

Abstract
On rat isolated pulmonary arteries, vasorelaxation by S-nitrosocaptopril (SNOcap) was compared with S-nitrosoglutathione (GSNO) and nitroprusside, and inhibition by SNOcap of contractions to angiotensin I was compared with the angiotensin converting enzyme (ACE) inhibitor, captopril.
SNOcap was equipotent as a vasorelaxant on main (i.d. 2–3 mm) and intralobar (i.d. 600 μm) pulmonary arteries (pIC50 values: 5.00 and 4.85, respectively). Vasorelaxant responses reached equilibrium rapidly (2–3 min).
Pulmonary vasorelaxant responses to SNOcap, like GSNO, were (i) partially inhibited by the soluble guanylate cyclase inhibitor, ODQ (1H-(1,2,4) oxadiazolo(4,3-a)-quinoxalin-1-one; 3 μM) whereas responses to nitroprusside were abolished and (ii) potentiated by hydroxocobalamin (HCOB; NO· free radical scavenger; 100 μM) whereas responses to nitroprusside were inhibited.
The relative potencies for pulmonary vasorelaxation compared with inhibition of platelet aggregation were: SNOcap 7 : 1; GSNO 25 : 1; nitroprusside >2000 : 1.
SNOcap, like captopril, concentration-dependently and time-dependently increased the EC50 for angiotensin I but not angiotensin II. The dependence on incubation time was independent of the presence of tissue but differed for SNOcap and captopril. This difference reflected the slow dissociation of SNOcap and instability of captopril, and precluded a valid comparison of the potency of the two drugs. After prolonged incubation (⩾5.6 h) SNOcap was more effective than captopril.
Thus, in pulmonary arteries SNOcap (i) possesses NO donor properties characteristic of S-nitrosothiols but different from nitroprusside and (ii) inhibits ACE at least as effectively as captopril. These properties suggest that SNOcap could be valuable in the treatment of pulmonary hypertension.
Keywords: S-nitrosocaptopril, captopril, nitroprusside, S-nitrosoglutathione, nitric oxide donors, angiotensin converting enzyme inhibitors, rat pulmonary artery, platelet aggregation
Introduction
Pulmonary hypertension is characterised by (i) abnormal pulmonary vasoconstriction, (ii) alterations in blood vessel structure (pulmonary vascular remodelling) and (iii) predisposition to pulmonary thromboembolism (Gaine & Rubin, 1998; Wanstall & Jeffery, 1998; McLaughlin, 2002). Hence drugs with pulmonary vasodilator, anti-remodelling and/or anti-thrombotic/anti-platelet properties are required to target these pathological features (Wanstall & Jeffery, 1998; Jeffery & Wanstall, 2001). None of the drugs in current use, with the possible exception of prostacyclin, possesses all of these properties. Therefore, an alternative for future therapy may be to use either (i) a combination of two or more drugs or (ii) a hybrid drug exhibiting two or more of these actions (Wanstall & Jeffery, 1998; McLaughlin, 2002).
Vasodilators that are currently used include calcium channel blockers, prostacyclin and nitric oxide (NO) gas. NO donor drugs may be an alternative to these vasodilators and moreover, some NO donor drugs may have the additional advantage of inhibiting platelet aggregation (Sogo et al., 2000b). There are no specific anti-remodelling drugs in current use in pulmonary hypertensive patients, but one group of drugs that show promise, based on studies in animal models of pulmonary hypertension, are angiotensin converting enzyme (ACE) inhibitors (Morrell et al., 1995; Jeffery & Wanstall, 1999).
S-Nitrosocaptopril (SNOcap), the subject of the present study, is a nitrosylated derivative of the ACE inhibitor, captopril (Jia et al., 1999). Hence it can be considered as a hybrid of a NO donor (an S-nitrosothiol) and an ACE inhibitor. Some of the pharmacological properties of SNOcap have been described in systemic vessels, in vitro (Loscalzo et al., 1989; Cooke et al., 1989) and in vivo (Shaffer et al., 1991; Nakae et al., 1995), but its effects have never been examined in pulmonary blood vessels.
The first aim of the present study was to examine the pulmonary vasorelaxant effects of SNOcap and compare these with other NO donors. Experiments were carried out on rat isolated pulmonary arteries, and the effects of (i) ODQ (1H-(1,2,4-)oxadiazolo(4,3-a)-quinoxalin-1-one), an inhibitor of soluble guanylate cyclase (Schrammel et al., 1996), and (ii) hydroxocobalamin (HCOB), a NO· free radical scavenger (Ellis et al., 2001) were examined on responses to the NO donors. In addition, pulmonary vasorelaxation was compared with inhibition of rat platelet aggregation. These pharmacological approaches are known to distinguish between NO donors from different classes, and hence the experiments were designed to test the hypothesis that SNOcap possesses various properties that are characteristic of S-nitrosothiols but not shared by traditional NO donor drugs such as nitroprusside.
The second aim was to compare the ACE inhibitory properties of SNOcap with those of captopril on pulmonary arteries. There are reports that NO possesses ACE inhibitor activity in rat carotid artery (Ackermann et al., 1998) and human platelets (Persson et al., 2000). Hence we hypothesised that, by virtue of its NO moiety, SNOcap would be more potent than captopril as an ACE inhibitor. To test this hypothesis the selective inhibition, by SNOcap and captopril, of contractile responses to angiotensin I was quantified on rat pulmonary arteries.
A preliminary report of these findings has been presented to a meeting of the Australasian Society of Clinical and Experimental Pharmacologists and Toxicologists, Dunedin, New Zealand, December 2001 (Tsui & Wanstall, 2001).
Methods
Animals
Male Wistar rats, aged 7–9 weeks (body weight 339±3.7 g, n=201) were used. They were anaesthetised with sodium pentobarbitone (90 mg kg−1, i.p) before dissecting out main or intralobar pulmonary arteries or removing blood for preparation of platelet rich plasma.
Main pulmonary artery rings
The main pulmonary artery was removed, cleared of any connective and fatty tissue, and from it a single ring preparation (3 mm in length; endothelium left intact; i.d. 2–3 mm) was obtained. The rings were mounted around two horizontal, stainless steel wires in a vertical organ bath containing physiological salt solution (PSS) at 37°C and bubbled with carbogen (95% O2 and 5% CO2). The composition of PSS was (mM); NaCl 118, KCl 5.9, CaCl2 1.5, MgSO4 0.72, NaHCO3 25 and glucose 11.7. The pulmonary artery preparation was set at a resting force of 10 mN. This force corresponds to a physiologically relevant transmural pressure of 10 mmHg (as previously determined from passive length/tension studies and the Laplace equation; Wanstall & O'donnell, 1990). The preparation was allowed to equilibrate for 1 h with replacement of PSS every 15 min. After equilibration, a sub-maximal contraction to phenylephrine (0.1 μM; a concentration corresponding to the EC55–EC65 for phenylephrine) was obtained. A relaxant response to acetylcholine (1 μM) was obtained to confirm the presence of functional endothelium (mean response=53±1.5% reversal of the phenylephrine contraction, n=186). After washing the tissue, the solution in the bath was replaced with K+-depolarising PSS (where 80 mM NaCl in PSS was replaced by 80 mM KCl) until the contraction reached equilibrium (approximately 15 min). This contraction was used as the reference contraction for the tissue. The tissues were then washed and used to measure either (i) relaxation responses or (ii) contractile responses, as described below. The responses were measured isometrically using a Statham Universal Transducer (UC3+UL5; Oxnard, CA, U.S.A.) attached to a micrometer (Mitutoyo, Tokyo, Japan).
Relaxation responses
Phenylephrine (0.1 μM; EC55–EC65) was added to the bath to induce a sub-maximal contraction. The magnitude of the contraction was 61±1.4% (n=70) of the reference contraction to potassium. When the contraction was stable, cumulative concentrations of one of the following drugs were added to assess the vasorelaxant properties of the drugs: SNOcap, S-nitrosoglutathione (GSNO), nitroprusside, spermine NONOate (Z-1-{N-[3-aminopropyl]-N-{4-(3-aminopropylammonio)butyl]-amino}diazen-1-ium-1,2-dioloate), glyceryl trinitrate, Angeli's salt (sodium trioxodinitrate; source of nitroxyl ion) or captopril. The tissue was washed and HCOB (100 μM, pre-incubation 3 min) or ODQ (3 μM, pre-incubation 30 min) was then added to the bath. A second concentration-response curve to the relevant drug was then obtained, with HCOB or ODQ still present. The phenylephrine contractions (as percentage of potassium contraction) in the presence of ODQ or HCOB were 73±5.5% (n=12) and 79±1.9 % (n=26), respectively. In a previous study on pulmonary artery we have presented data showing that the potency of NO donors is independent of the size of the submaximal contraction to 0.1 μM phenylephrine, within a range of contraction sizes from 11–79% of the potassium contraction (Homer & Wanstall, 2000). In the same study, increasing the concentration of phenylephrine to 0.3 μM (so that the contraction was greater than 80%) likewise had no effect on the position of the NO donor concentration-response curve. Based on this evidence it was not necessary in the present study to adjust the concentration of phenylephrine in the presence of ODQ or HCOB.
Parallel time-control experiments, in which the second curve to the NO donor was obtained after incubation with the vehicles for HCOB (water; 3 min) and ODQ (dimethylsulphoxide; DMSO; 30 min), were carried out. These experiments showed that repeated concentration-response curves on the same preparation were reproducible for all drugs with the exception of GSNO and Angeli's salt. For GSNO, there were small, significant (P<0.05) shifts in the curves to a higher concentration range (shifts in pIC50 in log units: 3 min, 0.37±0.03, n=4; 30 min, 0.34±0.12, n=4). In view of the consistency of these time-dependent shifts, the above values were used to correct the shifts obtained with HCOB and ODQ, respectively (as described in a previous study; Wanstall et al., 2001). For Angeli's salt, repeated curves were highly un-reproducible (i.e. responses in the second curve were markedly reduced, and by inconsistent amounts). Hence, instead of determining a correction factor, only one curve to Angeli's salt was obtained on each preparation, i.e. data in the absence and presence of inhibitor drug were obtained on separate preparations.
Relaxation responses were expressed as ‘per cent reversal' of the sub-maximal contraction to phenylephrine. Potency values were calculated as the negative logarithm of the IC50, (pIC50) where IC50 is the concentration producing 50% inhibition of the contraction to phenylephrine. The effects of HCOB and ODQ on the concentration–response curves were quantified as ‘log unit shifts' by using the formula: mean (pIC50 (control)−pIC50 (drug present)). Hence positive and negative values for ‘log unit shift' indicated inhibition or potentiation, respectively. In experiments with GSNO, the correction factors given above were subtracted from the values of ‘log unit shift'.
Contractile responses
A cumulative concentration-response (contraction) curve to angiotensin I or angiotensin II was obtained (control curve). After washing the tissue, SNOcap, captopril or GSNO was added to the bath for varying periods of time (see Results) and a second concentration-response (contraction) curve was then obtained to angiotensin I or angiotensin II.
Contractions were expressed as a percentage of the contraction obtained with K+-depolarising PSS. Potency values were calculated as the pEC50, where EC50 is the concentration producing 50% of the maximum response achieved. Effects of SNOcap, captopril and GSNO on contractile responses were then quantified as a ‘log unit shift' by using the formula: mean (pEC50 (control)−pEC50 (drug present)). In separate control experiments, it was shown that repeated concentration-response curves for angiotensin I and angiotensin II, after 30 min in the presence of vehicle, were reproducible (pEC50: angiotensin I, curve 1, 8.44±0.10, curve 2, 8.45±0.06, n=4; P>0.05; angiotensin II, curve 1, 8.66±0.07, curve 2, 8.71±0.08, n=4; P>0.05). Angiotensin I curves were also reproducible after intervals of 2.8 h (pEC50 curve 1, 7.91±0.08, curve 2, 7.93±0.10, n=3; P>0.05) or 5.6 h (pEC50 curve 1, 7.85±0.02, curve 2, 8.08±0.17, n=3; P>0.05).
Intralobar pulmonary artery rings
Third generation branches of pulmonary artery (intralobar pulmonary artery; 619±41 μm i.d. under tension) were dissected from the left lung lobe. Ring preparations (length 1.6-2.0 mm; endothelium-intact; three preparations from each rat) were mounted on two 40 μm diameter stainless steel wires in a small vessel myograph (Mulvany-Halpern type: Model 610M; JP Trading, Aarhus, Denmark). The tissue chamber contained PSS maintained at 37°C and bubbled with 95% O2/5% CO2. Force was measured isometrically. Preparations were set at a resting force of 0.5 mN mm−1, corresponding to physiologically relevant transmural pressure (Doggrell et al., 1999). After 30 min equilibration, preparations were contracted with the thromboxane-mimetic, U46619 (9,11 - dideoxy - 11α, 9α - epoxymethano - prostaglandin F2α; 100 nM) and a relaxation response to acetylcholine (10 μM) was obtained to confirm the presence of endothelium (relaxation response=61±4.5% reversal of the pre-contraction; n=12). U46619 was used instead of phenylephrine because α-adrenoceptor agonists give little or no contractile response in rat intralobar pulmonary arteries (Leach et al., 1992). A contraction to K+-depolarising PSS was then obtained. After washing the preparations, they were again contracted with U46619 (10–70 nM; EC50–EC60) and a cumulative concentration-response curve to SNOcap was obtained. One curve was obtained per preparation, and the three preparations from each rat were used to obtain (i) a control curve, (ii) a curve in the presence of HCOB (100 μM; 3 min incubation) or (iii) a curve in the presence ODQ (3 μM; 30 min incubation). The magnitudes of the pre-contractions (percentage of the K+ contraction; n=4) were: control 62±4.0%; HCOB present 71±6.1%; ODQ present 68±6.5%. Data were expressed as described for main pulmonary artery, except that the shift in the curve by ODQ was determined at the level of the IC30 rather than the IC50 because, in the presence of ODQ, the IC50 was not reached in every preparation.
Platelets
Seven ml blood were obtained from the abdominal aorta of each rat, via a heparinised cannula. The blood was collected into a syringe containing 105 IU of heparin sodium to give a final heparin concentration of 15 IU ml−1 blood.
Blood was centrifuged at 200×g for 10 min and the top layer, platelet rich plasma (PRP), was removed. PRP from two rats was pooled. Platelet poor plasma (PPP) was prepared by centrifuging the remaining sample at 4000×g for 15 min. The number of platelets in 1 μl of PRP was determined microscopically using a haemocytometer. PRP was diluted first with PPP to give 6×105 platelets μl−1 and then 1 : 1 with normal saline.
Platelet aggregation was determined in diluted PRP (see above) using a turbidimetric aggregometer (Chrono-log Corporation, PA, U.S.A.). Contents of the cuvettes in the aggregometer were maintained at 37°C and stirred constantly at 1200 r.p.m. Changes in light transmission through the diluted PRP were recorded on a chart recorder (Rikadenki, Tokyo, Japan). Light transmission through diluted PRP and PPP (diluted 1 : 1 with normal saline) were used for calibration, and represented minimum (0%) and maximum (100%) light transmission, respectively. A collagen concentration-response curve was first obtained to determine a concentration giving a just sub-maximal response (4.5 μg ml−1). The inhibitory effects of SNOcap (1 μM to 1 mM), GSNO (1–100 μM), nitroprusside (0.3–30 μM) or captopril (100 μM) on aggregation induced by this concentration of collagen were determined. These drugs (or the corresponding volume of vehicle) were added to the cuvette 2 min before the addition of collagen.
The response to the aggregating agent (collagen) was defined as the difference in light transmission through diluted PRP after and before addition of collagen and was expressed as a percentage of maximal light transmission, i.e. as a percentage of: (light transmission (PPP)–light transmission (PRP)). The inhibitory response to the NO donors or captopril was then calculated as:
Potency was defined as the pIC50 (where IC50 is the concentration producing 50% inhibition of platelet aggregation to collagen), and values were interpolated from individual concentration-response curves.
Drugs and solutions
Acetylcholine chloride (Sigma); Angeli's salt (sodium α-oxyhyponitrite; Cayman); angiotensin I (Auspep); angiotensin II (Sigma); captopril (Sigma); collagen (Helena Laboratories Aust Pty); dimethyl sulphoxide (DMSO; Sigma); glyceryl trinitrate (David Bull Laboratories; ampoules); heparin sodium (David Bull Laboratories), hydroxocobalamin (HCOB, Sigma); 1H-(1,2,4-)oxadiazolo(4,3-α)-quinoxalin-1-one (ODQ; Sigma); pentobarbitone sodium (Merial); L-phenylephrine hydrochloride (Sigma); S-nitrosocaptopril (SNOcap; Calbiochem); S-nitrosoglutathione (GSNO; Sigma); sodium nitroprusside (Sigma); spermine NONOate (Z-1-{N - [3 - aminopropyl] - N - {4 -(3-aminopropylammonio)butyl]-amino}diazen-1-ium-1,2-dioloate; Cayman); U46619 (9,11-dideoxy-11α,9α-epoxymethano-prostaglandin F2α; Sigma).
Stock solutions were prepared as follows (in mM): angiotensin I, 10, angiotensin II, 1, phenylephrine, 10 in 0.01 M hydrochloric acid; acetylcholine, 10, SNOcap, 100, GSNO, 100, nitroprusside, 100, captopril, 10, and HCOB, 100 in deionised water; ODQ, 10 in DMSO; Angeli's salt, 100 and spermine NONOate, 100 in 0.01 M sodium hydroxide; U46619, 10 in absolute ethanol. Ampoules of glyceryl trinitrate contained a concentration of 22 mM in ethanol. Dilutions of all drugs, when required, were made in PSS, except spermine NONOate and Angeli's salt which were diluted in 10 mM sodium hydroxide. Collagen (100 μg ml−1) and heparin (5000 IU ml−1) were obtained from the manufacturer in solution and were diluted in normal saline (0.9% NaCl in deionised water). All dilutions were kept on ice during the course of an experiment and discarded at the end of each experiment. Solutions of NO donors were protected from light.
Statistical tests
Mean values from a number (n) of different animals are given together with their standard errors (s.e.mean). Values of pEC50, pIC50, pIC30, maximum response, ‘log unit shift' or ‘time to equilibrium response' were compared, as appropriate, by paired or unpaired t-test (comparison of two values) or by one-way analysis of variance (ANOVA) followed by Student-Newman-Keuls post hoc test (comparison of more than two values).
Results
Pulmonary vasorelaxant effects of SNOcap: comparison with other NO donors
In phenylephrine pre-contracted main pulmonary artery preparations SNOcap, like GSNO and nitroprusside, caused concentration-dependent relaxation (Figures 1, 2 and 3). SNOcap was able to completely reverse the phenylephrine contraction but the potency (pIC50) was less than that of GSNO or nitroprusside (Table 1). The time to equilibrium response to 10 μM SNOcap (concentration equivalent to IC50) was 1.9±0.17 min (n=16). This was not different from the time to equilibrium response for 1 μM GSNO (2.1±0.24 min, n=16; P>0.05). Unlike the NO donors, captopril (1 nM–1 mM) did not relax the preparations, except at the two highest concentrations (⩽10% relaxation; Figure 3).
Figure 1.
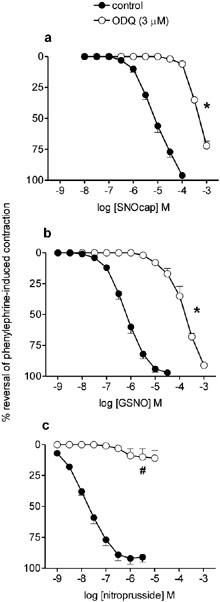
Mean concentration response curves to (a) S-nitrosocaptopril (SNOcap), (b) S-nitrosoglutathione (GSNO) and (c) nitroprusside on rat main pulmonary artery pre-contracted with phenylephrine in the absence (control; closed symbols) and then in the presence (open symbols) of ODQ (3 μM; pre-incubation 30 min). Relaxation responses are expressed as percentage reversal of the phenylephrine-induced contraction. Points are mean values (n=4) with s.e.mean shown by vertical bars except when smaller than the size of the symbols. *Significant parallel shift in curve to a higher concentration range (based on significant decrease in pIC50; P<0.05; see text for magnitude of shift expressed in log units). #Significant reduction in maximum response (P<0.05).
Figure 2.
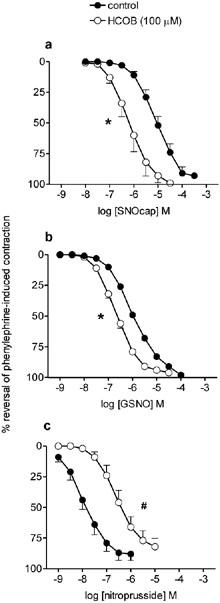
Mean concentration response curves to (a) S-nitrosocaptopril (SNOcap), (b) S-nitrosoglutathione (GSNO) and (c) nitroprusside on rat main pulmonary artery pre-contracted with phenylephrine in the absence (control; closed symbols) and then in the presence (open symbols) of hydroxocobalamin (HCOB; 100 μM; pre-incubation 3 min). Relaxation responses are expressed as per cent reversal of the phenylephrine-induced contraction. Points are mean values (n=4) with s.e.mean shown by vertical bars except when smaller than the size of the symbols. *Significant potentiation or #significant inhibition of responses in the presence of HCOB (P<0.05; based on significant changes in pIC50; see Table 2).
Figure 3.
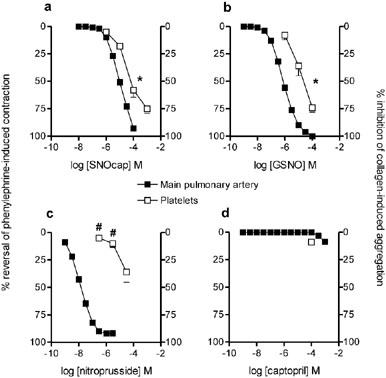
Comparison of the effects of (a) S-nitrosocaptopril (SNOcap), (b) S-nitrosoglutathione (GSNO), (c) nitroprusside and (d) captopril in rat main pulmonary artery (vasorelaxation; closed symbols) versus rat platelets (inhibition of aggregation; open symbols). Responses expressed as percentage reversal of the phenylephrine-induced contraction on main pulmonary artery (n=16; SNOcap, GSNO and nitroprusside; n=4 captopril) and percentage inhibition of collagen-induced aggregation in platelets (n=4; SNOcap, GSNO and nitroprusside; n=3 captopril). Points are mean values with s.e.mean shown by vertical bars except when smaller than the size of the symbols. *Potency on platelets significantly less than on pulmonary artery, based on pIC50 values in Table 1 (P<0.05). #Responses in platelets significantly less than responses to corresponding concentrations in pulmonary artery (P<0.05).
Table 1.
Potency (pIC50) of NO donors on main pulmonary artery and platelets and relative potency (pulmonary artery: platelets)

Effects of ODQ
ODQ (3 μM) caused parallel shifts in the SNOcap and GSNO concentration-relaxation curves to a higher concentration range (Figure 1a,b). The magnitudes of the shifts, measured in log units at the level of the IC50, were 1.81±0.09, n=4 and 2.03±0.09, n=4 for SNOcap and GSNO respectively; these shifts were not significantly different (P>0.05). In contrast, responses to nitroprusside were almost abolished in the presence of ODQ (Figure 1c). Because of the more pronounced effect of ODQ on responses to nitroprusside and the non-parallel shift in the curve, no log unit shift could be calculated for this particular NO donor.
Effects of HCOB
HCOB (100 μM) potentiated responses to SNOcap and GSNO (Figure 2a,b; Table 2). This effect of HCOB was in contrast to its inhibitory effect on responses to nitroprusside (Figure 2c; Table 2), glyceryl trinitrate and spermine NONOate (Table 2). For Angeli's salt, a source of nitroxyl ion (NO−), data in the absence and presence of HCOB were obtained on separate preparations (see Methods for reason). The pIC50 values in the absence (6.34±0.10, n=6) and presence (6.29±0.18, n=6) of HCOB were not significantly different (P>0.05), i.e. HCOB neither potentiated nor inhibited responses to Angeli's salt.
Table 2.
Effects of hydroxoxobalamin (HCOB) on concentration-response (relaxation) curves to NO donors on main pulmonary artery pre-contracted with 0.1 μM phenylephrine

Experiments on intralobar pulmonary artery
SNOcap also relaxed pre-contracted small intralobar pulmonary arteries (Figure 4) with a potency (pIC50: 4.85±0.21, n=4) not significantly different (P>0.05) from that obtained on main pulmonary artery (Table 1). The time to equilibrium relaxation for 10 μM SNOcap was 2.7±0.50 min (n=4; P>0.05 when compared with main pulmonary artery). As in main pulmonary artery, responses to SNOcap were inhibited by 3 μM ODQ (Figure 4; parallel shift in the curve; decrease in pIC30=2.04±0.30 log units, n=4) and potentiated by 100 μM HCOB (Figure 4; increase in pIC50=2.07±0.21 log units, n=4).
Figure 4.
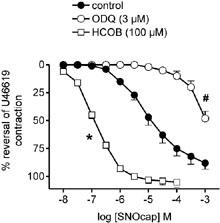
Mean concentration response curves to S-nitrosocaptopril (SNOcap) in intralobar pulmonary artery pre-contracted with U46619 in the absence (control; closed circles) or presence of ODQ (3 μM; pre-incubation 30 min; open circles) or HCOB (100 μM; pre-incubation 3 min; open squares). Relaxation responses are expressed as percentage reversal of the U46619-induced contraction. Control data and data in the presence of ODQ or HCOB were obtained in parallel in three artery preparations from the same rat. Points are mean values (n=4) with s.e.mean shown by vertical bars except when smaller than the size of the symbols. *Significant potentiation of responses in the presence of HCOB and #significant inhibition of responses in the presence of ODQ (P<0.05; based on significant changes in pIC50 and pIC30, respectively; see text for magnitude of shifts expressed in log units).
Comparison of vasorelaxation with inhibition of platelet aggregation
Collagen-induced aggregation of rat platelets was inhibited by SNOcap, GSNO and nitroprusside, but all three drugs were less potent on platelets than on main pulmonary artery (Figure 3; Table 1). Nevertheless, with SNOcap or GSNO, the concentration range for inhibition of platelet aggregation overlapped the concentration range for relaxation of pulmonary artery (Figure 3) and the relative potencies (pulmonary artery: platelets) were approximately one order of magnitude (Table 1). In contrast, nitroprusside inhibited platelet aggregation only at concentrations higher than those that caused maximal vasorelaxation (Figure 3) and had a relative potency value greater than three orders of magnitude (Table 1).
Together, these data on pulmonary artery and platelets showed that SNOcap resembles GSNO but differs from various non-nitrosothiol NO donors. Therefore in the next series of experiments, the only NO donor drug used for comparison with SNOcap was GSNO.
ACE inhibitory effect of SNOcap: comparison with captopril and GSNO on main pulmonary artery
In initial experiments in which captopril (1 μM) was incubated with the tissues for 10, 30 and 60 min, maximum inhibition of angiotensin I was achieved after 30 min incubation, i.e. 30 min was the optimal incubation time for this drug. Hence SNOcap, captopril and GSNO (each at 1 μM) were compared using this incubation time (Figure 5). SNOcap, like captopril, shifted the concentration-response curve to angiotensin I, but not that to angiotensin II, to a higher concentration range (Figure 5a,b,c,d). In addition, SNOcap, like GSNO, reduced the maximal response to both angiotensin I and angiotensin II (Figure 5a,b,e,f).
Figure 5.
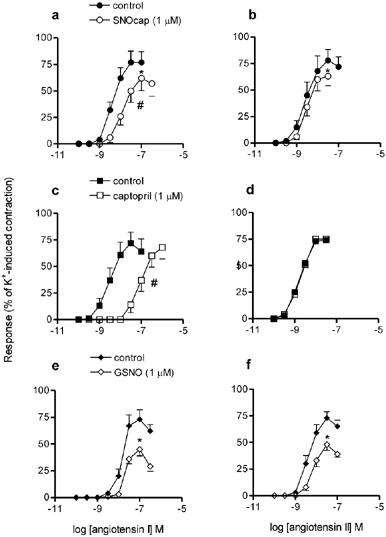
Mean concentration-response curves to angiotensin I (a, c, e) and angiotensin II (b, d, f) in the absence (control; closed symbols) and then in the presence (incubation 30 min; open symbols) of S-nitrosocaptopril (1 μM; top graphs), captopril (1 μM; centre graphs) or S-nitrosoglutathione (1 μM; bottom graphs) on rat main pulmonary artery. Responses expressed as percentage of the contraction to 80 mM K+PSS. Points are mean values (n=4) with s.e.mean shown by vertical bars except when smaller than the size of the symbols. #Significant shift in curve, based on differences in pEC50 values. *Significant depression in maximum response (P<0.05).
Subsequently, a range of concentrations of SNOcap and captopril were compared after 30 min incubation with the tissue. The magnitude of the shift in the angiotensin I curve, measured in log units at the level of the EC50, was concentration-dependent for both drugs, but SNOcap was approximately 10 fold less potent than captopril (Figure 6a). A possible reason for this potency difference was that, after 30 min, SNOcap may have only partially dissociated into captopril and NO. Therefore in further experiments SNOcap (at a single concentration; 1 μM) was pre-incubated at 37°C in oxygenated PSS for increasing times (2.3, 5.1 or 13.5 h), followed by 30 min incubation with the tissue (Figure 6b). Parallel experiments were carried out with captopril (1 μM). These experiments showed that, for both drugs, the magnitude of the shift in the angiotensin I curve (measured in log units) was time-dependent (Figure 6b). In particular, after 5.6 h the effect of SNOcap was significantly increased and greater than at any other time-point (Figure 6b). In contrast the effect of captopril significantly declined with time, and at 5.6 and 14 h SNOcap was more effective than captopril (Figure 6b).
Figure 6.
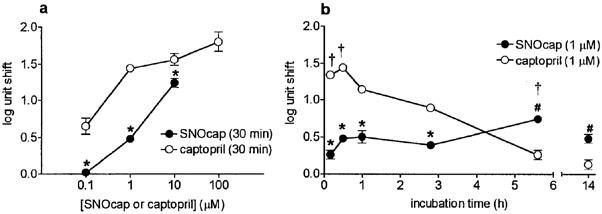
Inhibition of angiotensin I in rat main pulmonary artery. Comparison of S-nitrosocaptopril (SNOcap; closed symbols) and captopril (open symbols) at (a) various concentrations (30 min incubation) and (b) at a range of incubation times (concentration of both SNOcap and captopril, 1 μM). Incubation times 2.8, 5.6 and 14 h comprised pre-incubation periods (2.3, 5.1 and 13.5 h, respectively) in oxygenated PSS at 37°C in the absence of tissue, followed by 30 min in the presence of the tissue (see Results). Inhibition of angiotensin I is presented as ‘log unit shifts' defined as: mean (angiotensin I pEC50 (no drug)−angiotensin I pEC50 (drug present)). Data in the absence and presence of drug were obtained on the same preparation of main pulmonary artery. Points are mean values (n=4) with s.e.mean shown by vertical bars except when smaller than the size of the symbols. *Log unit shift for SNOcap significantly less than corresponding value for captopril (P<0.05). #Log unit shift for SNOcap significantly more than corresponding value for captopril (P<0.05). †Log unit shift significantly greater than at other incubation times (P<0.05).
To check whether the presence of the tissue would accelerate the dissociation of SNOcap, and hence increase its effectiveness at any given time point, additional experiments were carried out in which SNOcap was present with the tissue for 2.8 and 5.6 h (instead of the last 30 min only). However, the shifts obtained (2.8 h, 0.25±0.07 log units, n=4; 5.6 h, 0.90±0.09 log units, n=4) were not significantly different (P>0.05) from the corresponding values depicted in Figure 6b. Likewise captopril incubated with the tissue for 2.8 h gave log unit shifts (0.67 and 0.94 log units, n=2) no different from the corresponding value shown in Figure 6b.
Discussion
This study has characterized, for the first time, the vasorelaxant and ACE inhibitory properties of SNOcap specifically in pulmonary arteries. The findings extend, as well as complement, data obtained previously in non-pulmonary vessels (Cooke et al., 1989; Loscalzo et al., 1989). In particular, (i) in characterising the NO donor properties of the drug, variations in pharmacology between different types of NO donor have been taken into consideration and (ii) a detailed comparison with captopril has demonstrated some novel aspects of the ACE inhibitor profile of the drug.
Pulmonary vasorelaxation was demonstrated in isolated pulmonary artery rings in two ways, viz. (i) reversal of pre-induced phenylephrine contractions and (ii) depression of the maximal response in concentration-response (contraction) curves to angiotensin I and angiotensin II. Captopril did not exhibit either of these properties indicating that the vasorelaxation produced by SNOcap was due to the NO moiety of the molecule, i.e. due to SNOcap acting as a NO donor drug. It is known that NO donors with different types of chemical structure differ not only in the manner in which they produce NO (Gasco et al., 1996; Feelisch & Stamler, 1996) but also in their pharmacological profiles (Li & Rand, 1993; Feelisch et al., 1999; Homer et al., 1999; Wanstall et al., 2001). Since SNOcap belongs to the S-nitrosothiol group of NO donor drugs, its vasorelaxant properties were predicted to resemble those of other S-nitrosothiols, such as GSNO, rather than classical NO donors, such as nitroprusside. The data obtained supported this hypothesis. This conclusion was based on the results of experiments with two pharmacological tools. These were (i) ODQ, a soluble guanylate cyclase inhibitor, which was used to determine the degree of involvement of the soluble guanylate cyclase/guanosine 3′5′ cyclic monophosphate (cGMP) pathway and (ii) HCOB, which inhibits responses to drugs which generate NO· free radicals but, paradoxically, potentiates responses to some S-nitrosothiols (Li & Rand, 1993; Rand & Li, 1993). Further support for the hypothesis was obtained from data comparing the potency of SNOcap as a pulmonary vasorelaxant with its potency as an inhibitor of platelet aggregation.
This is the first study on the effects of ODQ on responses to SNOcap. Previously, on bovine coronary and femoral arteries the role of soluble guanylate cyclase was investigated using the less selective drug, methylene blue (Cooke et al., 1989). Methylene blue not only inhibits soluble guanylate cyclase but can also generate superoxide anions; hence inhibition of responses to NO donors by methylene blue could theoretically be due to inactivation of NO (Wolin et al., 1990; Marczin et al., 1992). ODQ was used at a concentration (3 μM) known to discriminate between spontaneous generators of NO (including S-nitrosothiols) and NO donors that require tissue activation (e.g. nitroprusside and organic nitrates) (Feelisch et al., 1999; Homer et al., 1999). At this concentration, ODQ only partially blocked responses to SNOcap or GSNO but, in contrast, abolished responses to nitroprusside. These data indicate either that SNOcap and GSNO cause vasorelaxation partially via mechanisms that are independent of the soluble guanylate cyclase/cGMP pathway (as reported for spermine NONOate and MAHMA NONOate in rat pulmonary artery; Homer & Wanstall, 2000) or, alternatively, that a sulfhydryl site, i.e. a non-haeme site, on soluble guanylate cyclase is involved (as reported for S-nitroso-N-acetylpenicillamine in rat aortic rings; Tseng et al., 2000).
In the experiments with HCOB, SNOcap was compared with five other NO donors. HCOB, which is a ‘scavenger' of NO· free radical, inhibited responses not only to nitroprusside but also glyceryl trinitrate and spermine NONOate, each of which is known to produce NO· free radicals (Feelisch & Stamler, 1996; Wanstall et al., 2001). Angeli's salt, which produces nitroxyl (NO−) ions rather than NO· free radical, was not inhibited. These data are in agreement with previous findings for these four NO donors in mouse aorta (Wanstall et al., 2001). The lack of effect of HCOB on responses to Angeli's salt is consistent with the selectivity of HCOB for NO in its free radical form. Responses to SNOcap and GSNO were not inhibited by HCOB, consistent with (i) data for S-nitrosothiols on rat anococcygeus muscle (Rand & Li, 1993) and (ii) the view that S-nitrosothiols act, biologically, via NO+ transfer (transnitrosation) or NO− rather than via NO· free radical (Feelisch & Stamler, 1996; Singh et al., 1996). Not only did HCOB fail to inhibit responses to SNOcap and GSNO on rat pulmonary artery but, in contrast, it caused potentiation. Potentiation of responses by HCOB has previously been described for GSNO on anococcygeus muscle (Rand & Li, 1993). Curiously, on this tissue no potentiation was seen with two other S-nitrosothiols, S-nitroso-N-acetylpenicillamine and S-nitrosocysteine, (Li & Rand, 1993; Rand & Li, 1993). The mechanism for the potentiation, and the reason why this occurs with some S-nitrosothiols but not others, is currently not known.
One further similarity between SNOcap and GSNO (and difference from nitroprusside) was observed when their potencies on pulmonary artery and platelets were compared. SNOcap and GSNO were only one order of magnitude less potent on platelets than on pulmonary artery, in marked contrast to nitroprusside where the difference in potency was more than three orders of magnitude. Data obtained in human tissues have shown differences in the relative potencies (blood vessels : platelets) for S-nitrosothiols compared with nitroprusside. For example, two S-nitrosothiols, GSNOand RIG200 (N-(S-nitroso-N-acetylpenicillamine)-2-amino-2-deoxy-1,3,4,6-tetra-O-acetyl-β-D-glucopyranose), were more-or-less equipotent on human saphenous vein and human platelets whereas nitroprusside was less potent on platelets than on the blood vessel preparation (Sogo et al., 2000a, b).
SNOcap, like captopril, exhibited ACE inhibitor properties on rat pulmonary artery in that it increased the EC50 for angiotensin I but not angiotensin II. We had hypothesised that SNOcap would be more potent than captopril (for reason, see Introduction). However, it became apparent that a simple comparison of the potencies of SNOcap and captopril was not possible. This was because the effectiveness of both drugs exhibited a time-dependence that was not the same for the two drugs. The time-dependence studies showed that at short incubation times (⩽2.8 h), SNOcap was less effective than captopril. This presumably reflected the slow dissociation of SNOcap into captopril and NO, since the half-life of SNOcap in oxygenated PSS at 37°C is 2.8 h (Loscalzo et al., 1989). The requirement for SNOcap to dissociate before it can inhibit ACE is to be anticipated. This is because (i) in undissociated SNOcap it is the sulfhydryl group of captopril that is occupied by NO and (ii) the sulfhydryl group is important for the binding of captopril to zinc in the active site of ACE (Mackaness, 1985). Therefore in undissociated SNOcap binding to ACE would be reduced. It was found that a longer incubation time (corresponding to two half-lives and hence commensurate with greater dissociation of SNOcap) led to an increase in the effectiveness of SNOcap, as expected. However, under these prolonged incubation conditions the effectiveness of captopril significantly declined. The reason for the decline in potency of captopril is not certain but the conditions of incubation used would tend to destabilise captopril since, in oxygenated aqueous solutions, captopril is vulnerable to oxidation, especially above room temperature and at pH >4 (Timmins et al., 1982; Pramar et al., 1992).
The results of this detailed study suggest that at any given time point, the effectiveness of SNOcap was governed by both (i) the amount of captopril released from the parent compound and (ii) the degree of degradation of free captopril. The effectiveness of captopril, on the other hand, would have been governed by the latter variable only. As a result, a meaningful comparison of the potencies of SNOcap and captopril could not be made. Nevertheless the experiments showed that SNOcap could inhibit ACE in pulmonary artery, consistent with previous findings in non-pulmonary vessels (Cooke et al., 1989; Loscalzo et al., 1989). However our results differed quantitatively from those reported on bovine femoral artery, where 1 μM SNOcap was as effective as 1 μM captopril in inhibiting angiotensin I after a comparatively short incubation time (45 min; Cooke et al., 1989). This discrepancy between our results and those of Cooke et al. (1989) remains unexplained. Mathews & Kerr (1993) have suggested that some, but not all, blood vessels have the ability to accelerate the decomposition of S-nitrosothiols and it is possible that bovine femoral artery has this ability while rat pulmonary artery does not. Certainly there was no evidence that rat pulmonary artery hastened the decomposition of SNOcap since experiments using prolonged incubation times gave identical results whether the blood vessel preparation was present or not.
Experiments showing concentration-dependency (as opposed to time-dependency) in inhibiting angiotensin I were carried out at only one incubation time (i.e. 30 min, the optimal time for captopril). It was noted that there was a flattening of the concentration-response curve for captopril at the highest concentration tested. A possible explanation is that conversion of angiotensin I to angiotensin II is catalysed by not only ACE but also chymase, which would not be blocked by captopril (Iwamoto et al., 2001; Richard et al., 2001). The concentrations of SNOcap used were not sufficiently high to determine whether or not there was a comparable flattening of the curve for this drug.
In view of the slow dissociation of SNOcap into captopril and NO even in the presence of tissue (as established in the ACE inhibitor studies), it was interesting that direct vasorelaxant responses to SNOcap reached equilibrium in as little as 2 min. Pulmonary vasodilator responses in vivo after i.v. administration of SNOcap reach equilibrium equally rapidly (Wanstall; unpublished data). The simplest explanation for this is that vasorelaxation does not require prior dissociation of NO, in one of its forms, from the parent molecule. This is curious in view of the involvement of activation of soluble guanylate cyclase in the response. It is known that some of the biological actions of S-nitrosothiols occur as a result of transnitrosation reactions that do not require the prior release of NO (Feelisch & Stamler, 1996) and that occur very rapidly (Arnelle & Stamler, 1995). However it could be argued that transnitrosation would still be associated with the appearance of free captopril; yet in the ACE inhibitor experiments there was no functional evidence for much free captopril even after 10 min. One possible, though speculative, explanation for this apparent paradox is that after transnitrosation (an initial rapid event) captopril may exist largely as captopril disulphide, which would not bind to ACE, whereas after dissociation of SNOcap into captopril and NO (a more gradual process) the sulfhydryl group of captopril is free to bind to ACE.
Not all vasoactive drugs necessarily have pharmacological profiles that are identical in large and small pulmonary arteries (Leach et al., 1992). However in the present study the vasorelaxant properties of SNOcap on intralobar arteries were consistent with those on main pulmonary artery, viz. (i) SNOcap had the same potency as on main pulmonary artery, (ii) vasorelaxant responses reached equilibrium rapidly, (iii) responses were potentiated by HCOB and (iv) inhibition by ODQ was seen as a parallel shift in the concentration-response curve. Thus with respect to vasorelaxation, the properties of SNOcap on main pulmonary artery are indicative of its properties on intralobar pulmonary arteries. A previous study in main and intralobar pulmonary arteries has shown that this is also true with respect to the inhibition of contractions to angiotensin I by ACE inhibitors (Jeffery & Wanstall, 1999).
In summary, the present investigation has shown that SNOcap is an effective vasorelaxant of rat pulmonary arteries and also a potent inhibitor of rat platelet aggregation. This study highlighted the similarities between SNOcap and GSNO and the differences between SNOcap and nitroprusside (and other traditional NO donors). This was evident not only from comparisons of potency between pulmonary artery and platelets but also from experiments with ODQ and HCOB, two pharmacological tools that have not previously been studied in conjunction with SNOcap previously. The comparative effectiveness of SNOcap and captopril as ACE inhibitors on the pulmonary vasculature depended on the incubation time. However after prolonged incubation SNOcap was more effective than captopril. The two properties of SNOcap described above, namely ACE inhibition and vasorelaxation, are potentially valuable in the treatment of pulmonary hypertension. ACE inhibitors such as captopril (Morrell et al., 1995) and perindopril (Jeffery & Wanstall, 1999) have been shown to possess anti-remodelling properties when administered for several weeks to pulmonary hypertensive rats. It would be valuable in the future to carry out chronic studies with SNOcap in pulmonary hypertensive animals. This would enable assessment of the combined benefits of the drug's pulmonary vasodilatory and platelet inhibitory effects (residing in the NO moiety of the molecule) and likely anti-remodelling effects, which can be predicted from the drug's ACE inhibitor properties in pulmonary vessels.
Acknowledgments
The financial support of the National Health and Medical Research Council of Australia is gratefully acknowledged. J.C. Wanstall is an NHMRC Senior Research Fellow. We are grateful to Kerry Homer for helpful discussions.
Abbreviations
- ANOVA
analysis of variance
- DMSO
dimethylsulphoxide
- GSNO
S-nitrosoglutathione
- HCOB
hydroxocobalamin
- NO−
nitroxyl ion
- ODQ
1H-(1,2,4-)oxadiazolo(4,3-a)-quinoxalin-1-one
- PPP
platelet poor plasma
- PRP
platelet rich plasma
- PSS
physiological salt solution
- SNOcap
S-nitrosocaptopril
- spermine NONOate
Z-1-{N-[3-aminopropyl]-N-{4-(3-aminopropylammonio)butyl]amino}diazen-1-ium-1,2-diolate
References
- ACKERMANN A., FERNANDEZ-ALFONSO M.S., SANCHEZ DE ROJAS R., ORTEGA T., PAUL M., GONZALEZ C. Modulation of angiotensin-converting enzyme by nitric oxide. Br. J. Pharmacol. 1998;124:291–298. doi: 10.1038/sj.bjp.0701836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARNELLE D.R., STAMLER J.S. NO+, NO· and NO− donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch. Biochem. Phys. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- COOKE J.P., ANDON N., LOSCALZO J. S-Nitrosocaptopril. II. Effects on vascular reactivity. J. Pharmacol. Exp. Ther. 1989;249:730–734. [PubMed] [Google Scholar]
- DOGGRELL S.A., WANSTALL J.C., GAMBINO A. Functional effects of 4-aminopyridine (4-AP) on pulmonary and systemic vessels from normoxic control and hypoxic pulmonary hypertensive rats. Naunyn-Schmeideberg's Arch. Pharmacol. 1999;360:317–323. doi: 10.1007/s002109900064. [DOI] [PubMed] [Google Scholar]
- ELLIS A., LU H., LI C.G., RAND M.J. Effects of agents that inactivate free radical NO (NO•) on nitroxyl anion-mediated relaxations, and on the detection of NO• released from the nitroxyl anion donor Angeli's salt. Br. J. Pharmacol. 2001;134:521–528. doi: 10.1038/sj.bjp.0704287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEELISCH M., KOTSONIS P., SIEBE J., CLEMENT B., SCHMIDT H.H.H.W. The soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a] quinoxalin-1-one is a nonselective heme protein inhibitor of nitric oxide synthase and other cytochrome P-450 enzymes involved in nitric oxide donor bioactivation. Mol. Pharmacol. 1999;56:243–253. doi: 10.1124/mol.56.2.243. [DOI] [PubMed] [Google Scholar]
- FEELISCH M., STAMLER J.S.Donors of nitrogen oxides Methods in Nitric Oxide Research. 1996Chichester: Wiley and Sons; 71–115.eds. Feelisch, M. & Stamler, J.S. [Google Scholar]
- GAINE S.P., RUBIN L.J. Primary pulmonary hypertension. The Lancet. 1998;352:719–725. doi: 10.1016/S0140-6736(98)02111-4. [DOI] [PubMed] [Google Scholar]
- GASCO A., FRUTTERO R., SORBA G. NO-donors: an emerging class of compounds in medicinal chemistry. Il Farmaco. 1996;51:617–635. [PubMed] [Google Scholar]
- HOMER K.L., FIORE S.A., WANSTALL J.C. Inhibition by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) of responses to nitric oxide-donors in rat pulmonary artery: influence of the mechanism of nitric oxide generation. J. Pharm. Pharmacol. 1999;51:135–139. doi: 10.1211/0022357991772240. [DOI] [PubMed] [Google Scholar]
- HOMER K.L., WANSTALL J.C. Cyclic GMP-independent relaxation of rat pulmonary artery by spermine NONOate, a diazeniumdiolate nitric oxide donor. Br. J. Pharmacol. 2000;131:673–682. doi: 10.1038/sj.bjp.0703613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IWAMOTO Y., SONG K., TAKAI S., YAMADA M., JIN D., SAKAGUCHI M., UEDA H., KATSUOKA Y., MIYAZAKI M. Multiple pathways of angiotensin I conversion and their functional role in the canine penile corpus cavernosum. J. Pharmacol. Exp. Ther. 2001;298:43–48. [PubMed] [Google Scholar]
- JEFFERY T.K., WANSTALL J.C. Perindopril, an angiotensin converting enzyme inhibitor, in pulmo+nary hypertensive rats: comparative effects on pulmonary vascular structure and function. Br. J. Pharmacol. 1999;128:1407–1418. doi: 10.1038/sj.bjp.0702923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JEFFERY T.K., WANSTALL J.C. Pulmonary vascular remodelling: a target for therapeutic intervention in pulmonary hypertension. Pharmacol. Therap. 2001;92:1–20. doi: 10.1016/s0163-7258(01)00157-7. [DOI] [PubMed] [Google Scholar]
- JIA L., YOUNG X., GUO W. Physicochemistry, pharmacokinetics, and pharmacodynamics of S-nitrosocaptopril crystals, a new nitric oxide donor. J. Pharm. Sci. 1999;88:981–986. doi: 10.1021/js990108g. [DOI] [PubMed] [Google Scholar]
- LEACH R.M., TWORT C.H.C., CAMERON I.R., WARD J.P. A comparison of the pharmacological and mechanical properties in vitro of large and small pulmonary arteries of the rat. Clin. Sci. 1992;82:55–62. doi: 10.1042/cs0820055. [DOI] [PubMed] [Google Scholar]
- LI C.G., RAND M.J. Effects of hydroxocobalamin and haemoglobin on NO-mediated relaxations in the rat anococcygeus muscle. Clin. Exp. Pharmacol. Physiol. 1993;20:633–640. doi: 10.1111/j.1440-1681.1993.tb01645.x. [DOI] [PubMed] [Google Scholar]
- LOSCALZO J., SMICK D., ANDON N., COOKE J. S-Nitrosocaptopril. I. Molecular characterization and effects on the vasculature and on platelets. J. Pharmacol. Exp. Ther. 1989;249:726–729. [PubMed] [Google Scholar]
- MACKANESS G.B. The future of angiotensin-converting enzyme inhibitors. J. Cardiovasc. Pharmacol. 1985;7 Suppl 1:S30–S34. doi: 10.1097/00005344-198507001-00007. [DOI] [PubMed] [Google Scholar]
- MARCZIN N., RYAN U.S., CATRAVAS J.D. Methylene blue inhibits nitrovasodilator- and endothelium-derived relaxing factor-induced cyclic GMP accumulation in cultured pulmonary arterial smooth muscle cells via generation of superoxide anion. J. Pharmacol. Exp. Ther. 1992;263:170–179. [PubMed] [Google Scholar]
- MATHEWS W.R., KERR S.W. Biological activity of S-nitrosothiols: the role of nitric oxide. J. Pharmacol. Exp. Ther. 1993;267:1529–1537. [PubMed] [Google Scholar]
- MCLAUGHLIN V.V. Medical management of primary pulmonary hypertension. Expert. Opin. Pharmacother. 2002;3:159–165. doi: 10.1517/14656566.3.2.159. [DOI] [PubMed] [Google Scholar]
- MORRELL N.W., MORRIS K.G., STENMARK K.R. Role of angiotensin-converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am. J. Physiol. 1995;269:H1186–H1194. doi: 10.1152/ajpheart.1995.269.4.H1186. [DOI] [PubMed] [Google Scholar]
- NAKAE I., TAKAHASHI M., KINOSHITA T., MATSUMOTO T., KINOSHITA M. The effects of S-nitrosocaptopril on canine coronary circulation. J. Pharmacol. Exp. Ther. 1995;274:40–46. [PubMed] [Google Scholar]
- PERSSON K., WHISS P.A., NYHLEN K., JACOBSSON-STRIER M., GLINDELL M., ANDERSSON R.G.G. Nitric oxide donors and angiotensin-converting enzyme inhibitors act in concert to inhibit human angiotensin-converting enzyme activity and platelet aggregation in vitro. Eur. J. Pharmacol. 2000;406:15–23. doi: 10.1016/s0014-2999(00)00647-6. [DOI] [PubMed] [Google Scholar]
- PRAMAR Y., DAS GUPTA V., BETHEA C. Stability of captopril in some aqueous systems. J. Clin. Pharm. Ther. 1992;17:185–189. doi: 10.1111/j.1365-2710.1992.tb01291.x. [DOI] [PubMed] [Google Scholar]
- RAND M.J., LI C.G. Differential effects of hydroxocobalamin on relaxations induced by nitrosothiols in rat aorta and anococcygeus muscle. Eur. J. Pharmacol. 1993;241:249–254. doi: 10.1016/0014-2999(93)90210-9. [DOI] [PubMed] [Google Scholar]
- RICHARD V., HUREL-MERLE S., SCALBERT E., FERRY G., LALLEMAND F., BESSOU J.P., THUILLEZ C. Functional evidence for a role of vascular chymase in the production of angiotensin II in isolated human arteries. Circulation. 2001;104:750–752. doi: 10.1161/hc3201.094971. [DOI] [PubMed] [Google Scholar]
- SCHRAMMEL A., BEHRENDS S., SCHMIDT K., KOESLING D., MAYER B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol. Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- SHAFFER J.E., LEE F., THOMSON S., HAN B.J., COOKE J.P., LOSCALZO J. The hemodynamic effects of S-nitrosocaptopril in anesthetized dogs. J. Pharmacol. Exp. Ther. 1991;256:704–709. [PubMed] [Google Scholar]
- SINGH R.J., HOGG N., JOSEPH J., KALYANARAMAN B. Mechanism of nitric oxide release from S-nitrosothiols. J. Biol. Chem. 1996;271:18596–18603. doi: 10.1074/jbc.271.31.18596. [DOI] [PubMed] [Google Scholar]
- SOGO N., CAMPANELLA C., WEBB D.J., MEGSON I.L. S-Nitrosothiols cause prolonged, nitric oxide-mediated relaxation in human saphenous vein and internal mammary artery: therapeutic potential in bypass surgery. Br. J. Pharmacol. 2000a;131:1236–1244. doi: 10.1038/sj.bjp.0703700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOGO N., MAGID K.S., SHAW C.A., WEBB D.J., MEGSON I.L. Inhibition of human platelet aggregation by nitric oxide donor drugs: relative contribution of cGMP-independent mechanisms. Biochem. Biophys. Res. Commun. 2000b;279:412–419. doi: 10.1006/bbrc.2000.3976. [DOI] [PubMed] [Google Scholar]
- TIMMINS P., JACKSON I.M., WANG Y.J. Factors affecting captopril stability in aqueous solution. Int. J. Pharmaceut. 1982;11:329–336. [Google Scholar]
- TSENG C.-M.L., TABRIZI-FARD M.A., FUNG H.-L. Differential sensitivity among nitric oxide donors toward ODQ-mediated inhibition of vascular relaxation. J. Pharmacol. Exp. Ther. 2000;292:737–742. [PubMed] [Google Scholar]
- TSUI D., WANSTALL J.C.S-Nitrosocaptopril: Effects on pulmonary artery and platelets in normal rats and pulmonary artery pressure in pulmonary hypertensive rats Proc. Aust. Soc. Clin. Exp. Pharmacol. Toxicol. 2001963Abstract [Google Scholar]
- WANSTALL J.C., JEFFERY T.K. Recognition and management of pulmonary hypertension. Drugs. 1998;56:989–1007. doi: 10.2165/00003495-199856060-00004. [DOI] [PubMed] [Google Scholar]
- WANSTALL J.C., JEFFERY T.K., GAMBINO A., LOVREN F., TRIGGLE C.R. Vascular smooth muscle relaxation mediated by nitric oxide donors: a comparison with acetylcholine, nitric oxide and nitroxyl ion. Br. J. Pharmacol. 2001;134:463–472. doi: 10.1038/sj.bjp.0704269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANSTALL J.C., O'DONNELL S.R. Endothelin and 5-hydroxytryptamine on rat pulmonary artery in pulmonary hypertension. Eur. J. Pharmacol. 1990;176:159–168. doi: 10.1016/0014-2999(90)90524-a. [DOI] [PubMed] [Google Scholar]
- WOLIN M.S., CHERRY P.D., RODENBURG J.M., MESSINA E.J., KALEY G. Methylene blue inhibits vasodilation of skeletal muscle arterioles to acetylcholine and nitric oxide via the extracellular generation of superoxide anion. J. Pharmacol. Exp. Ther. 1990;254:872–876. [PubMed] [Google Scholar]