

Abstract
Adrenaline (ADR) and noradrenaline (NA) can simultaneously activate inhibitory α2- and stimulatory β-adrenoceptors (AR). However, ADR and NA differ significantly in that ADR is a potent β2-AR agonist while NA is not. Only recently has the interaction resulting from the simultaneous activation of α2- and β2-AR been examined at the cellular level to determine the mechanisms of α2-AR regulation following concomitant activation of both α2- and β2-ARs by chronic ADR.
This study evaluates β2-AR regulation of α2A-AR signalling following chronic ADR (300 nM) and NA (1 and 30 μM) treatments of BE(2)-C human neuroblastoma cells that natively express both β2- and α2A-ARs.
Chronic (24 h) treatment with ADR (300 nM) desensitized the response to the α2A-AR agonist, brimonidine, in BE(2)-C cells. Addition of the β-AR antagonist, propranolol, blocked the ADR-induced α2A-AR desensitization. Unlike ADR, chronic NA (1 μM) treatment had no effect on the α2A-AR response. However if NA was increased to 30 μM for 24 h, α2A-AR desensitization was observed; this desensitization was partially reversed by propranolol.
Chronic ADR (300 nM) treatment reduced α2A-AR binding levels, contributing to the α2A-AR desensitization. This decrease was prevented by addition of propranolol during ADR treatment. Chronic NA (30 μM), like ADR, treatment lowered specific binding, whereas 1 μM NA treatment was without effect.
Chronic ADR treatment produced a significant increase in GRK3 levels and this was blocked by propranolol or GRK2/3 antisense DNA treatment. This antisense DNA, common to both GRK2 and GRK3, also blocked chronic ADR-induced α2A-AR desensitization and down-regulation.
Acute (1 h) ADR (300 nM) or NA treatment (1 μM) produced α2A-AR desensitization. The desensitization produced by acute treatment was β-AR independent, as it was not blocked by propranolol.
We conclude that chronic treatment with modest levels of ADR produces α2A-AR desensitization by mechanisms that involve up-regulation of GRK3 and down-regulation of α2A-AR levels through interactions with the β2-AR.
Keywords: BE(2)-C cells, desensitization, down-regulation, receptor binding, adenylyl cyclase, alpha2-adrenoceptor, beta-adrenoceptor, human neuroblastoma
Introduction
Adrenaline (ADR) and noradrenaline (NA) are endogenous catecholamines that activate three different families of adrenoceptors (ARs): α1-ARs (α1A, 1B, and 1D) that activate phospholipase C, α2-ARs (α2A, 2B, and 2C) that inhibit adenylate cyclase, and β-ARs (β1, β2, and β3) that activate adenylate cyclase. ADR and NA share similar efficacies and potencies for all these receptors except the β2-AR, where ADR has a higher affinity than NA.
The interaction between β- and α2-ARs is especially interesting because these ARs mediate opposing actions on adenylate cyclase. The interplay between these two ARs is important for maintaining homeostasis of various physiological mechanisms and, in some cases, causing the manifestation of pathophysiological conditions. In peripheral tissues, α2 and β-ARs play important roles in modulating uterine smooth muscle tone (Lécrivain et al., 1998), insulin release from pancreatic islet cells (John et al., 1990), cytokine release (Kalinichenko et al., 1999) and vasomotor tone. In addition, neuronal α2- and β2-ARs play an important role in the modulation of noradrenergic neurotransmission in the human CNS and PNS. Neuronal α2-ARs are considered potential therapeutic targets for the treatment of major cardiovascular disorders, such as hypertension and cerebral ischaemia, and CNS disorders, such as pain and depression (Lanier et al., 1996). Hence the interaction between α2- and β-ARs in a single cell after exposure to ADR or NA has potentially great physiological and pathophysiological significance (Meana et al., 1992; Shi et al., 1996; Kalinichenko et al., 1999).
The influence of cross talk between receptors that exert reciprocal effects on a common signal transduction pathway within a single cell has been examined previously. For example, Malbon and co-workers determined that chronic (24 h) activation of adenylyl cyclase by β2-ARs resulted in a 3 fold increase in the expression of the inhibitory G protein Gαi2 in S49 lymphoma cells (Hadcock et al., 1990). Moreover, maximal inhibition of forskolin-stimulated adenylyl cyclase activity by somatostatin was increased from 35 to 65% after 24 h isoprenaline (ISO) treatment. In a subsequent report, this same group described cross-talk in the opposite direction (Hadcock et al., 1991): chronic stimulation of the inhibitory pathway by somatostatin resulted in an increased expression of the stimulatory G protein Gαs and an increased efficacy of ISO on adenylyl cyclase activity. In summary, this suggests that stimulation of one signalling pathway results in sensitization of the opposing pathway.
The results described above clearly illustrate the potential for cross talk between inhibitory and stimulatory receptors in a single cell. However, there is a paucity of information regarding the consequences of chronic simultaneous activation of both the stimulatory and inhibitory pathways in the same cell. This information is important, since in vivo studies suggest that α2-AR desensitization may occur more readily when the inhibitory α2-AR and stimulatory β2-AR are chronically and simultaneously activated by ADR (Schwartz & Eikenburg, 1988; Apparsundaram & Eikenburg, 1996). In contrast, chronic NA treatment that activates only the α2-AR in vivo does not produce α2-AR desensitization (Eikenburg, 1990). Studies utilizing β2-AR antagonists during chronic ADR treatment suggest a role for the β2-AR in ADR-induced α2-AR desensitization (Apparsundaram & Eikenburg, 1996). These observations are particularly noteworthy when considered with in vitro evidence that the α2-AR is more resistant to desensitization, requiring much higher agonist concentrations for desensitization than other ARs, such as the β2-AR (Eason & Ligget, 1992; Lanier et al., 1996). Therefore, the present study was undertaken to examine the differential regulation of α2-AR signalling by modest concentrations of ADR (300 nM) and 1 μM NA in a neuronal cell line (BE(2)-C) that endogenously expresses both α2A- and β2-ARs. The objective was to determine why long-term simultaneous α2- and β2-AR activation by ADR (300 nM) produces α2-AR desensitization, while α2-AR activation alone by 1 μM NA fails to do so. For the purpose of this study, desensitization is defined as simply a loss of agonist response.
Methods
Materials
The following drugs were purchased or obtained from the indicated sources: (−)adrenaline (ADR), isoprenaline bitartrate (ISO), p-aminoclonidine (PAC), guanabenz, oxymetazoline, phenylephrine, yohimbine (RBI, Natick, MA, U.S.A.); ARC-239 (Tocris Cookson, St. Louis, MO, U.S.A.); (±)noradrenaline (NA), agmatine, idazoxan, sodium ascorbate, adrenal cortex acetone powder, IBMX, cAMP, forskolin, hydroxyapatite (Sigma Chemical Co., St. Louis, MO, U.S.A.); oligodeoxynucleotides (ODNs; Sigma-Genosys, Woodlands, TX, U.S.A.); [3H]-rauwolscine (60–80 Ci mmol−1), [3H]-RX821002 (56–60 Ci mmol−1), [3H]-cAMP (35–45 Ci mmol−1) and [125I]-CYP (2000 Ci mmol−1; Amersham Corp., Arlington Heights, IL, U.S.A.); Liquiscint scintillation fluor, (National Diagnostics, Atlanta, GA, U.S.A.); cell culture media (Gibco, Grand Island, NY, U.S.A.); foetal bovine serum (Atlanta Biologicals, Norcross, GA, U.S.A.); antibiotics (Mediatech, Inc., Herndon, VA, U.S.A.); Gαi1/2 (AS/7), Gαi3 (EC/2, New England Nuclear Corp., Boston MA, U.S.A.), Gαo, GRK2 and GRK3 (H-222), GRK2 (C-15) GRK3 (C-14) and GRK5 (C-20) primary antibodies and horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, U.S.A.); and glyceraldehyde-3-phosphate dehydrogenase antibody (GADPH, Research Diagnostics, Inc., Flanders, NJ, U.S.A.).
Brimonidine was provided by RBI as part of the Chemical Synthesis Program of the National Institute of Mental Health, Contract N01MH30003. Rauwolscine was a generous gift from CarlRoth (GmbH, Karlsruhe, Germany).
Cell culture
BE(2)-C (passages 16–58) human neuroblastoma cells (Dr Robert A. Ross, Fordham University, Bronx, NY, U.S.A.) were maintained in a humidified atmosphere (6% CO2 : 94% air) in a 1 : 1 mixture of Eagle's minimum essential medium with non-essential amino acids and Ham's F-12 containing 10% foetal bovine serum, 100 U ml−1 penicillin G and 0.1 mg ml−1 streptomycin sulphate. Plates of cells greater than 60% confluence were used throughout the study, and lifted after incubation for 5 min at room temperature with phosphate buffered saline containing 1 mM EGTA. Intact cells were sedimented by centrifugation at 1000×g for 5 min, and resuspended in the appropriate buffer or reagent, as described below.
Receptor binding
Preparation of cell membranes
Cells were homogenized with a polytron (setting 3, 10 s) in 20 volumes of Tris-HCl buffer (50 mM, pH 7.4) containing (mM) NaCl 100, Na2 EDTA 1 and PMSF 0.1, incubated for 15 min at 25°C, and the membranes sedimented by centrifugation (30 min, 34,000×g) at 4°C. Pellets were resuspended in 0.32 M sucrose, and aliquots of the membrane fractions were stored frozen (−80°C) until use.
Saturation binding
Levels of α2-ARs in BE(2)-C cell membranes (0.5 mg ml−1) were determined with various concentrations of either [3H]-rauwolscine (0.3–12 nM) or [3H]-RX821002 (0.1–10 nM) in a total volume of 1–2 ml in potassium phosphate buffer (50 mM, pH 7.4) containing MgSO4 (5 mM) at 37°C for 45 min. Thereafter, 2 ml Tris-HCl (5 mM, pH 7.4, 4°C) was added to the mixture to terminate the binding reaction before filtration over no.32 glass fibre filter strips (Schleicher & Schuell, Keene, NH, U.S.A.) using a PHD cell harvester (Cambridge Technology, Cambridge, MA, U.S.A.). The reaction tubes and the filter strips were rinsed twice with a further 2–3 ml of buffer. Levels of radioactivity were determined by scintillation spectroscopy in a Beckman LS6000 liquid scintillation counter. Assays were performed in triplicate, and specific binding was determined by subtracting the binding in the presence of yohimbine or phentolamine (10 μM; non-specific) from the binding in its absence; specific binding comprised 50–75% of total binding. Saturation studies indicated that agonist treatments did not alter the Kd of the radioligand for the α2-AR, so a single concentration (2 nM) of either [3H]-rauwolscine or [3H]-RX821002 was used to determine levels of binding following catecholamine treatment.
In some studies, plated cells were washed (4×5 ml) with serum-free media and treated with an unmodified phosphodiester antisense ODN common to both GRK2 and GRK3: 5′-ACC GCC TCC AGG TCC GCC AT-3′ (20 μM; Shih & Malbon, 1994) for 4 h, before adding sera back for the remainder of the treatment period. Cells treated with ODNs corresponding to the complementary common GRK sense (20 μM) served as controls in those experiments.
β-AR levels were determined as described (Standifer et al., 1989) with [125I]-CYP (2-300 pM) and 0.3 mg ml−1 of membrane protein at 37°C for 60 min in a total volume of 250 μl. Non-specific binding was determined in the presence of 10 μM alprenolol, while specific binding was determined by subtracting the binding in the presence of alprenolol from the binding in its absence.
Competitive binding
Cell membrane fractions were incubated as described above, except that the concentration of [3H]-rauwolscine was fixed (2 nM), and various (5–9) concentrations of unlabelled drugs were included. For β-AR displacement studies, multiple concentrations of competitor were incubated with 100 pM [125I]-CYP.
cAMP accumulation
To determine the effects of α2-AR agonists on forskolin-induced cAMP accumulation, intact cells were incubated for 5 min at 37°C in HBSS buffer (in mM): NaCl 137, KCl 5, Na2HPO4 0.6, KH2PO4 0.4, NaHCO3 4, D-glucose 6, MgCl2 0.5, MgSO4 0.4 and CaCl2 1, containing the phosphodiesterase inhibitor IBMX (0.5 mM). In some experiments, antagonists were also included in this step. To prohibit oxidation, sodium ascorbate (0.11 mM) was included when assaying catecholamines. Upon addition of forskolin (10 μM) and agonist, assay tubes were incubated for an additional 10 min at 37°C. Removing the tubes to a boiling water bath for 5 min terminated the assay. All assays were performed in duplicate in a total volume of 0.5 ml. After boiling, samples were centrifuged for 5 min at 14,000×g, and cAMP levels from the supernatant fractions were determined in a [3H]-cAMP (0.8 pmol) binding assay as previously described (Standifer et al., 1994). β-AR-mediated stimulation of cAMP accumulation was performed in the same manner, except that forskolin was not included in the assay mixture.
Immunoblotting
Membrane proteins (30 μg) were resolved by SDS–PAGE through 10% gels. Proteins were transferred to PVDF membrane, blocked with 5% non-fat dried milk in PBS containing 0.1% Tween (PBST) and incubated overnight at 4°C with dilutions of a rabbit polyclonal antibody directed against Gαi3 (1 : 1000), Gαi1/2 (1 : 500), Gαo (1 : 500), GRK2 and GRK3 (1 : 1000), GRK2 (1 : 1000), GRK3 (1 : 1000) or GRK5 (1 : 500). Blots were subjected to four washes before incubating for 60 min at room temperature with a goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (1 : 2000) in PBST. Immunoreactive bands were visualized by enhanced chemiluminescence (Amersham Corp., Arlington Heights, IL, U.S.A.). The intensity of each immunoreactive band was determined using a Nucleovision Imaging Workstation (Nucleotech Corp., San Carlos, CA, U.S.A.), and normalized to the GAPDH loading control (1 : 8000).
Protein determination
Bovine serum albumin was used as a standard in the determination of protein levels in intact cells and cell membranes as described (Lowry et al., 1951).
Data analysis
Kd, Bmax, IC50 and LogEC50 values were determined by nonlinear regression analysis using GraphPad Prism version 3.0 for Windows 95 and 98 (GraphPad Software, San Diego, CA, U.S.A., www.graphpad.com). Ki values were calculated according to the Cheng–Prusoff equation (Cheng & Prusoff, 1973) in which Ki=(IC50) [(1+S)]−1, where S=[concentration of radioligand] [KD of radioligand]−1. Comparisons between groups were made by two-way Student's t-tests or ANOVA and Tukey's or Dunnett's post hoc test (where appropriate; GraphPad Software), and groups were considered significantly different if P⩽0.05.
Results
Chronic ADR induces α2A-AR desensitization and down-regulation
To test the hypothesis that α2-AR regulation by chronic ADR is β-AR-dependent, α2-AR function following chronic 300 nM ADR or 1 μM NA pretreatment (16–24 h) was assayed in BE(2)-C cells (passage <50) by measuring the ability of the α2-AR agonist, brimonidine, to inhibit forskolin (10 μM)-stimulated cAMP accumulation. Chronic 300 nM ADR exposure desensitized the α2-AR, significantly reducing brimonidine efficacy (P<0.0001) while reducing brimonidine potency only 2 fold (Figure 1A). The decrease in brimonidine efficacy was prevented when the nonselective β-AR antagonist propranolol (30 nM) was added to the chronic ADR treatment (Figure 1A); 30 nM propranolol alone had no effect. To confirm that 300 nM ADR was sufficient to modulate β-AR activity, the ability of a maximally stimulating concentration of ISO (250 nM) to increase cAMP accumulation after chronic ADR treatment was also determined. Pretreatment with 1 μM ISO desensitized the β-AR response and was included as a positive control. Chronic 300 nM ADR pretreatment desensitized the β-AR response (Figure 2; P<0.05), indicating that ADR was activating both α2- and β-ARs.
Figure 1.
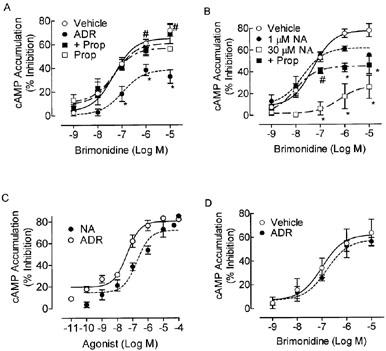
Chronic 300 nM ADR and 30 μM NA desensitizes the α2-AR response in BE(2)-C cells. The α2-AR response, as determined by the ability of brimonidine to inhibit forskolin (10 μM)-stimulated cAMP accumulation in intact BE(2)-C human neuroblastoma cells, was measured 16–24 h after pretreatment with any or all of the following: ADR (300 nM), propranolol (30 nM or 1 μM), ADR+30 nM propranolol, 1 μM NA, 30 μM NA, 30 μM NA+1 μM propranolol, ISO (1 μM) or vehicle (0.1 mM ascorbate). Forskolin increased cAMP levels over 10 fold (287±109 pmol mg−1 protein) compared to the basal state (21.8±3.5 pmol mg −1protein), and levels of both were unaltered by any pretreatments. (A) ADR (300 nM) treatment reduced brimonidine efficacy (n=7; *P<0.05 by ANOVA) compared to vehicle in BE(2)-C cells cultured up to passage 50. Alpha2-AR desensitization was not noted if the β-AR antagonist propranolol (30 nM) was included during chronic ADR exposure (n=6; #P<0.05 as compared to ADR by ANOVA). Propranolol (30 nM) alone (n=5) had no effect on α2-AR signalling. (B) Chronic 1 μM NA treatment did not alter the α2-AR signal as compared to vehicle (n=6–9) but 30 μM NA treatment reduced brimonidine potency and efficacy (n=3; *P<0.05). Inclusion of a higher propranolol concentration (1 μM) blocked 30 μM NA-induced loss of α2-AR potency, but not efficacy. Neither 1 μM ISO nor 1 μM propranolol alone altered the α2-AR response (data not shown). (C) To better visualize the ability of different concentrations of ADR and NA to activate α2-ARs in BE(2)-C cells, the concentration-response curves from which the values in Table 2 were derived for each agonist are shown. Each point represents the mean±s.e.mean of three (ADR) or five (NA) independent observations, performed in duplicate. (D) Chronic 300 nM ADR treatment does not produce a significant reduction of the brimonidine response as compared to vehicle (n=3) in higher passage cells in which β-AR binding and responsiveness has dropped off (Table 1).
Figure 2.
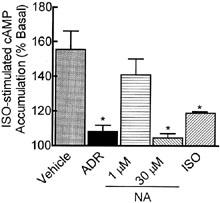
Chronic 300 nM ADR, 30 μM NA, and 1 μM ISO desensitize the β-AR response in BE(2)-C cells. BE(2)-C cells were incubated for 16–24 h with vehicle (0.1 mM ascorbate), 300 nM ADR, 1 μM NA, 30 μM NA or 1 μM ISO. After washing, intact cells were assessed for ISO-stimulated (250 nM) cAMP accumulation. Chronic 300 nM ADR, 30 μM NA, and 1 μM ISO (n=2–9; *P<0.05), but not 1 μM NA (n=6), pretreatment desensitized the β-AR response to ISO compared to the corresponding vehicle-treated control. None of the pretreatments altered basal cAMP levels (27.6±6.1 pmol mg−1 protein). Data represent mean±s.e.mean; comparisons were made by ANOVA with Dunnett's post-hoc test.
Unlike 300 nM ADR, 1 μM NA pretreatment had no effect on brimonidine potency or efficacy (Figure 1B). This concentration of NA also failed to desensitize the β-AR response (Figure 2), suggesting that a concentration of NA producing only activation of the α2-AR alone was not sufficient to produce α2-AR desensitization in this cell line. However, while 300 nMM ADR produces a near maximal α2-AR response (72% inhibition of forskolin-stimulated cAMP accumulation; Figure 1C), 1 μM NA inhibits forskolin-stimulated cAMP accumulation a bit less (53.8%). Also, if the population of β-AR in the cells were primarily of the β2-AR subtype, NA would be much less effective than ADR at activating those receptors. To achieve equivalent α2-AR activation as ADR (Figure 1C) and ensure activation of β-ARs, chronic pretreatment was also performed with a higher concentration of NA (30 μM; Figure 1B). This concentration reduced brimonidine efficacy to levels produced by 300 nM ADR treatment, produced a 10 fold loss of brimonidine potency, and desensitized the β-AR response to the same extent as ADR pretreatment (Figure 2). A higher propranolol concentration (1 μM) was only partially effective in preventing the α2-AR desensitization induced by 30 μM NA pretreatment (Figure 1B), as it reversed the loss of brimonidine potency but not the loss of efficacy. Whether this reflects (1) a significantly higher α2-AR occupancy than 300 nM ADR, (2) the fact that NA was present in 30 fold excess compared to propranolol, so that complete β-AR blockade was not achieved, or (3) a combination of both is not clear. It is unlikely that differential α2-AR occupancy played a big role because 300 nM ADR produces an α2-AR response that is 90% of maximal (Figure 1C, Table 2). It is more likely that there was an insufficient concentration of propranolol to completely antagonize the actions of NA at the β-AR. Unfortunately, higher concentrations of propranolol could not be used without producing nonspecific effects. While both ADR and NA also can activate α1-ARs, it is unlikely that their ability to desensitize that α2-AR response in this cell line involves α1-ARs: the α1-AR agonist phenylephrine failed to stimulate IP3 formation in BE(2)-C cells at concentrations up to 100 μM (data not shown) confirming the lack of functional α1-ARs in this cell line.
Table 2.
Characteristics of α2-ARs in BE(2)-C cells

To verify the involvement of the β-AR in the process of α2-AR desensitization by 300 nM chronic ADR, we took advantage of the fact that the β-AR number in our cells significantly declines with increasing passage number while α2-AR levels and responsiveness are maintained. After passage 50, β-AR responsiveness wanes. By passage 55, both β-AR responsiveness and number are reduced over 80% (Table 1). In BE(2)C cells>passage 50, chronic ADR treatment does not alter brimonidine efficacy, underscoring the contribution of the β-AR to α2-AR desensitization by ADR (Figure 1D).
Table 1.
Effect of cell passage number on α2- and β-ARs in BE(2)-C cells
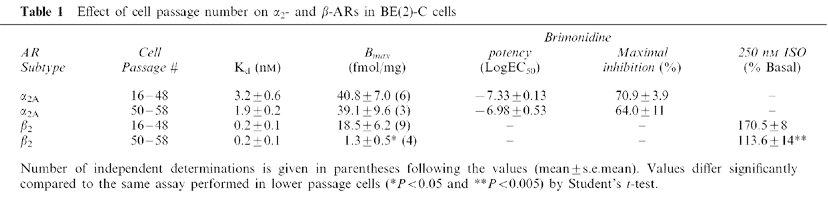
To establish that α2-AR desensitization was not produced by activation of the β-AR alone, and that it required simultaneous activation of both α2- and β-ARs, cells were pretreated with the β-AR-specific agonist, ISO. ISO pretreatment (1 μM) had no effect on brimonidine potency (log M EC50 vehicle −7.44±0.14 vs ISO −7.59±0.23) or efficacy (Imax value: ISO 70.2±2.65; vehicle 69.7±7.3; n=2–6). None of the pretreatments altered basal or forskolin-stimulated levels of cAMP accumulation compared to vehicle-treated controls. Together, these results confirm previous reports that treatment with very high concentrations of catecholamines are required to desensitize the α2-AR response (Eason & Ligget, 1992; Lanier et al., 1996) if no β-ARs are present or activated, but that much lower concentrations of catecholamines can produce α2-AR desensitization if activating both α2-AR and β-ARs simultaneously.
To confirm the β2-AR classification, the β-AR response to 100 nM ISO was dose-dependently reduced by the β2-selective antagonist, ICI 118,551, while the β1-selective antagonist, CGP 20712A was inactive at the highest concentration tested (1 μM; Figure 3). These results indicate that ISO increases cAMP accumulation through β2-ARs in BE(2)-C cells. In addition, propranolol (30 nM) also blocked β-AR stimulation of cAMP accumulation, supporting its effectiveness at blocking the β-AR in the desensitization studies (Figure 3). Binding studies (Altememi et al., 1997; data not shown) further supported the β2-AR subtype distinction.
Figure 3.
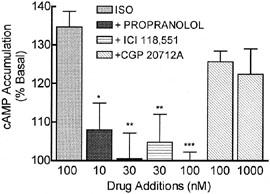
The ISO response in BE(2)-C cells is mediated through β2-ARs. Cells were assayed for ISO-stimulated accumulation of cAMP in the presence and absence of three different β-AR antagonists. The non-selective antagonist propranolol and the β2-selective antagonist ICI 118,551 antagonized the stimulatory effect of ISO (100 nM), while the β1-selective antagonist CGP 20712A was without effect (*P<0.05, **P<0.01, ***P<0.001 by ANOVA; n=3–13). Antagonists alone had no effect on basal cAMP levels at these concentrations (data not shown).
To determine whether ADR-induced desensitization of α2-ARs was a result of receptor down-regulation, changes in levels of specific α2-AR binding were measured using a single concentration of radioligand. This was sufficient for accurate assessment of changes in receptor number since α2-AR down-regulation does not change radioligand affinity as observed by our own (Kd values: vehicle 0.95±.03; ADR 0.71±.01 nM) and others' studies (Eason & Liggett, 1992). Alpha2-AR levels were reduced 60% following chronic 300 nM ADR treatment (Figure 4; P<0.05), consistent with the functional deficits noted (Figure 1). Furthermore, inclusion of 30 nM propranolol to block activation of β2-ARs during chronic ADR treatment also prevented the α2-AR down-regulation; propranolol treatment alone had no effect. Alpha2-AR levels were also reduced following 24 h pretreatment with 30 μM NA (Figure 4; P<0.05), but were unaltered by 1 μM NA or ISO treatments that also failed to produce α2-AR desensitization.
Figure 4.
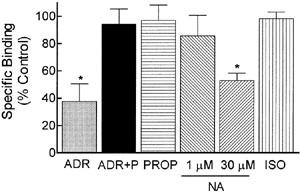
Chronic ADR treatment down-regulates α2-ARs. BE(2)-C cells were incubated for 16–24 h with vehicle (0.1 mM ascorbate), 300 nM ADR, 30 nM propranolol, ADR+30 nM propranolol, 1 μM NA, 30 μM NA or 1 μM ISO. Specific binding to cell membrane homogenates was calculated by subtracting the binding of a single concentration of radioligand (2 nM) in the presence of phentolamine (10 μM) from the binding in its absence. ADR profoundly down-regulated α2-AR levels, but this down-regulation was blocked by the β-AR antagonist, propranolol (*P<0.05 by ANOVA). Chronic 30 μM NA treatment also reduced α2-AR levels while ISO, 1 μM NA or propranolol alone had no effect. Data are presented as mean±s.e.mean of 3–5 independent experiments; α2-AR binding in control cells was 1143±283 c.p.m. mg−1 protein.
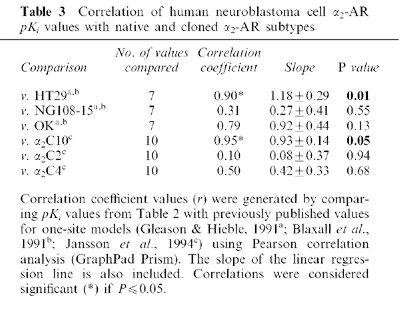
Since down-regulation of the α2A-AR subtype requires pretreatment with higher agonist concentrations than that of α2B- and α2C-ARs (Heck & Bylund, 1997; Deupree et al., 2002), various binding and antagonism studies were conducted to determine the α2-AR subtype expressed in BE(2)-C cells. The nonselective α2-AR antagonists, rauwolscine and yohimbine, competed for specific [3H]-rauwolscine binding to membranes with high affinity (Table 2; Figure 5A); the two α2B/C-selective antagonists, ARC-239 and prazosin, displaced [3H]-rauwolscine from binding sites with 100–400 fold lower affinity than rauwolscine. Hill slope values of antagonist binding did not differ from unity, consistent with binding to a single site (Table 2). The various α-AR agonists showed significant differences in their ability to competitively displace [3H]-rauwolscine from its binding site (Table 2), with the more α2A-AR-preferring agonists (oxymetazoline, brimonidine, guanabenz) and nonselective agonists (ADR, p-aminoclonidine) exhibiting higher affinities than the α2B/C-AR-preferring agonist, agmatine or the α1-AR agonist, phenylephrine. Unlike the α2-AR agonists, phenylephrine exhibited only weak, partial agonist actions in cyclase assays (Table 2). To further distinguish between the different α2-AR subtypes that could be expressed in BE(2)-C cells, another process was employed comparing the affinity ratios of prazosin to OXY or OXY to yohimbine (Hieble et al., 1997; Table 2). Prazosin : OXY (141) and OXY : yohimbine (1.54) ratios were within the range reported for native and recombinant α2A-ARs, and differ by at least 10 fold from α2C-AR values (from Hieble et al., 1997). Apparent Ki values were generated for comparison with previously reported values from cells natively expressing α2A-, α2B, or α2C-ARs (HT29, NG108-15, OK; Gleason & Hieble, 1991; Blaxall et al., 1991) or cell lines expressing cloned α2C10, α2C2, and α2C4 (Jansson et al., 1994; Jasper et al., 1998). Values obtained from our binding studies correlated significantly with those derived from native and cloned α2A-AR-containing cell membranes only (Table 3), with no correlation in cells or membranes containing α2B or α2C-ARs.
Figure 5.
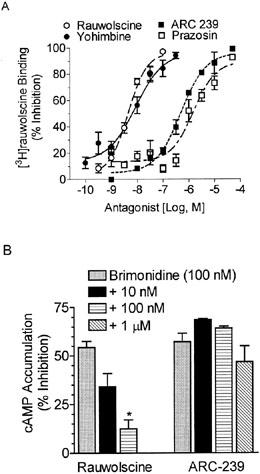
Alpha2A-AR antagonists displace [3H]-rauwolscine binding and block brimonidine-mediated inhibition of cAMP accumulation. (A) Binding assays were performed as described in ‘Methods' using [3H]-rauwolscine in the presence of 6–8 concentrations of rauwolscine, yohimbine, ARC 239, or prazosin in BE(2)-C cell membrane homogenates. Curves were best fit by nonlinear regression analysis (GraphPad Prism, San Diego, CA, U.S.A.). Each point is the mean±s.e.mean of triplicate determinations from 3–8 experiments. (B) Rauwolscine dose-dependently and significantly antagonized the ability of brimonidine (100 nM) to inhibit cAMP accumulation, while the α2B/C-AR-selective antagonist, ARC-239, had no effect. The results represent the mean±s.e. of 3–9 experiments, performed in duplicates.
Table 3.
Correlation of human neuroblastoma cell α2-AR pKi values with native and cloned α2-AR subtypes
The classification of α2-AR is further borne out by functional antagonism studies. Inhibition of cAMP accumulation by brimonidine (100 nM; Figure 5B) in BE(2)-C cells was reversed by rauwolscine in a concentration-dependent manner, whereas the α2B/C-selective antagonist ARC-239 failed to reverse the actions of the agonist. This further supports the classification of the functional α2-AR subtype in this neuroblastoma cell line as α2A.
Previous studies have shown that chronic activation of the α2-AR leads to down-regulation of inhibitory G-proteins (Gαi; Jewell-Motz et al., 1998; Eason & Liggett, 1992). We observed a small reduction in Gαi3 levels following chronic 300 nM ADR treatment, but this effect never quite achieved significance (Table 4).
Table 4.
G-protein levels following treatment in BE(2)-C cells

Chronic ADR up-regulates GRK3 facilitating α2A-AR desensitization and down-regulation
β-AR-dependent α2-AR desensitization could occur via β2-AR activation of protein kinase A (PKA; Reutter et al., 1998), G-protein-coupled receptor kinase 2 or 3 (GRK2/3; Benovic et al., 1987) or PKC (Shih & Malbon, 1994). Cyclic AMP levels in the cell would need to be increased by ADR treatment in order to activate the cAMP-dependent enzyme, PKA. In the absence of forskolin, ADR (300 nM) alone has little effect on cAMP levels (13.8±10.5% inhibition of basal; n=3), even though β2-, as well as α2-ARs, are activated. This suggests that α2-AR desensitization is not PKA-dependent.
Agonist-occupied α2A-AR is a substrate for GRK2 and GRK3, but not for GRK5 or GRK6 (Jewell-Motz & Liggett, 1996). To examine the role of GRKs in β2-AR-faciliated α2A-AR desensitization, we utilized antisense DNA that is common to a coding sequence found in both GRK2 and GRK3 (GRK 2/3), but not present in GRK5 or GRK6. Cells were pretreated with either the GRK2/3 antisense or the corresponding sense DNA for 4 h in serum free media (Shih & Malbon, 1994), before adding sera back for the remainder of the treatment period (24 h±ADR). Under these conditions, 300 nM ADR-induced α2-AR desensitization was blocked by GRK2/3 antisense, but not sense, treatment (Figure 6A). Antisense and sense treatments alone were without effect on the α2-AR response. Pretreatment with GRK2/3 antisense also ablated chronic 300 nM ADR-induced α2-AR down-regulation (Figure 6B), suggesting that GRK regulates both desensitization and downregulation of the α2-AR following prolonged agonist treatment. This is consistent with the idea that α2-AR desensitization results from α2-AR down-regulation. Cell lysates from these experiments were resolved on SDS–PAGE to insure that the GRK 2/3 antisense ODN reduced GRK 2/3 protein levels. Using antibodies recognizing both GRK2 and GRK3, we observed that chronic 300 nM ADR pretreatment alone up-regulated GRK 2/3 (Figure 6A, insert) and GRK 2/3 antisense blocked the up-regulation. Utilizing antibodies specific for GRK2, GRK3, or GRK5 isoforms, we found that chronic ADR preferentially up-regulated GRK3 (Figure 7A); levels of GRK6 in these cells are too low to consistently detect by Western blotting. Inclusion of propranolol (30 nM; Figure 7A) blocked this up-regulation. Neither chronic 1 μM NA nor 250 nM ISO treatment altered GRK3 levels, but after increasing the NA concentration 30 fold (Figure 7B), GRK3 levels were up-regulated as noted with ADR pretreatment. Chronic 30 μM NA-induced GRK3 up-regulation was partially ablated by the addition of 1 μM propranolol, similar to the desensitization results (Figure 1B), though this ablation failed to achieve significance by ANOVA. Levels of GRK5 were neither up-regulated by chronic treatments (Figure 7), nor altered by GRK2/3 antisense treatment (data not shown).
Figure 6.
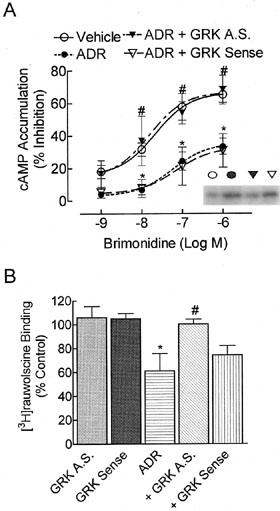
GRK antisense ODN treatment inhibits chronic 300 nM ADR-induced α2-AR desensitization and down-regulation. BE(2)-C cells were washed with serum-free media and incubated in the presence or absence of an antisense (AS) or sense ODN recognizing sequences common to both GRK2 and 3 (20 μM) for 4 h. Serum was returned to the media for an additional 24 h with ADR (300 nM) or vehicle. (A) Again, 300 nM ADR treatment reduced brimonidine potency and efficacy (n=8; *P<0.001 by ANOVA) as compared to vehicle-treated cells. Treatment with GRK antisense (n=5), but not GRK sense (n=3) ODNs reversed these effects and prevented ADR-induced α2-AR desensitization (P<0.05). Treatment with GRK antisense (n=3) or sense (n=4) ODNs alone had no effect (data not shown). Insert: 30 μg of whole cell lysate from each treatment group was resolved by SDS–PAGE through a 10% gel as indicated by the representative blot. Chronic 300 nM ADR increased GRK levels using antisera common to GRK2 and GRK3 (n=4–5; *P⩽0.05) compared to vehicle-treated controls. GRK antisense ODN treatment blocked ADR-induced increases in GRK expression as determined by densitometric analysis of immunoblots normalized to a GAPDH loading control. (B) Specific binding to cell membrane homogenates was calculated by subtracting the binding of [3H]-rauwolscine (2 nM) in the presence of phentolamine (10 μM) from the binding in its absence. GRK antisense ODN ablated chronic ADR-induced α2-AR down-regulation (P<0.05 by ANOVA) while GRK sense ODN had no effect. Neither GRK antisense nor sense ODN alone altered α2-AR levels. Data are presented as mean±s.e.mean of four independent experiments; α2-AR binding in control cells was 2815±525 c.p.m. mg−1 protein.
Figure 7.
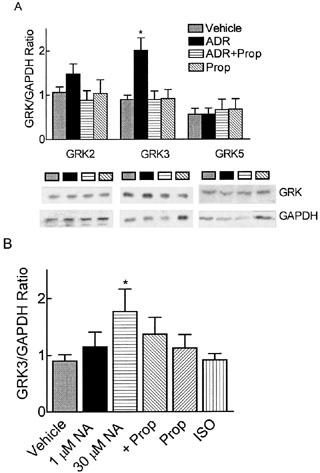
β2-AR-dependent GRK3 up-regulation. BE(2)-C cells were treated with vehicle (0.1 mM ascorbate), 300 nM ADR, propranolol (30 nM or 1 μM), ADR+30 nM propranolol, 1 μM NA, 30 μM NA, 30 μM NA+1 μM propranolol, or 250 nM ISO for 24 h. Thirty μg of whole cell lysate from each treatment group was resolved by SDS–PAGE through a 10% gel. Immunoreactive bands were normalized to the GAPDH loading control. (A) GRK isoform-specific antibodies indicated that the GRK3 isoform, but not GRK2 or GRK5, was significantly up-regulated following chronic 300 nM ADR treatment (n=4; *P⩽0.05) compared to vehicle-treated controls. Addition of 30 nM propranolol ablated ADR-induced GRK3 up-regulation while 30 nM propranolol alone had no effect. (B) Chronic 30 μM NA treatment up-regulated GRK3 (*P⩽0.05) similar to ADR treatment while treatment with 1 μM NA, 250 nM ISO, or 1 μM propranolol alone had no effect on GRK3 levels. Though inclusion of 1 μM propranolol partially prevented 30 μM NA-induced GRK3 up-regulation, it was not significantly different from 30 μM NA by ANOVA with Tukey's post-hoc test.
Acute 300 nM ADR and 1 μM NA induce α2-AR desensitization
In the present study, chronic co-activation of α2- and β2-AR by 300 nM ADR induces α2-AR desensitization while activation of α2-AR alone by 1 μM NA has no effect. To determine whether the same mechanism regulates α2-AR signalling with acute agonist treatment, cells were treated with ADR (300 nM) or NA (1 μM) for only 60 min. Unlike chronic treatments, both acute 300 nM ADR and 1 μM NA pretreatments reduced brimonidine efficacy as compared to vehicle (P<0.05; Figure 8) in BE(2)-C cells. As noted with chronic ADR treatment, brimonidine potency was reduced by only 2 fold. Addition of 30 nM propranolol to either acute 300 nM ADR- (Figure 8) or 1 μM NA-treated cells (data not shown) failed to block α2-AR desensitization. Therefore, α2-ARs in these cells can desensitize following acute exposure to both 300 nM ADR and 1 μM NA suggesting that co-activation of the β2-AR is not necessary for acute α2-AR desensitization by submaximal concentrations of catecholamines. These results also indicate the 1 μM NA is sufficient to produce α2-AR desensitization, but that mechanisms of α2-AR desensitization vary with different times of agonist exposure.
Figure 8.
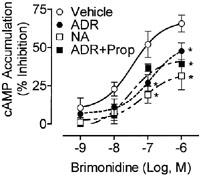
Acute 300 nM ADR and 1 μM NA induce β2-AR-independent α2-AR desensitization. The α2-AR response as determined by the ability of brimonidine to inhibit forskolin (10 μM)-stimulated cAMP accumulation in intact BE(2)-C human neuroblastoma cells was measured 1 h after pretreatment with any or all of the following: vehicle (0.1 mM ascorbate), 300 nM ADR, 30 nM propranolol, ADR+30 nM propranolol, or 1 μM NA. Both 300 nM ADR and 1 μM NA treatment reduced brimonidine potency and efficacy (n=5–6; *P<0.05 by ANOVA) compared to vehicle in BE(2)-C cells cultured up to passage 50. Addition of 30 nM propranolol to either ADR or NA (data not shown) treatment failed to block α2-AR desensitization.
Discussion
Cross-regulation of two opposing signalling cascades activated by the same endogenous neurotransmitter is a complex physiological event. Several in vivo studies suggested that α2-AR desensitization following prolonged agonist exposure was regulated by β2-AR activation (Schwartz & Eikenburg, 1988; Apparsundaram & Eikenburg, 1996), however the mechanism of β2-AR-faciliated α2-AR desensitization was not examined. Further elucidation of the cellular mechanisms responsible for this cross-talk required the study of this process in a cell system expressing both β2- and α2-ARs, preferably at physiological levels. Therefore, we set out to determine how simultaneous activation of natively expressed β2- and α2-ARs by chronic 300 nM ADR in BE(2)-C cells produces α2-AR desensitization, while activation of the α2-AR alone by 1 μM NA fails to do so.
β2-AR involvement in α2-AR desensitization is clearly implicated by several results in the present study. First, α2-AR desensitization occurred following chronic 300 nM ADR and 30 μM NA, but not 1 μM NA or ISO treatments. Lands et al. (1967) established that ADR has a higher potency at β2-ARs than NA, while ADR and NA have equal potencies at each α2-AR subtype (Lanier et al., 1996; Jasper et al., 1998; Deupree et al., 2002). Therefore, chronic 300 nM ADR desensitized the α2-AR response by activating both α2- and β2-ARs simultaneously, while 1 μM NA or ISO activated only α2- or β2-ARs, respectively. Only when a higher concentration of NA was used (30 μM), a concentration that activates both α2- and β-ARs, was α2-AR desensitization noted. Second, α2-AR desensitization produced by modest levels of ADR was observed only when the cells expressed both α2- and β2-ARs. When BE(2)-C cells are cultured beyond 50 passages, β-AR expression is lost and 300 nM ADR fails to produce α2-AR desensitization. Finally, the β-AR antagonist, propranolol prevents chronic ADR (300 nM)-promoted α2-AR desensitization and partially antagonizes α2-AR desensitization produced by the higher NA concentration (30 μM). Collectively, these observations illustrate that chronic simultaneous activation of α2- and β2-ARs enables α2-AR desensitization at lower concentrations of ADR than NA. Moreover, since the degree of α2-AR desensitization produced by the higher concentration of NA is partially reduced by β-AR blockade, it suggests that the β-AR may also enhance α2-AR desensitization at higher concentrations of NA that also activate both α2- and β-AR.
Acute 300 nM ADR treatment also desensitized the α2-AR, but unlike chronic treatment, the desensitization was independent of β2-AR activation as indicated by several results. First, both acute 300 nM ADR (co-activating α2- and β2-ARs) and 1 μM NA (activating only α2-ARs) treatments induced α2-AR desensitization to a similar extent. Second, inclusion of 30 nM propranolol during 300 nM ADR or 1 μM NA treatments failed to block α2-AR desensitization. The fact that 1 μM NA desensitizes the α2-AR after acute, but not chronic, treatment underscores the difference in mechanisms responsible for α2-AR regulation following short- and long-term agonist exposure.
The present study is the first to provide evidence for the mechanisms that contribute to β2- AR-dependent α2-AR desensitization after chronic ADR exposure. When recombinant α2-ARs were expressed in the absence of β-ARs, long-term (24 h) agonist-induced α2-AR desensitization was attributed to α2-AR (Pleus et al., 1993; Heck & Bylund, 1997; Jewell-Motz et al., 1997) and Gαi/o down-regulation (Eason & Liggett, 1992; Liggett et al., 1992; Jewell-Motz et al., 1998). β2-AR-dependent α2-AR desensitization appears to involve α2- AR down-regulation more than reduction of Gαi/o. Since the three α2-AR subtypes have different propensities to down-regulate (Pleus et al., 1993), binding and functional studies were utilized to establish the α2-AR subtype expressed in BE(2)-C cells. Several results support the α2A-AR designation. First, rauwolscine and yohimbine compete for [3H]-rauwolscine binding with much higher affinity than the α2B/C-AR selective antagonists ARC-239 and prazosin, that displayed only a weak displacement capability. Second, when apparent pKi values for various α2-AR agonists and antagonists binding to BE(2)-C membrane homogenates were compared with previously reported values in cell lines expressing a single α2-AR subtype, a significant correlation was observed only with those cells that expressed α2A-AR. Finally, the inhibitory effect of α2-AR agonists on cAMP production is readily reversed in a concentration-dependent fashion by the antagonist rauwolscine. The failure of the selective α2B/C-AR antagonist ARC-239 to produce any antagonism of brimonidine is consistent with activation of α2A-AR in BE(2)-C cells. Alpha2A-ARs require exposure to higher agonist concentration to induce receptor down-regulation than α2B or α2C-ARs (Deupree et al., 2002). In the present study, we report down-regulation of the α2A-AR by ADR after exposure to much lower agonist concentrations than previously reported for this subtype (Eason & Liggett, 1992; Pleus et al., 1993; Jewell-Motz et al., 1997). Whether this is due to more physiological AR levels in our system, a more physiological concentration of ADR (300 nM), a β2-AR-facilitated effect permitting down-regulation of the α2-AR at lower ADR concentrations, or a combination of these effects is not clear. Nevertheless, our results suggest the β2-ARs on BE(2)-C cells regulate the α2-AR response, in part, by directly modulating the level of α2A-ARs.
The results of the present study differ from previous reports where the effect of β-AR activation on inhibitory receptor number/function has been studied in other cells/tissues. For example, in HT-29 cells, 24 h stimulation of adenylyl cyclase with forskolin or cAMP analogues resulted in an increase in inhibitory α2A-AR expression (Sakaue & Hoffman, 1991). That response was attributed, in part, to increased α2A-AR gene expression by cAMP response elements in the AR gene. A major difference between our study and these previous studies is that we simultaneously activated both the stimulatory (β2-AR) and inhibitory (α2-AR) receptors during chronic treatment. This difference is significant for two reasons. First, chronic simultaneous activation of α2- and β2-ARs in BE(2)-C cells by ADR resulted in α2A-AR down-regulation. Second ADR treatment does not induce cAMP accumulation even though both α2- and β2-AR are simultaneously activated. Therefore it seems unlikely that cAMP or PKA contributed to the response in the present study. The mechanism most likely to contribute to the β2-AR-dependent α2-AR desensitization is G protein coupled receptor kinases (GRKs).
Recently several reports have implicated GRKs in regulation of receptor signalling following prolonged agonist treatment (McGraw et al., 1998; Thakker & Standifer, 2002). Several lines of evidence support a role for GRKs in the present study. First, chronic ADR treatment of BE(2)-C cells resulted in up-regulation of GRK3 expression. Since the α2A-AR is a substrate for GRK3 when agonist-occupied (Benovic et al., 1987; Jewell-Motz & Liggett, 1996), an increase in the expression of GRK3 could render the α2-AR more readily desensitized upon re-challenge with the α2-AR agonist (brimonidine). In support of this suggestion, injection of recombinant GRK3 into isolated chick dorsal root ganglion neurons increased GRK3 levels in those cells and rendered the α2-AR more susceptible to homologous desensitization (Diverse-Pierluissi et al., 1996). This effect was specific for GRK3 as increasing levels of GRK2 failed to facilitate α2-AR desensitization. Second, β-AR blockade eliminates α2-AR desensitization as well as the up-regulation of GRK3 by ADR. Moreover, antisense ODNs, which block GRK3 up-regulation, also prevent ADR-induced α2-AR desensitization and down-regulation. A recent study by Deupree et al. (2002) reports that intact GRK phosphorylation sites are required for the down-regulation of the α2C-AR, suggesting that GRK phosphorylation regulates α2C-AR down-regulation. Similarly in the present study up-regulation of GRK3 is required for the profound α2-AR down-regulation induced by modest ADR concentrations. In fact, in the present study, 300 nM ADR produced desensitization and down-regulation of the α2A-AR at least as marked as that reported at 30–300 fold higher concentrations of ADR by other investigators (Eason & Liggett, 1992; Jewell-Motz et al., 1997). Therefore, we suggest that chronic simultaneous activation of α2- and β2-ARs by ADR renders the α2-AR more sensitive to homologous desensitization and α2-AR down-regulation via GRK3 up-regulation.
Acknowledgments
The authors appreciate the thoughtful comments of Drs Brian Knoll and Richard Bond, and the technical assistance of Katherine Nguyen, California Do and Deepak Thakker. This work was supported, in part, by grants from the Department of Health and Human Services (DA10738), the University of Houston (P.E.E.R.), and the Texas Advanced Research Program (003652-0114-1999 and 003652-0114-2001) to KMS and the American Heart Association, Texas Affiliate to D.C. Eikenburg. Portions of this work were previously presented in abstract form: Altememi, G., Su, W. and Standifer, K.M. (1997). Pharmacological characterization of α- and β-adrenoceptors in BE(2)-C human neuroblastoma cells. The Pharmacologist 39, 461; and Bawa, T., Altememi, G., Eikenburg, D.C. and Standifer, K.M. (2000). Modulation of alpha2-adrenoceptor signalling by the beta-adrenoceptor: Evidence for cross-talk in human neuronal cell lines, Soc. Neurosci. 26, 115.
Abbreviations
- AR
adrenoceptor
- ARC-239
2-(2,4-(O-methoxyphenyl)-piperazin-1-yl)-ethyl- 4,4dimethyl-1,3-(2H,4H)-isoquinolindione
- brimonidine
5-Bromo-N- (4,5-dihydro-1H-imidazole-2-yl)-6-quinoxalinamine (or UK14,304)
- Gαi
alpha subunit of inhibitory guanine nucleotide binding proteins
- Gαs
alpha subunit of stimulatory guanine nucleotide binding proteins
- Gβγ
beta and gamma subunits of guanine nucleotide binding proteins
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GRK
G protein coupled receptor kinase
- HBSS
Hank's balanced salt solution
- IBMX
3-isobutyl-1- methylxanthine
- I-CYP
iodo-cyanopindolol
- ODN
oligodeoxynucleotide
- PKA
cAMPdependent protein kinase
- PKC
phospholipase C-dependent protein kinase
- PMSF
phenylmethyl sulfonylflouride
- PNS
peripheral nervous system
- PVDF
polyvinylidene diflouride
- SDS–PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- TBS/T
tris-buffered saline/tween
References
- ALTEMEMI G., SU W., STANDIFER K.M. Pharmacological characterization of α- and β-adrenoceptors in BE(2)-C human neuroblastoma cells. The Pharmacologist. 1997;39:461–. [Google Scholar]
- APPARSUNDARAM S., EIKENBURG D.C. Prejunctional alpha adrenoceptor desensitization in rat heart after chronic adrenaline treatment. J. Pharmacol. Exp. Ther. 1996;278:862–870. [PubMed] [Google Scholar]
- BLAXALL H.S., MURPHY T.J., BAKER J.C., RAY C., BYLUND D.B. Characterization of the alpha-2C adrenergic receptor subtype in the opossum kidney and in the OK cell line. J. Pharmacol. Exp. Ther. 1991;259:323–329. [PubMed] [Google Scholar]
- BENOVIC J.L., REGAN J.W., MATSUI H., MAYOR F., COTECCHIA S., LEEB-LUNDBERG L.M., CARON M.G., LEFKOWITZ R.J. Agonist-dependent phosphorylation of the alpha2-adrenergic receptor by the beta-adrenergic receptor kinase. J. Biol. Chem. 1987;262:7251–7253. [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- DEUPREE J.D., BORGESON C.D., BYLUND D.B. Down-regulation of the alpha-2C adrenergic receptor: involvement of a serine/threonine motif in the third cytoplasmic loop. BMC Pharmacol. 2002;2:9. doi: 10.1186/1471-2210-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIVERSE-PIERLUISSI M., INGLESE J., STOFFEL R.H., LEFKOWITZ R.J., DUNLAP K. G protein-coupled receptor kinase mediates desensitization of norepinephrine-induced Ca+2 channel inhibition. Neuron. 1996;16:579–585. doi: 10.1016/s0896-6273(00)80077-x. [DOI] [PubMed] [Google Scholar]
- EASON M.G., LIGGETT S.B. Subtype-selective desensitization of α2- adrenergic receptors. Different mechanisms control short and long term agonist-promoted desensitization of α2C10, α2C4, and α2C2. J. Biol. Chem. 1992;267:25473–25479. [PubMed] [Google Scholar]
- EIKENBURG D.C. Effects of chronic noradrenaline administration on sympathetic neurotransmission in the isolated perfused rat kidney. J. Pharmacol. Exp. Ther. 1990;255:1053–1059. [PubMed] [Google Scholar]
- GLEASON M.M., HIEBLE J.P. Ability of SK&F 104078 and SK&F 104856 to identify α2-adrenoceptor subtypes in NCB20 cells and guinea pig lung. J. Pharmacol. Exp. Ther. 1991;259:1124–1132. [PubMed] [Google Scholar]
- HADCOCK J.R., PORT J.D., MALBON C.C. Cross-regulation between G-protein- mediated pathways. Activation of the inhibitory pathway of adenylylcyclase increaseas the expression of beta 2-adrenergic receptors. J. Biol. Chem. 1991;266:11915–11922. [PubMed] [Google Scholar]
- HADCOCK J.R., ROS M., WATKINS D.C., MALBON C.C. Cross-regulation between G-protein-mediated pathways. Stimulation of adenylyl cyclase increases expression of the inhibitory G-protein, Gi alpha 2. J. Biol. Chem. 1990;265:14784–14790. [PubMed] [Google Scholar]
- HECK D.A., BYLUND D.B. Mechanism of down-regulation of alpha-2 adrenergic receptor subtypes. J. Pharmacol. Exp. Ther. 1997;282:1219–1227. [PubMed] [Google Scholar]
- HIEBLE J.P., RUFFOLO R.R., JR, STARKE K.Identification, characterization and subclassification of α2-adrenoceptors: an overview α2-Adrenergic Receptors: Structure, Function and Therapeutic Implications 1997Amsterdam: Harwood Academic Publishers; 1–18.eds Lanier, S.M. & Limbird. L.E. [Google Scholar]
- JANSSON C.C., MARJAMAKI A., LUOMALA K., SAVOLA J.-M., SCHEININ M., AKERMAN K.E.O. Coupling of human α2-adrenoceptor subtypes to regulation of cAMP production in transfected S115 cells. Eur. J. Pharmacol.-Mol. Pharmacol. 1994;266:165–174. doi: 10.1016/0922-4106(94)90106-6. [DOI] [PubMed] [Google Scholar]
- JASPER J.R., LESNICK J.D., CHANG L.K., YAMANISHI S.S., CHANG T.K., HSU S.A.O., DAUNT D.A., BONHAUS D.W., EGLEN R.M. Ligand efficacy and potency at recombinant α2 adrenergic receptors: agonist-mediated [35S]GTPγS binding. Biochem. Pharmacol. 1998;55:1035–1043. doi: 10.1016/s0006-2952(97)00631-x. [DOI] [PubMed] [Google Scholar]
- JEWELL-MOTZ E.A., DONNELLY E.T., EASON M.G., LIGGETT S.B. Role of the amino terminus of the third intracellular loop in agonist-promoted down-regulation of the α2A-adrenergic receptor. Biochemistry. 1997;36:8858–8863. doi: 10.1021/bi970487x. [DOI] [PubMed] [Google Scholar]
- JEWELL-MOTZ E.A., DONNELLY E.T., EASON M.G., LIGGETT S.B. Agonist-mediated down-regulation of Gai via the α2-adrenergic receptor is targeted by receptor-Gi interaction and is independent of receptor signaling and regulation. Biochemistry. 1998;37:15720–15725. doi: 10.1021/bi980999r. [DOI] [PubMed] [Google Scholar]
- JEWELL-MOTZ E.A., LIGGETT S.B. G protein-coupled receptor kinase specificity for phosphorylation and desensitization of α2A-adrenergic receptor subtypes. J. Biol. Chem. 1996;271:18082–18087. doi: 10.1074/jbc.271.30.18082. [DOI] [PubMed] [Google Scholar]
- JOHN G.W., DOXEY J.C., WALTER D.S., REID J.L. The role of alpha- and beta-adrenoceptor subtypes in mediating the effects of catecholamines on fasting glucose and insulin concentrations in the rat. Br. J. Pharmacol. 1990;100:699–704. doi: 10.1111/j.1476-5381.1990.tb14078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KALINICHENKO V.V., MOKYR M.B., GRAF L.H., JR, COHEN R.L., CHAMBERS D.A. Noradrenaline-mediated inhibition of antitumor cytotoxic T lymphocyte generation involves a beta-adrenergic receptor mechanism and decreased TNF-alpha gene expression. J. Immunol. 1999;163:2492–2499. [PubMed] [Google Scholar]
- LANDS A.M., ARNOLD A., MCAULIFF J.P., LUDUENO F.P., BROWN T.G. Differentiation of receptor systems activated by sympathomimetic amines. Nature (London) 1967;214:597–598. doi: 10.1038/214597a0. [DOI] [PubMed] [Google Scholar]
- LANIER S.M., LAFONTAN M., LIMBIRD L.E., PARIS H. Summary of the ASPET-sponsored colloquium: Alpha-2 adrenergic receptors: Structure, Function, and Therapeutic Implications, October 25-27, 1995. J. Pharmacol. Exp. Ther. 1996;277:10–16. [PubMed] [Google Scholar]
- LÉCRIVAIN J.-L., MHAOUTY-KODJA S., COHEN-TANNOUDJI J., MALTIER J.-P., LEGRAND C. In vivo stimulation of the β2-adrenergic pathway increases expression of the Gi proteins and the α2A-adrenergic receptor genes in the pregnant rat myometrium. J. Endocrinol. 1998;156:379–387. doi: 10.1677/joe.0.1560379. [DOI] [PubMed] [Google Scholar]
- LIGGETT S.A., OSTROWSKI J., CHESTNUT L.C., KUROSE H., RAYMOND J.R., CARON M.G., LEFKOWITZ R.J. Sites in the third intracellular loop of the alpha2A-adrenergic receptor confer short term agonist-promoted desensitization. Evidence for a receptor kinase-mediated action. J. Biol. Chem. 1992;267:4740–4746. [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MCGRAW D.W., DONNELLY E.T., EASON M.G., GREEN S.A., LIGGETT S.B. Role of βARK in long-term agonist-promoted desensitisation of the β2-adrenergic receptor. Cell. Signal. 1998;10:197–204. doi: 10.1016/s0898-6568(97)00112-5. [DOI] [PubMed] [Google Scholar]
- MEANA J.J., BARTUREN F., CARCIA-SEVILLA J.A. Alpha 2- adrenoceptors in the brain of suicide victims: increased receptor density associated with major depression. Biol. Psychiatry. 1992;31:471–490. doi: 10.1016/0006-3223(92)90259-3. [DOI] [PubMed] [Google Scholar]
- PLEUS R.C., SHREVE P.E., TOEWS M.L., BYLUND D.B. Down-regulation of α2-adrenoceptor subtypes. Eur. J. Pharmacol.-Mol. Pharmacol. Section. 1993;244:181–185. doi: 10.1016/0922-4106(93)90024-4. [DOI] [PubMed] [Google Scholar]
- REUTTER M.A., RICHARDS E.M., SUMNERS C. Regulation of α2A-adrenergic receptor expression by adrenaline in cultured astroglia from rat brain. J. Neurochem. 1998;70:86–95. doi: 10.1046/j.1471-4159.1998.70010086.x. [DOI] [PubMed] [Google Scholar]
- SAKAUE M., HOFFMAN B.B. cAMP regulates transcription of the α2A adrenergic receptor gene in HT-29 cells. J. Biol. Chem. 1991;266:5743–5749. [PubMed] [Google Scholar]
- SCHWARTZ D.D., EIKENBURG D.C. Enhanced endogenous neurotransmitter overflow in the isolated perfused rat kidney after chronic adrenaline administration: Lack of a prejunctional beta adrenoceptor influence. J. Pharmacol. Exp. Ther. 1988;244:11–18. [PubMed] [Google Scholar]
- SHI C.L., SEHLIN J., TALJEDAL I.B. Effects of UK 14,304, noradrenaline, and propranolol on insulin release from transplanted mouse islets. Eur. J. Endocrinol. 1996;135:724–728. doi: 10.1530/eje.0.1350724. [DOI] [PubMed] [Google Scholar]
- SHIH M., MALBON C.C. Oligodeoxynucleotides antisense to mRNA encoding protein kinase A, protein kinase C, and β-adrenergic kinase reveal distinctive cell-type-specific roles in agonist-induced desensitization. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12193–12197. doi: 10.1073/pnas.91.25.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STANDIFER K.M., CHENG J., BROOKS A.I., HONRADO C.P., SU W., VISCONTI L.M., BIELDER J.L., PASTERNAK G.W. Biochemical and pharmacological characterization of Mu, Delta and Kappa3 opioid receptors expressed in BE(2)-C neuroblastoma cells. J. Pharmacol. Exp. Ther. 1994;270:1246–1255. [PubMed] [Google Scholar]
- STANDIFER K.M., PITHA J., BAKER S.P. Carbostyril-based beta-adrenergic agonists: evidence for long lasting or apparent irreversible receptor binding and activation of adenylate cyclase activity in vitro. Naunyn-Schmiedeberg's Arch. Pharmacol. 1989;339:129–137. doi: 10.1007/BF00165134. [DOI] [PubMed] [Google Scholar]
- THAKKER D.R., STANDIFER K.M. Induction of G protein-coupled receptor kinases 2 and 3 contributes to the cross-talk between opioid receptor-like 1 and μ opioid receptors following prolong agonist exposure. Neuropharmacology. 2002;43:979–990. doi: 10.1016/s0028-3908(02)00145-4. [DOI] [PubMed] [Google Scholar]