium concentration and the presence or the absence of serum in the culture medium.
The amount of cadmium uptaken by the neurons after the incubation with 100 μM of cadmium in the absence of serum was about six times higher than the uptake measured in the presence of serum. These results indicate that serum may protect neurons from cadmium toxicity. The mechanism through which serum would protect the neurons might be due to the high affinity of cadmium to the sulfhydryl groups of serum proteins (Savolainen, 1995; Jungmann et al., 1993). In this case, the serum proteins would sequester cadmium, so that the free cadmium concentration in a medium with serum would be lower.
Since cadmium enters the neurons and is implicated in neurological disorders (Pihl & Parkes, 1977) we wanted to study its possible toxic effect on these cells. An XTT test showed that cadmium affects the neuronal viability in both a dose- and serum-dependent way. The results obtained with this test indicated that after 24 h of treatment with cadmium, the loss of neuronal viability was clear at concentrations of 1 μM cadmium or higher in a serum-free medium and from 10 μM to 100 μM cadmium in a medium supplemented with serum. These results suggest once more the protective effect of serum against cadmium toxicity. Time- and dose-dependent cadmium-induced toxicity has been described by some authors in other cell types (Figueiredo-Pereira et al., 1998; Warren et al., 2000; Almazán et al., 2000).
Cadmium also affected the cell morphology after 6 h of treatment in a serum-free medium, while in a medium with serum the morphological changes were clearly shown at 24 h, indicating once more that serum protects against cadmium damage. These morphological changes were mainly located in the neural extensions (axons and dendrites), which almost disappeared after 24 h of treatment with 1 μM of cadmium in a serum-free medium. Such morphological changes induced by cadmium have also been described in other cells (Bucio et al., 1995). These results indicate that cadmium induces cytotoxicity on cortical neurons although we could not distinguish between necrosis and apoptosis. In order to clarify whether the cadmium-induced cytotoxic effect was due to a necrotic and/or apoptotic mechanism, several parameters associated with both mechanisms were studied. Our results performed with propidium iodide showed a higher staining in neurons treated with 100 μM of cadmium than in non-treated cells, indicating alterations in the plasma membrane and death by a necrotic process. An increase in the propidium iodide signal induced by cadmium has also been described by Koizumi et al. (1996) in hepatocytes and by Almazán et al. (2000) in oligodendrocytes. The cadmium-induced LDH release also suggests a necrotic process. The LDH release was higher when the exposure to cadmium was performed in the absence of serum. Under these conditions, the exposure to 1 μM of cadmium for 24 h was enough to release LDH, while in the presence of serum, concentrations of 50 μM or higher were required. The induction of necrosis by cadmium in cortical neurons was dependent on the cadmium concentration. The cadmium-induced necrosis accompanied by LDH release has been found in several cell types (Bucio et al., 1995; Koizumi et al., 1996; Zimmerhackl et al., 1998).
In addition, the bioenergetic depletion induced by high cadmium concentrations (evidenced by a decrease in the intracellular ATP levels and the ATP release to the extracellular medium) is also a hallmark of necrosis. The high ATP release accompanied by the decrease in the ATP levels suggests an important damage in the cell plasma membrane, responsible for the cell death by necrosis. Decrease in the intracellular ATP levels besides LDH release induced by cadmium was also described by Zimmerhackl et al. (1998) in LLC-PK1 cells and Muller-Taubenberger et al. (1988), who indicated that cadmium doses of 10 μM and 100 μM decreased the ATP/ADP ratio and increased the lipid peroxidation in cultured rat hepatocytes.
Our results obtained by flow cytometry, showed that cortical neurons exposed to 100 nM to 1 μM of cadmium in the absence of serum for 24 h underwent apoptosis. However, higher cadmium concentrations did not increase the apoptosis with regard to the control neurons. When the cadmium treatment was performed in the presence of serum, there was a dose-dependent apoptotic increase from 100 nM to 50 μM of cadmium.
On the other hand, neurons exposed to 1 μM cadmium in the absence of serum exhibited high apoptosis (measured by flow cytometry) and slight LDH release, but they also showed both translocation of phosphatidylserine to the outer side of the plasma membrane (assessed by using Annexin-V FLUOS) and DNA staining with propidium iodide, which indicate apoptosis and necrosis, respectively. This dual mechanism induced by cadmium is not surprising because as Nicotera et al. (1999) comment, classical apoptosis and necrosis represent only the extreme ends of a wide range of possible morphological and biochemical deaths and these two death types can occur simultaneously in tissues and cell cultures exposed to the same stimulus and, often, the intensity of the same initial insult decides the prevalence of either apoptosis or necrosis. In our case, the cadmium concentration of 1 μM seems to represent the switch between apoptosis and necrosis.
Presence of phosphatidylserine on the outer face of membrane induced by cadmium has also been evidenced in U-937 cells from human lymphoma (Li et al., 2000) and in rat fibroblasts (Kim et al., 2000). The induction of apoptosis mediated by low cadmium concentrations (1 μM) seems to involve caspase-3 activation. This process is not accompanied by a decrease of the intracellular ATP levels, which means that the energetic depletion in cortical neurons only occurs in a necrotic death or in an advanced phase of the apoptosis. These results agree with those found by Eguchi et al. (1997), who suggested that ATP is critical to induce several apoptotic events. On the other hand, Nicotera et al. (2000) indicate that ATP depletion during the early phases of apoptosis can preclude caspase activation, and consequently switch execution of cell death towards necrosis. The participation of the caspase pathway in the cadmium-induced apoptosis in cortical neurons is supported by the fact that when cortical neurons were exposed to this cation in the presence of a caspase-3 inhibitor, there was a reversal of the caspase-3 activation and the apoptosis was prevented. Induction of apoptosis mediated by cadmium through a caspase-3-related mechanism has been found for several authors in different cell preparations (Li et al., 2000; Kim et al., 2000).
The decrease in caspase-3 activity mediated by the highest cadmium concentration employed, may be due to the cadmium inhibition of the enzyme activity. This cation can bind to the sulfhydryl groups of proteins and peptides (Rastogi et al., 1977), and caspases are cysteine-proteases with −SH groups in their active sites (Earnshaw et al., 1999). The oxidation of these −SH groups could alter the enzyme, and subsequently inhibit its activity. Caspase-3 inhibition by cadmium has been shown by Yuan et al. (2000) in CHO K1-BH4 cells.
Cadmium may be responsible for several disturbances in neurons. Thus, it might bind to cytoskeletal proteins, since several authors have already found that cadmium produces cytoskeletal disruption and subsequently cell death (Wang & Templeton, 1996).
In summary, the data presented here clearly demonstrate: (1) that cadmium is a potent toxin for cortical neurons, since very low concentrations (100 nM) are able to induce cell death by apoptosis and higher concentrations (10–100 μM) produce necrotic death of 50% of the neurons in a medium free of serum and a 30% in a medium with serum, judging by the viability tests; (2) that serum protects neurons against cadmium toxicity probably by a mechanism in which cadmium may be sequestered by its binding to the serum proteins; (3) that the high ATP release mediated by cadmium at the highest doses of cadmium (10–100 μM) may probably be due to a necrotic process; and (4) that the cadmium-induced apoptosis in cortical neurons in the absence of serum involves the caspase pathway.
Acknowledgments
The CAYCIT 98-0121 and CAM 08.8/0012/1998 supported this work. E. López and S. Figueroa are recipients of Fellowships from Ministerio de Educación y Ciencia and UCM, respectively. We thank M. García Mauriño for helping us in the culture preparations.
Abbreviations
- Ac. DEVD-AMC
N-Acethyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin
- AMC
7-amino-4-methylcoumarin
- EMEM
Eagle's Minimum Essential Medium
- FAU
fluorescent arbitrary units
- FCS
foetal calf serum
- FITC
fluorescein isothiocyanate isomer
- GFAP
glial fibrillar acidic protein
- HBSS
Hank's balanced salt solution
- HS
horse serum
- PBS
phosphate buffered saline
- PS
phosphatidylserine
References
- AKIYAMA K., SUTOO D., REID D.G. A 1H-NMR comparison of calmodulin activation by calcium and by cadmium. J. Pharmacol. 1990;53:393–401. doi: 10.1254/jjp.53.393. [DOI] [PubMed] [Google Scholar]
- ALMAZÁN G., LIU H.N., KHORCHID A., SUNDARAJAN S., MARTÍNEZ-BERMÚDEZ A.K., CHEMTOB S. Exposure of developing oligodendrocytes to cadmium causes HSP72 induction, free radical generation, reduction in glutathione levels, and cell death. Free Radic. Biol. Med. 2000;29:858–869. doi: 10.1016/s0891-5849(00)00384-1. [DOI] [PubMed] [Google Scholar]
- ANDERSSON H., PETERSSON-GRAWE K., LINDQVIST E., LUTHMAN J., OSKARSSON A., OLSON L. Low-level cadmium exposure of lactating rats causes alterations in brain serotonin levels in the offspring. Neurotoxicol. Teratol. 1997;19:105–115. doi: 10.1016/s0892-0362(96)00218-8. [DOI] [PubMed] [Google Scholar]
- ANDREE H.A., REUTELINGSPERGER C.P., HAUPTMANN R., HEMKER H.C., HERMENS W.T., WILLEMS G.M. Binding of vascular anticoagulant alpha (VAC alpha) to planar phospholipid bilayers. J. Biol. Chem. 1990;265:4923–4928. [PubMed] [Google Scholar]
- ANKARCRONA M., DYPBUKT J.M., BONFOCO E., ZHIVOTOVSKY B., ORRENIUS S., LIPTON S.A., NICOTERA P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- ANKARCRONA M., DYPBUKT J.M., ORRENIUS S., NICOTERA P. Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Lett. 1996;394:321–324. doi: 10.1016/0014-5793(96)00959-3. [DOI] [PubMed] [Google Scholar]
- BABITCH J.A.Cadmium neurotoxicity Metal Neurotoxicity 1988Boca Raton, Florida: CRC Press Inc; 141–166.eds. Bondy, S. & Prasad, K. [Google Scholar]
- BONFOCO E., KRAINC D., ANKARCRONA M., NICOTERA P., LIPTON S.A. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRADFORD M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- BUCIO L., SOUZA V., ALBORES A., SIERRA A., CHÁVEZ E., CARABEZ A., GUTIÉRREZ-RUÍZ M.C. Cadmium and mercury toxicity in a human foetal hepatic cell line (WRL-68 cells) Toxicology. 1995;102:285–299. doi: 10.1016/0300-483x(95)03095-w. [DOI] [PubMed] [Google Scholar]
- COHEN G., WERNER P. Neurodegenerative Diseases 1994Philadelphia: W.B. Sanders Co; 139–161.eds. Cone, D. & Saunders, W.B. [Google Scholar]
- COYLE J.T., PUTTFARCKEN P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- CHOUDHURI S., LIU V.L., BERMAN N.E., KLAASSEN C.D. Cadmium accumulation and metallothionein expression in brain of mice at different stages of development. Toxicol. Lett. 1996;84:127–133. doi: 10.1016/0378-4274(95)03444-7. [DOI] [PubMed] [Google Scholar]
- DE SMET H., BLUST R., MOENS L. Cadmium-binding to tranferrin in the plasma of the common carp Cyprinus carpio. Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 2001;128:45–53. doi: 10.1016/s1532-0456(00)00171-x. [DOI] [PubMed] [Google Scholar]
- EARNSHAW W.C., MARTINS L.M., KAUFMANN S.H. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- EGUCHI Y., SHIMIZU S., TSUJIMOTO Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- EL AZZOUZI B., TSANGARIS G.T., PELLEGRINI O., MANUEL Y., BENVENISTE J., THOMAS Y. Cadmium induces apoptosis in a human T cell line. Toxicology. 1994;88:127–139. doi: 10.1016/0300-483x(94)90115-5. [DOI] [PubMed] [Google Scholar]
- FADOK V.A., SAVILL J.S., HASLETT C., BRATTON D.L., DOHERTY D.E., CAMPBELL P.A., HENSON P.M. Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognise and remove apoptotic cells. J. Immunol. 1992;149:4029–4035. [PubMed] [Google Scholar]
- FIGUEIREDO-PEREIRA M.E., YAKUSHIN S., COHEN G. Disruption of the intracellular sulfhydryl homeostasis by cadmium-induced oxidative stress leads to protein thiolation and ubiquitination in neuronal cells. J. Biol. Chem. 1998;273:12703–12709. doi: 10.1074/jbc.273.21.12703. [DOI] [PubMed] [Google Scholar]
- GALÁN A., GARCÍA-BERMEJO M.L., TROYANO A., VILABOA N.E., BLAS E., KAZANIETZ M.G., ALLER P. Stimulation of p38 mitogen-activated kinase is an early regulatory event for the cadmium-induced apoptosis in human promonocytes cells. J. Biol. Chem. 2000;275:11418–11424. doi: 10.1074/jbc.275.15.11418. [DOI] [PubMed] [Google Scholar]
- GOUMAKOS W., LAUSSAC J.P., SARKAR B. Binding of cadmium (II) and zinc (II) to human and dog serum albumins. An equilibrium dialysis and 113 CXd-NMR study. Biochem. Cell. Biol. 1991;69:802–820. doi: 10.1139/o91-121. [DOI] [PubMed] [Google Scholar]
- HABEEBU S.M., LIU J., KLAASSEN C.D. Cadmium-induced apoptosis in mouse liver. Toxicol. Appl. Pharmacol. 1998;149:203–209. doi: 10.1006/taap.1997.8334. [DOI] [PubMed] [Google Scholar]
- HINKLE P.M., KINSELLA P.A., OSTERHOUDT K.C. Cadmium uptake and toxicity via voltage-sensitive calcium channels. J. Biol. Chem. 1987;262:16333–16337. [PubMed] [Google Scholar]
- HINKLE P.M., OSBORNE M.E. Cadmium toxicity in rat pheochromocytoma cells: studies on the mechanism of uptake. Toxicol. Appl. Pharmacol. 1994;124:91–98. doi: 10.1006/taap.1994.1012. [DOI] [PubMed] [Google Scholar]
- HINKLE P.M., SHANYSHALA E.D., NELSON E.J. Measurement of intracellular cadmium with fluorescent dyes. Further evidence for the role of calcium channels in cadmium uptake. J. Biol. Chem. 1992;267:25553–25559. [PubMed] [Google Scholar]
- ISHIDO M., TOHYAMA C., SUZUKI T. Cadmium-bound metallothionein induces apoptosis in rat kidneys, but not in cultured kidney LLC-PK1 cells. Life Sci. 1999;64:797–804. doi: 10.1016/s0024-3205(98)00621-3. [DOI] [PubMed] [Google Scholar]
- JIN P., RINGERTZ N.R. Cadmium induces transcription of proto-oncogenes c-jun and c-myc in rat L6 myoblasts. J. Biol. Chem. 1990;265:14061–14064. [PubMed] [Google Scholar]
- JONES M.M., XU C., LADD P.A. Selenite suppression of cadmium-induced testicular apoptosis. Toxicology. 1997;116:169–175. doi: 10.1016/s0300-483x(96)03541-x. [DOI] [PubMed] [Google Scholar]
- JUNGMANN J., REINS H.A., SCHOBERT C., JENTSCH S. Resistance to cadmium mediated by ubiquitin-dependent proteolysis. Nature. 1993;361:369–371. doi: 10.1038/361369a0. [DOI] [PubMed] [Google Scholar]
- KAJI T., YAMAMOTO C., TSUBAKI S., OHKAWARA S., SAKAMOTO M., SATO M., KOZUKA H. Metallothionein induction by cadmium, cytokines, thrombin and endothelin-1 in cultured vascular endothelial cells. Life Sci. 1993;53:1185–1191. doi: 10.1016/0024-3205(93)90536-c. [DOI] [PubMed] [Google Scholar]
- KANTHASAMY A.G., ISOM G.E., BOROWITZ J.L. Role of intracellular Cd2+ in catecholamine release and lethality in PC12 cells. Toxicol. Lett. 1995;81:151–157. doi: 10.1016/0378-4274(95)03425-0. [DOI] [PubMed] [Google Scholar]
- KAZANTZIS G., BLANKS R.G., SULLIVAN K.R. Is cadmium a human carcinogen. IARC Sci. Publ. 1992;118:435–446. [PubMed] [Google Scholar]
- KIM M.S., KIM B.J., WOO H.N., KIN K.W., KIN I.K., JUNG Y.K. Cadmium induces caspase-mediated cell death: suppression by Bcl-2. Toxicology. 2000;145:27–37. doi: 10.1016/s0300-483x(99)00176-6. [DOI] [PubMed] [Google Scholar]
- KOIZUMI T., LI Z.G. Role of oxidative stress in single-dose, cadmium induced testicular cancer. J. Toxicol. Environ. Health. 1992;37:25–36. doi: 10.1080/15287399209531654. [DOI] [PubMed] [Google Scholar]
- KOIZUMI T., SHIRAKURA H., KUMAGAI H., TATSUMOTO H., SUKUZI K.T. Mechanism of cadmium-induced cytotoxicity in rat hepatocytes: cadmium-induced active oxygen-related permeability changes of the plasma membrane. Toxicology. 1996;114:125–134. doi: 10.1016/s0300-483x(96)03477-4. [DOI] [PubMed] [Google Scholar]
- KONDOH M., ARARAGI S., SATO K., HIGHASHIMOTO M., TAKIGUCHI M., SATO M. Cadmium induces apoptosis partly via caspase-9 activation in HL-60 cells. Toxicology. 2002;170:111–117. doi: 10.1016/s0300-483x(01)00536-4. [DOI] [PubMed] [Google Scholar]
- LI M., KONDON T., ZHAO Q.L., LI F.J., TANABE K., ARAI Y., ZHOU Z.C., KASUYA M. Apoptosis induced by cadmium in human lymphoma U937 cells through Ca2+-calpain and caspase mitochondria dependent pathways. J. Biol. Chem. 2000;275:39702–39709. doi: 10.1074/jbc.M007369200. [DOI] [PubMed] [Google Scholar]
- LIMAYE D.A., SHAIKH Z.A. Cytotoxicity of cadmium and characteristics of its transport in cardiomyocytes. Toxicol. Appl. Pharmacol. 1999;154:59–66. doi: 10.1006/taap.1998.8575. [DOI] [PubMed] [Google Scholar]
- MARLOWE M., COSSAIRT A., MOON C., ERRERA J., MACNEEL A., PEAK R., RAY J., SCHROEDER C. Main and interaction effects of metallic toxins on classroom behaviour. J. Abnorm. Child. Psychol. 1985;13:185–198. doi: 10.1007/BF00910641. [DOI] [PubMed] [Google Scholar]
- MASTERS B., KELLY E.J., QUAIFE C.J., BRINSTER R.L., PALMITER R.D. Targeted disruption of metallothionein I and II genes increases sensitivity to cadmium. Proc. Natl. Acad. Sci. U.S.A. 1994;91:584–588. doi: 10.1073/pnas.91.2.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLER-TAUBENBERGER A., HAGMANN J., NOEGEL A., GERISCH G. Ubiquitin gene expression in Dictyostelium is induced by heat and cold shock, cadmium, and inhibitors of protein synthesis. J. Cell. Sci. 1988;90:51–58. doi: 10.1242/jcs.90.1.51. [DOI] [PubMed] [Google Scholar]
- NICOTERA P., LEIST M., FAVA E., BERLIOCCHI L., VOLBRACHT C. Energy requirement for caspase activation and neuronal cell death. Brain Pathol. 2000;10:276–282. doi: 10.1111/j.1750-3639.2000.tb00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICOTERA P., LEIST M., FERRANDO-MAY E. Apoptosis and necrosis: different execution of the same death. Biochem. Soc. Symp. 1999;66:69–73. doi: 10.1042/bss0660069. [DOI] [PubMed] [Google Scholar]
- NORDBERG M. General aspects of cadmium: transport, uptake and metabolism by the kidney. Environ. Health. Perspect. 1984;54:13–20. doi: 10.1289/ehp.845413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKUDA B., IWAMOTO Y., TACHIBANA H., SUGITA M. Parkinsonism after acute cadmium poisoning. Clin. Neurol. Neurosurg. 1997;99:263–265. doi: 10.1016/s0303-8467(97)00090-5. [DOI] [PubMed] [Google Scholar]
- PERRY D.K., SMYTH M.J., STENNICKE H.R., SALVESEN G.S., DURIEZ P., POIRIER G.G., HANNUN Y.A. Zinc is a potent inhibitor of the apoptosis protease, caspase-3. J. Biol. Chem. 1997;272:18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- PIHL R.O., PARKES M. Hair element content in learning disabled children. Science. 1977;198:204–266. doi: 10.1126/science.905825. [DOI] [PubMed] [Google Scholar]
- QUAMME G.A. Free cadmium activity in renal epithelial cells is enhanced by Mg2+ depletion. Kidney Int. 1992;41:1237–1244. doi: 10.1038/ki.1992.185. [DOI] [PubMed] [Google Scholar]
- RASTOGI R.B., MERALI Z., SINGHAL R.L. Cadmium alters behaviour and biosynthetic capacity for catecholamines and serotonin in neonatal rat brain. J. Neurochem. 1977;28:789–794. doi: 10.1111/j.1471-4159.1977.tb10629.x. [DOI] [PubMed] [Google Scholar]
- RISSO-DE FAVERNEY C., DEVAUX A., LAFAURIE M., GIRARD J.P., BAILLY B., RAHMANI R. Cadmium induced apoptosis and genotoxicity in rainbow trout hepatocytes through generation of reactive oxygen species. Aquat. Toxicol. 2001;53:65–76. doi: 10.1016/s0166-445x(00)00154-5. [DOI] [PubMed] [Google Scholar]
- SAVOLAINEN H. Cadmium-associated renal disease. Ren. Fail. 1995;17:483–487. doi: 10.3109/08860229509037613. [DOI] [PubMed] [Google Scholar]
- SEGAL M. Rat hippocampal neurons in culture: responses to electrical and chemical stimuli. J. Neurophysiol. 1983;50:1249–1264. doi: 10.1152/jn.1983.50.6.1249. [DOI] [PubMed] [Google Scholar]
- SHIBUYA I., DOUGLAS W.W. Calcium channels in rat melanotrophs are permeable to manganese, cobalt, cadmium, and lanthanum, but not to nickel: evidence provided by fluorescence changes using fura-2-loaded cells. Endocrinology. 1992;131:1936–1941. doi: 10.1210/endo.131.4.1327724. [DOI] [PubMed] [Google Scholar]
- SHIMADA H., SHIAO Y.H., SHIBATA M., WAALKES M.P. Cadmium suppressed apoptosis induced by chromium. J. Toxicol. Environ. Health A. 1998;54:159–168. doi: 10.1080/009841098158980. [DOI] [PubMed] [Google Scholar]
- SHUKLA G.S., HUSSAIN T., CHANDRA S.V. Possible role of regional, superoxide dismutase activity and lipid peroxide levels in cadmium neurotoxicity: in vivo and in vitro studies in growing rats. Life Sci. 1987;41:2215–2221. doi: 10.1016/0024-3205(87)90518-2. [DOI] [PubMed] [Google Scholar]
- SHUKLA G.S., HUSSAIN T., SRIVASTAVA R.S., CHANDRA S.V. Glutathione peroxidase and catalase in liver, kidney, testis and brain regions of rats following cadmium exposure and subsequent withdrawal. Ind. Health. 1989;29:59–69. doi: 10.2486/indhealth.27.59. [DOI] [PubMed] [Google Scholar]
- SHUKLA A., SHUKLA G.S., SRIMAL R.C. Cadmium-induced alterations in blood-brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum. Exp. Toxicol. 1996;15:400–405. doi: 10.1177/096032719601500507. [DOI] [PubMed] [Google Scholar]
- SOUZA V., BUCIO L., JAY D., CHÁVEZ E., GUTIÉRREZ-RUÍZ M.C. Effect of cadmium on calcium transport in a human fetal hepatic cell line (WRL-68 cells) Toxicology. 1996;112:97–104. doi: 10.1016/0300-483x(96)03335-5. [DOI] [PubMed] [Google Scholar]
- STADTMAN E.R. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- SUDO J., HAYASHI T., KIMURA S., KAKUNO K., TERUI J., TAKASHIMA K., SOYAMA M. Mechanism of nephrotoxicity induced by repeated administration of cadmium chloride in rats. J. Toxicol. Environ. Health. 1996;48:333–348. doi: 10.1080/009841096161230. [DOI] [PubMed] [Google Scholar]
- SUTOO D. Disturbances of brain function by exogenous cadmium. New York: Plenum Press; Vulnerable Brain and Environmental Risks. 1994;Vol 3:281–300. [Google Scholar]
- TSANGARIS G.T., TZORTZATOU-STATHOPOULOU F. Cadmium induces apoptosis differentially on immune system cell lines. Toxicology. 1998;128:143–150. doi: 10.1016/s0300-483x(98)00032-8. [DOI] [PubMed] [Google Scholar]
- USAI C., BARBERIS A., MOCCAGATTA L., MARCHETTI C. Pathways of cadmium influx in mammalian neurons. J. Neurochem. 1999;72:2154–2161. doi: 10.1046/j.1471-4159.1999.0722154.x. [DOI] [PubMed] [Google Scholar]
- VERMES I., HAANEN C., STEFFENS-NAKKEN H., REUTELINGSPERGER C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin-V. J. Immunol. Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- WAALKES M.P., COOGAN T.P., BARTER R.A. Toxicological principles of metal carcinogenesis with special emphasis on cadmium. Crit. Rev. Toxicol. 1992;22:175–201. doi: 10.3109/10408449209145323. [DOI] [PubMed] [Google Scholar]
- WANG Z., TEMPLETON D.M. Cellular factors mediate cadmium-dependent actin depolymerization. Toxicol. Appl. Pharmacol. 1996;139:115–121. doi: 10.1006/taap.1996.0149. [DOI] [PubMed] [Google Scholar]
- WARREN S., PATEL S., KAPRON C.M. The effect of vitamin E exposure on cadmium toxicity in mouse embryo cells in vitro. Toxicology. 2000;142:119–126. doi: 10.1016/s0300-483x(99)00132-8. [DOI] [PubMed] [Google Scholar]
- WILLIAMS L.R. Oxidative stress, age-related neurodegeneration, and the potential for neurotrophic treatment. Cerebrovasc. Brain Metab. Rev. 1995;7:55–73. [PubMed] [Google Scholar]
- WONG K.L., KLAASSEN C.D. Neurotoxic effect of cadmium in young rats. Toxicol. Appl. Pharmacol. 1982;63:330–337. doi: 10.1016/0041-008x(82)90261-7. [DOI] [PubMed] [Google Scholar]
- XU C., JOHNSON J.E., SINGH P.K., JONES M.M., YAN H., CARTER C.E. In vivo studies of cadmium-induced apoptosis in testicular tissue of the rat and its modulation by chelating agents. Toxicology. 1996;107:1–8. doi: 10.1016/0300-483x(95)03195-l. [DOI] [PubMed] [Google Scholar]
- YUAN C., KADIISKA M., ACHANZAR E.E., MASON R.P., WAALKES M.P. Possible role of caspase-3 inhibition in cadmium-induced blockage of apoptosis. Toxicol. Appl. Pharmacol. 2000;164:321–329. doi: 10.1006/taap.2000.8921. [DOI] [PubMed] [Google Scholar]
- ZAK I., STEIBERT E., LESZCZYNSKA E. Effect of cadmium on serum level of total protein and glycoprotein carbohydrate components in rats. Med. Pr. 1978;29:1–7. [PubMed] [Google Scholar]
- ZHOU Y., HU X., DOU C., LIU H., WANG S., SHEN P. Structural studies on metal-serum albumin. IV. The interaction of Zn(II), Cd(II) and Hg(II) with HAS and BSA. Biophys. Chem. 1992;42:201–211. doi: 10.1016/0301-4622(92)85010-2. [DOI] [PubMed] [Google Scholar]
- ZIMMERHACKL L.B., MOMM F., WIEGELE G., BRANDIS M. Cadmium is more toxic to LLC-PK1 cells than to MDCK cells acting on the cadherin-catenin complex. Am. J. Physiol. 1998;275:F143–F153. doi: 10.1152/ajprenal.1998.275.1.F143. [DOI] [PubMed] [Google Scholar]