

Abstract
We have examined possible mechanisms of cross-talk between the Gq/11-linked M3 muscarinic acetylcholine (mACh) receptor and the Gi/o-linked M2 mACh receptor by stable receptor coexpression in Chinese hamster ovary (CHO) cells. A number of second messenger (cyclic AMP, Ins(1,4,5)P3) and mitogen-activated protein kinase (ERK and JNK) responses stimulated by the mACh receptor agonist methacholine were examined in CHO-m2m3 cells and compared to those stimulated in CHO-m2 and CHO-m3 cell-lines, expressing comparable levels of M2 or M3 mACh receptors.
Based on comparisons between cell-lines and pertussis toxin (PTx) pretreatment to eliminate receptor-Gi/o coupling, evidence was obtained for (i) an M2 mACh receptor-mediated contribution to the predominantly M3 mACh receptor-mediated Ins(1,4,5)P3 response and (ii) a facilitation of the inhibitory effect of M2 mACh receptor on forskolin-stimulated cyclic AMP accumulation by M3 mACh receptor coactivation at low agonist concentrations (MCh 10−9–10−6 M).
The most profound cross-talk effects were observed with respect to ERK activation. Thus, while MCh stimulated ERK activation in both CHO-m2 and CHO-m3 cells (pEC50 values: 5.64±0.09 and 5.57±0.16, respectively), the concentration–effect relation was approx 50-fold left-shifted in CHO-m2m3 cells (pEC50: 7.17±0.07). In addition, the ERK response was greater and more sustained in CHO-m2m3 cells. In contrast, only minor differences were seen in the time-courses and concentration-dependencies of JNK activation in CHO-m3 and CHO-m2m3 cells.
Costimulation of endogenous P2Y2 purinoceptors also caused an approx 10-fold left-shift in the MCh-stimulated ERK response in CHO-m2 cells, suggesting that the Gq/11/Gi/o interaction to affect ERK activation is not specific to muscarinic receptors.
PTx pretreatment of cells had unexpected effects on ERK activation by MCh in both CHO-m2m3 and CHO-m3 cells. Thus, in CHO-m3 cells PTx pretreatment caused a marked left-shift in the MCh concentration–effect curve, while in PTx-treated CHO-m2m3 cells the maximal responsiveness was decreased, but the potency of MCh was only slightly affected.
The data presented here strongly suggest that cross-talk between M2 and M3 mACh receptors occurs at the level of both second messenger and ERK regulation. Further, these data provide novel insights into the involvement of Gi/o proteins in both positive and negative modulation of ERK responses evoked by G protein-coupled receptors.
Keywords: Muscarinic acetylcholine receptor; receptor-G protein coupling; extracellular signal-regulated kinase (ERK); c-Jun N-terminal kinase (JNK); mitogen-activated protein kinase (MAPK); radioligand binding; cyclic AMP; inositol 1,4,5-trisphosphate
Introduction
Acetylcholine, as a neurotransmitter, and as a possible non-neuronal mediator (Wessler et al., 1998), exerts many of its actions via interaction with one or more of the five mammalian muscarinic acetylcholine (mACh) receptor subtypes (Bonner et al., 1987; Caulfield & Birdsall, 1998). The mACh receptors can be grouped into two subsets, based on sequence similarities and receptor-G protein-effector coupling preferences (Wess, 1993; Caulfield & Birdsall, 1998). Thus, M1, M3 and M5 mACh receptors preferentially couple via Gq/11 proteins to the activation of phospholipase C (PLC), while M2 and M4 mACh receptors link to downstream effectors via Gi/o protein activation (e.g. to inhibit adenylyl cyclase). Despite such apparent exclusivity between the signalling pathways activated by M1/M3/M5 and M2/M4 mACh receptors, there are many opportunities for ‘cross-talk' (Eglen et al., 1994; Challiss & Blank, 1997; Selbie & Hill, 1998). For example, M2/M4 mACh receptors can affect signalling mediated by M1/M3/M5 mACh receptors through Gi-derived βγ-subunit effects on PLCs (Rhee, 2001) or modulate changes in intracellular Ca2+ concentrations through altering K+-conductance (Kurachi, 1995), or activating nonselective cation channels (Zholos & Bolton, 1997; Wang et al., 1999). However, interpretation of potential cross-talk is complicated by the demonstration that M1/M3/M5 mACh receptors can also couple to pertussis toxin (PTx) -sensitive G proteins (Offermanns et al., 1994; Akam et al., 2001) and the reported stimulatory effects of M1 (and M3/M5) mACh receptors on adenylyl cyclase activity probably result from direct receptor coupling to Gs proteins (Burford & Nahorski, 1996).
Compelling evidence has accumulated in recent years linking a wide range of G protein-coupled receptors (GPCRs) to the control of mitogen-activated protein kinases (MAPKs) and, as a consequence, the regulation of cell growth and proliferation (Dhanasekaran et al., 1995; Gutkind, 1998; Gudermann et al., 2000). In the case of mACh receptors, activation of MAPK pathways has been demonstrated for both the M1/M3/M5 and M2/M4 subfamilies (Winitz et al., 1993; Crespo et al., 1994; Mitchell et al., 1995), suggesting the potential for mACh receptor cross-talk in the regulation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) cascades (Schaeffer & Weber, 1999).
Understanding the mechanisms by which cross-talk between mACh receptors may occur is clearly an important objective, as many cells and tissues express more than one mACh receptor subtype. For example, a wide variety of smooth muscle types express both M2 and M3 mACh receptors (see Eglen et al., 1994,1996) and the respective roles of each subtype have been the subject of much investigation and speculation. The role of the M3 mACh receptor in smooth muscle contraction is well established (Eglen et al., 1994); however, the role of the usually more abundant M2 mACh receptor subpopulation is less clear (Eglen et al., 1994,1996). With respect to the regulation of muscle contraction, pharmacological (Hegde et al., 1997; Sawyer & Ehlert, 1999) and genetic/gene knockout (Matsui et al., 2000; Stengel et al., 2000,2002) approaches have shown either small direct effects of the M2 population on contraction or, more commonly, an indirect role through inhibition of the actions of relaxants. To date, very little work has been carried out to analyse the potential for cross-talk between signalling pathways following M2 and M3 mACh receptor coactivation.
In the present study we have utilized Chinese hamster ovary (CHO) cells recombinantly expressing M2 and M3 mACh receptors, either alone or in combination, to investigate the possible cross-talk between the second messenger-generating and ERK- and JNK-signalling pathways downstream of these receptors.
Methods
Materials
1-[N-methyl-3H]-scopolamine (80–85 Ci mmol−1), [2,8-3H]-cyclic AMP (30–50 Ci mmol−1), [methyl-3H]-thymidine (25 Ci mmol−1) and [γ-32P]-ATP (3000 Ci mmol−1) were obtained from Amersham Biosciences U.K. Ltd. (Little Chalfont, U.K.). Guanosine 5′-(γ-thio-[35S])triphosphate (1250 Ci mmol−1) and D-[inositol-1-3H(N)]-1,4,5-trisphosphate (30 Ci mmol−1) were obtained from NEN Life Science Products (Zaventem, Belgium). PTx, methacholine (MCh), carbachol (CCh) and forskolin were from Sigma-Aldrich Co. Ltd. (Poole, U.K.). All other reagents were of analytical grade and were obtained from the sources given by Wylie et al. (1999) and Akam et al. (2001).
Cell culture and transfection
CHO cell-lines expressing human M2 (CHO-m2) or M3 (CHO-m3) mACh receptors were originally obtained from Dr N.J. Buckley (then of the National Institute for Medical Research, Mill Hill, London) and were grown in minimal essential medium (MEMα) supplemented with newborn calf serum (10%), penicillin (100 U ml−1), streptomycin (100 μg ml−1) and amphotericin B (2.5 μg ml−1). To generate M2/M3-coexpressing cell-lines, cDNA encoding the human m3-mACh receptor was inserted into a pCEP4 vector (Invitrogen BV, Groningen, The Netherlands). Low passage CHO-m2 cells were transfected with this plasmid by calcium phosphate–DNA coprecipitation (Sambrook et al., 1989). After 24 h, cells were harvested and seeded into Petri dishes at various cell densities and grown in the constant presence of 400–800 μg ml−1 hygromycin B to select for antibiotic-resistant colonies. Colonies were selected after 21–28 days and expression of M3 mACh receptor initially assessed by Western blotting using an anti-m3-mACh receptor-specific antibody (Tobin & Nahorski, 1993). M3 mACh receptor-positive clones were expanded and radioligand binding and Ins(1,4,5)P3 mass determinations made to verify expression and function of this receptor, while M2 mACh receptor function was assessed by inhibition of forskolin-stimulated adenylyl cyclase (see below). Based on these criteria, three cell-lines were archived and the B2 clone selected for the most extensive further characterization.
[3H]-NMS saturation and displacement radioligand binding
CHO cell-lines expressing M2 and/or M3 mACh receptors were grown to confluence in 175 cm2 flasks and harvested using HBS–EDTA (10 mM HEPES, 0.9% NaCl, 0.02% EDTA, pH 7.4). A cell-membrane fraction was then prepared by homogenization and centrifugation, and [3H]-NMS saturation binding performed as described previously (Burford & Nahorski,1996; Akam et al., 2001), except that GTP was omitted and membranes were incubated with the radioligand for 90 min at 37°C before separating bound and free fractions by rapid vacuum filtration. Nonspecific binding was defined in the presence of 1 μM atropine. Displacement analysis for tripitramine was performed using approx 0.8 nM [3H]-NMS and a wide range of antagonist concentrations (0.1 nM–100 μM); again incubations were for 90 min at 37°C.
[35S]-GTPγS binding and Gqα immunoprecipitation
Membranes prepared from CHO-m2, -m3 and -m2m3 cells were incubated with approx 1 nM [35S]-GTPγS in a buffer containing 100 mM NaCl, 1 μM GDP±agonist for 2 min at 30°C. Residual binding in the presence of 10 μM GTPγS was considered nonspecific (NSB) and subtracted from basal and agonist-stimulated total counts. Incubations were terminated, membranes solubilized and G proteins immunoprecipitated using an ‘in-house' Gqα-specific antibody (raised against the C-terminal sequence CLQLNLKEYNLV) exactly as described previously by Akam et al. (2001). Scintillation fluid was added to thoroughly washed protein A-sepharose-Gqα-[35S]-GTPγS complexes and radioactivity determined.
Incubation methods and Ins(1,4,5)P3 and cyclic AMP determination
For experiments in intact cells, the different CHO cell-lines were grown to confluence in 24-well multiwells. Where cells were pretreated with PTx, it was added (100 ng ml−1) for 20–24 h before experimentation. For all experiments, monolayers were washed with HEPES-buffered Krebs–Henseleit buffer (KHB: composition in mM: NaCl, 118; KCl, 4.7; NaHCO3, 25; MgSO4, 1.2; NaH2PO4, 1.2, CaCl2, 1.3; D-glucose, 11; HEPES, 10; pH 7.4 after equilibration with O2/CO2 95 : 5). Cells were challenged with the concentrations of agonists, and for the times indicated in the Results section. Incubations were terminated by aspiration and rapid addition of 250 μl ice-cold 0.5 M trichloroacetic acid (TCA) and transferal of the multiwell plate to an icebath. After extraction for 30 min, TCA extracts were collected and neutralized using tri-n-octylamine/freon (Challiss et al., 1988). Cyclic AMP and Ins(1,4,5)P3 were determined using the radioreceptor assays of Brown et al. (1971) and Challiss et al. (1988), respectively.
ERK and JNK activity determinations
CHO-m2, -m3 or -m2m3 cells were grown in six-well plates. Where cells were pretreated with PTx, it was added (100 ng ml−1) for 20–24 h before experimentation. Incubations were performed in KHB as described above. Incubations were terminated by aspiration and washing each well with ice-cold phosphate-buffered saline. Cells were processed for ERK and JNK assay exactly as described previously (Wylie et al., 1999) with 200 μl (ERK) and/or 400 μl (JNK) aliquots of supernatant being taken for subsequent activity assays. ERK proteins were immunoprecipitated and activity assays performed exactly as described previously (Wylie et al., 1999). GST-c-Jun beads were prepared and used to determine JNK activity exactly as previously described (Wylie et al., 1999).
Data analysis
All data are presented as means±s.e. mean for the indicated number of separate experiments performed with the indicated individual experiment replication. Radioligand binding data and agonist/antagonist concentration–effect curves were analysed using a commercially available program (GraphPad Prism version 3.0; GraphPad Software, San Diego, CA, U.S.A.). Where responses to MCh were biphasic (see cyclic AMP data), concentration–response relations were fitted by a sum of two logistic equations:
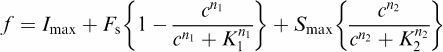 |
where Imax is the maximum inhibition of the forskolin-stimulated adenylyl cyclase activity by MCh (Fs) with IC50 value K1 and Hill coefficient n1; Smax is the maximum stimulation of cAMP accumulation by high concentrations of MCh with EC50 value and Hill coefficient K2 and n2, respectively, and c is the concentration of MCh. Fitting was done by least-squares minimization using the above equation and GraphPad Prism (version 3.0).
Statistical differences between datasets were assessed by one-way analysis of variance followed by Duncan's multiple-range test at P<0.05 using SPSS (version 10, SPSS, Chicago, IL, U.S.A.).
Results
Establishing CHO-m2m3 cell-lines
CHO cells stably expressing M2 mACh receptors (CHO-m2; Bmax 858±51 pmol mg−1 protein) were transfected with a plasmid encoding the M3 mACh receptor. Hygromycin-resistant clones were initially assessed by Western blotting using a M3-specific antibody (Tobin & Nahorski, 1993) and cells expressing M3 mACh receptor immunoreactivity screened for second messenger responses by assessing the ability of MCh to stimulate Ins(1,4,5)P3 generation and inhibit forskolin-stimulated cyclic AMP accumulation. Two cell-lines (termed B2 and B7) emerged from this screening and were evaluated for total mACh receptor expression by performing [3H]-NMS binding on suspensions of intact cells. In addition, to subdivide the M2/M3 subpopulations in B2 and B7 cell-lines, displacement of specific [3H]-NMS binding from membranes by the highly M2-selective antagonist tripitramine (Melchiorre et al., 1993) was assessed. [3H]-NMS displacement analysis in membranes derived from CHO-m2 and CHO-m3 cells demonstrated tripitramine to be highly discriminating (approx 400 fold) between M2 (pKi 9.4) and M3 (pKi 6.8) mACh receptors. The results of these binding studies are summarized in Table 1. It can be seen that for both the B2 and B7 cell-lines there is an approx 3 : 1 M3 : M2 ratio. The CHO-m2m3 B2 cell-line was selected for further study. Key findings of this study were reproduced in the B7 cell-line to provide evidence against clonal variation underlying any of the observed effects.
Table 1.
Assessment of receptor expression levels in CHO-m2, -m3 and -m2m3 cell-lines by [3H]-NMS saturation binding and quantitation of M2 and M3 mACh receptor subpopulations in B2 and B7 clones
| Cell-line | Bmax (pmol mg−1 protein) | Ratio M2 : M3 | No. expts. (n) |
|---|---|---|---|
| CHO-m2 | 0.86±0.05 | 100 : 0 | 4 |
| CHO-m3 | 1.48±0.12 | 0 : 100 | 4 |
| CHO-m2m3 (B2) | 2.16±0.21 | 27 : 73 | 6 |
| CHO-m2m3 (B7) | 2.28±0.26 | 30 : 70 | 3 |
Bmax values are expressed as means±s.e. mean for the indicated number (n) of experiments. M2 : M3 ratios were determined by two-site analysis (GraphPad Prism) of tripitramine displacement isotherms for a single concentration of [3H]-NMS (approx 0.8 nM).
[35S]-GTPγS binding and Gα-subunit-selective immunoprecipitation to assess receptor–Gq/11 coupling
Receptor coupling to Gq proteins was initially investigated in membranes prepared from CHO-m2, -m3 or -m2m3 (B2) cell-lines by [35S]-GTPγS binding and subsequent immunoprecipitation using a Gqα-specific antibody (Akam et al., 2001). Membranes prepared from CHO-m3, but not -m2 cells showed robust agonist-stimulated increases in [35S]-GTPγS associated with immunoprecipitated Gqα (Figure 1). A comparable increase in [35S]-GTPγS binding to Gqα was observed in CHO-m2m3 membranes and MCh stimulated this response with similar potencies in the two preparations (pEC50 values: CHO-m3, 4.49±0.34; CHO-m2m3, 4.29±0.37). PTx pretreatment prior to membrane preparation only appeared to affect receptor-G protein coupling in the case of the CHO-m2m3 cell-line. Thus, MCh stimulated an approx 70% greater accumulation of [35S]-GTPγS–Gqα complexes in membranes prepared from PTx pretreated CHO-m2m3 cells (Figure 1), despite the increase in Emax, the potency of MCh was similar in PTx-treated M3-expressing cell-lines (pEC50 values: CHO-m3, 4.29±0.31; CHO-m2m3, 4.20±0.14).
Figure 1.

Effects of PTx pretreatment on basal and MCh-stimulated [35S]-GTPγS binding to Gq/11α in membranes prepared from CHO-m2, -m3 and -m2m3 cells. CHO cells were preincubated in the presence of PTx (100 ng ml−1; 24 h) or vehicle before preparation of membranes. MCh (1 mM) or buffer additions to membranes were for 2 min at 30°C and, following solubilization, [35S]-GTPγS-Gq/11α complexes were immunoprecipitated and radioactivity assessed as described in the Methods section. Data are shown as means±s.e. mean for four separate experiments performed in duplicate. For both CHO-m3 and CHO-m2m3 membranes, a significant (*P<0.05) increase in Gq/11α-associated [35S]-GTPγS binding was stimulated by MCh irrespective of PTx pretreatment. In addition, following PTx pretreatment a greater agonist-stimulated Gq/11α-[35S]-GTPγS binding was observed (#P<0.05) in CHO-m2m3, but not CHO-m2 or -m3 membranes.
Comparison of Ins(1,4,5)P3 responses in CHO-m2, -m3 and -m2m3 cell-lines
MCh (1 mM) stimulated robust increases in Ins(1,4,5)P3 accumulation in the CHO-m3 and -m2m3 cell-lines (Figure 2a), but not in CHO-m2 cells where no agonist-stimulated increase in this second messenger was observed. As can be seen in Figure 2, the amplitude of the increase in Ins(1,4,5)P3 accumulation was greater in the CHO-m2m3 cell-line, with both a greater peak (25±9%) and plateau response being consistently observed. Examination of the concentration-dependency of the initial peak response (at 15 s; Figure 2b) confirmed the greater peak increase in Ins(1,4,5)P3 in the CHO-m2m3 cell-line and also demonstrated a three-fold decrease in the EC50 value in this cell-line (pEC50 values: CHO-m3, 5.21±0.10; CHO-m2m3, 5.67±0.11; P<0.05). As we have shown previously (Wylie et al., 1999) PTx pretreatment of CHO-m3 cells had no effect on either the time-course (data not shown) or concentration-dependency of Ins(1,4,5)P3 accumulation (Figure 2c). Interestingly, PTx treatment of CHO-m2m3 cells did affect the Ins(1,4,5)P3 response, generating Emax and EC50 values in this cell-line that were indistinguishable from those obtained in control CHO-m3 cells (Figure 2d).
Figure 2.

Comparison of time- and concentration-dependent agonist-stimulated Ins(1,4,5)P3 accumulations in CHO-m3 and -m2m3 cells. Cell monolayers were stimulated with either MCh (1 mM) for the times indicated (a), or for 15 s with different concentrations of MCh (b). CHO-m3 (c) or CHO-m2m3 (d) cells were pretreated with PTx (100 ng ml−1, 24 h) or vehicle before challenge with the indicated concentrations of MCh for 15 s. Incubations were terminated and extracts prepared for Ins(1,4,5)P3 mass determination as described in the Methods section. Data are shown as means±s.e. mean for three (panels a and c) or four (panels b and d) separate experiments performed in duplicate.
Effects of PTx pretreatment on basal and agonist-stimulated cyclic AMP responses in CHO-m2m3 cells
MCh (1 mM) stimulated a robust (eight-fold) increase in cyclic AMP accumulation in CHO-m2m3 cells and PTx pretreatment affected this second messenger response (Figure 3a). Thus, the cyclic AMP response to MCh was approx two-fold greater in PTx pretreated CHO-m2m3 cells, without a change in the concentration-dependency assessed at the 5 min peak (pEC50 values: −PTx, 4.24±0.22; +PTx, 4.38±0.19; Figure 3b). The data shown in Figure 2 and Figure 3 provide evidence consistent with a M2 mACh receptor-Gi modulation of both Ins(1,4,5)P3 and cyclic AMP responses in the CHO-m2m3 cell-line.
Figure 3.

Effects of PTx pretreatment on time- and concentration-dependencies of MCh-stimulated cyclic AMP accumulations in CHO-m2m3 cells. CHO-m2m3 cell monolayers were pretreated with PTx (100 ng ml−1, 24 h) or vehicle before challenge with either 1 mM MCh for the times indicated (a), or with different concentrations of MCh for 5 min (b). Incubations were terminated and extracts prepared for cyclic AMP mass determination as described in the Methods section. Data are shown as means±s.e. mean for three separate experiments performed in duplicate.
Modulatory effects of mACh receptor activation on forskolin-stimulated adenylyl cyclase activity in CHO-m2, -m3 and -m2m3 cells
The adenylyl cyclase activator forskolin (10 μM) stimulated comparable, large (>100 fold) increases in cyclic AMP accumulations in CHO-m2, -m3 and -m2m3 cell-lines. The effects of increasing concentrations of MCh on forskolin-stimulated cyclic AMP responses in CHO-m2, -m3 and -m2m3 cells are shown in Figure 4. In CHO-m2 cells, MCh concentration-dependently inhibited forskolin-stimulated cyclic AMP accumulation (pIC50: 6.60±0.06) with a maximally effective concentration of agonist causing a >90% inhibition. In both CHO-m3 and -m2m3 cells, a biphasic modulation of forskolin-stimulated adenylyl cyclase activity by MCh was observed (Figure 4). At low agonist concentrations, inhibitory effects were observed that were maximal between 0.1 and 1 μM MCh for both cell-lines, whereas at higher MCh concentrations this effect was superseded by an enhancement of the forskolin response. Although the modulatory effects of MCh were similar in the CHO-m3 and -m2m3 cell-lines, subtle differences could be observed. Thus, analysis of the curves (using the equation given in the Methods) revealed that K1 values (see Data analysis section for K1 definition) for MCh-mediated inhibitions were 22±5 versus 38±5 nM in CHO-m2m3 and -m3 cells, respectively, and MCh caused a significantly greater maximum inhibitory effect (66±3% versus 42±4%; P<0.05) in CHO-m2m3 compared to CHO-m3 cells over the 10−9–10−6 M concentration range (see Figure 4 inset).
Figure 4.

Effects of M2 and/or M3 mACh receptor activation on forskolin-stimulated cyclic AMP accumulations in CHO-m2, -m3 and -m2m3 cells. The indicated concentrations of MCh were added to cell monolayers for 10 min before challenge with forskolin (10 μM). After a further period of 10 min incubations were terminated and cyclic AMP mass determined as described in the Methods section. Data are shown as means±s.e. mean for three (CHO-m2) or four (CHO-m3 and -m2m3) separate experiments performed in duplicate. The inset focuses on the inhibitory effects of 10−9–10−6 M MCh in the three cell-lines.
Further evidence that both M2 and M3 mACh receptors shape the modulatory effects of MCh on forskolin-stimulated adenylyl cyclase activity in the CHO-m2m3 cell-line is provided in Figure 5. Thus, while PTx treatment of CHO-m3 cells has no significant effect on the MCh modulation of cyclic AMP levels, a marked attenuation of the inhibitory effect seen at low MCh concentrations was observed in the CHO-m2m3 cell-line (Figure 5). These data together strongly suggest that M2 and M3 mACh receptors cooperate in the regulation of adenylyl cyclase activity at low agonist concentrations, whereas at high agonist concentration the adenylyl cyclase stimulatory effect mediated by the M3 mACh receptor predominates.
Figure 5.

Effects of PTx pretreatment on the concentration-dependent modulatory actions of MCh on forskolin-stimulated cyclic AMP accumulations in CHO-m3 and -m2m3 cells. (a) CHO-m3 and (b) CHO-m2m3 cell monolayers were pretreated with PTx (100 ng ml−1, 24 h) or vehicle before challenge with the indicated concentrations of MCh for 10 min before addition of forskolin (10 μM). After a further period of 10 min, incubations were terminated and cyclic AMP mass determined as described in the Methods section. Results are expressed relative to the cyclic AMP responses to forskolin alone (CHO-m3: −PTx, 1008±41; +PTx, 433±50; CHO-m2m3: −PTx, 908±110; +PTx, 526±51 pmol mg−1 protein), which are set to 100% for each condition. Data are shown as means±s.e. mean for four (CHO-m3) or five (CHO-m2m3) separate experiments performed in duplicate. PTx did not significantly affect the modulatory effect of MCh in CHO-m3 cells, but significant differences were seen in toxin-treated CHO-m2m3 cells (*P<0.05).
Time- and concentration-dependent effects of M2 and M3 mACh receptor stimulation on ERK and JNK activities
MCh stimulation of CHO-m2 and -m3 cells resulted in rapid, robust (15–20 fold-over-basal) increases in ERK activity that peaked at 5 min and had returned to close to basal levels within 30 min (Figure 6a). The stimulatory effect of MCh was concentration-dependent (assessed at 5 min) generating EC50 values of 2–3 μM for each cell-line (pEC50 values: CHO-m2, 5.64±0.09; CHO-m3, 5.57±0.16; Figure 6b). Strikingly, two potentially important differences were observed in the CHO-m2m3 cell-line. Firstly, the magnitude of the ERK response was greater and the increase in activity sustained over a longer period (Figure 6a). Thus, whereas ERK activity had returned to close to basal levels in CHO-m2 and -m3 cells (2.4±0.4 and 2.5±0.6-fold-over-basal, respectively) after 30 min exposure to agonist, it was still substantially elevated in CHO-m2m3 cells (8.2±0.8-fold-over-basal; P<0.05 compared to CHO-m2 and -m3 cells, Figure 6a). Secondly, the concentration–effect curve for ERK activation by MCh was >25-fold left-shifted (pEC50: 7.17±0.07; P<0.05 compared to CHO-m2 and -m3 cells, Figure 6b). These key differences were also seen in parallel experiments using the CHO-m2m3 B7 clone (data not shown).
Figure 6.

Time- and concentration-dependent increases in extracellular signal-regulated kinase (ERK) activity stimulated by MCh in CHO-m2, -m3 and -m2m3 cells. (a) Confluent monolayers of CHO cells were stimulated with MCh (100 μM) for the times indicated, (b) or with different concentrations of MCh for 5 min. Incubations were terminated, cell lysates prepared and kinase assays performed as described in the Methods section. Data are expressed as either a ‘fold' increase over basal ERK activity (a) or as an enzymic activity (expressed as fmol phosphate incorporated into the EGF receptor fragment substrate per min per mg of cell protein (b)). Data are presented as means±s.e. mean for four to eight separate experiments (panel a), or, for data shown in panel b, four (CHO-m2), five (CHO-m3) or six (CHO-m2m3) separate experiments.
With respect to JNK activity, MCh addition to CHO-m2 cells failed to cause a reproducible increase in the activity of this MAPK, whereas stimulation of CHO-m3 and -m2m3 cells produced time- and concentration-dependent increases in JNK activity (Figure 7). MCh stimulated similar maximal five to six-fold increases in JNK activity in both CHO-m3 and -m2m3 cells at 45 min after agonist addition, although basal JNK activity was consistently higher in the coexpressing cell-line (CHO-m3, 1.01±0.38; CHO-m2m3, 2.25±0.05 pmol phosphate incorporated into c-Jun min−1 mg−1 protein; P<0.05). The concentration-dependency of JNK activation by MCh was 2.5-fold left-shifted in the coexpressing cell-line (pEC50 values: CHO-m3, 6.11±0.11; CHO-m2m3, 6.55±0.13; P<0.05) compared to the >25-fold shift noted for ERK activation in the coexpressing cell-line.
Figure 7.

Concentration-dependent increases in c-Jun N-terminal kinase (JNK) activity stimulated by MCh in CHO-m3 and -m2m3 cells. Confluent monolayers of CHO cells were stimulated with the indicated concentrations of MCh for 30 min. Incubations were then terminated, cell lysates prepared and JNK assays performed as described in the Methods section. Data are expressed as enzymic activities (expressed as fmol phosphate incorporated into c-Jun fusion protein per min per mg of cell protein) and represent means±s.e. mean for four (CHO-m3) or six (CHO-m2m3) separate experiments. The inset shows a representative experiment to illustrate a typical time-course of MCh (100 μM)-stimulated JNK activation in the two cell-lines.
Coactivation of P2Y and M2 mACh receptors also facilitates ERK activation
CHO cells possess endogenous P2Y2 purinoceptors that can mediate an ERK response (Dickenson et al., 1998). This Gq/11-coupled receptor caused a modest concentration-dependent increase in ERK activity in the CHO-m2 cell-line (pEC50: 5.40±0.34; Emax, 5.0±0.5-fold-over-basal) in response to UTP (Figure 8 inset). Stimulation of CHO-m2 cells with MCh in the presence of UTP (10 μM) produced a concentration–effect curve for ERK activation, which was approx 10-fold left-shifted (pEC50 values: −UTP, 5.53±0.24; +UTP, 6.51±0.31; P<0.05) without significantly affecting the maximal response (Figure 8). Thus, both the native UTP receptor and recombinant M3 mACh receptor increase the apparent potency of MCh at the M2 mACh receptor to cause ERK activation.
Figure 8.

Effects of coincident P2Y/M2 receptor stimulation on ERK activity in CHO-m2 cells. Confluent CHO-m2 monolayers were challenged with the indicated concentrations of MCh in the absence or presence of UTP (10 μM) for 5 min. Incubations were terminated, cell lysates prepared and kinase assays performed as described in the Methods section. Data are expressed as a ‘fold' increase over basal ERK activity and are presented as means±s.e. mean for three separate experiments. The inset shows the concentration-dependency of ERK activation by UTP (n=3 for all data, except 300 μM UTP point where n=2).
Effects of PTx on the concentration-dependent activation of ERK by MCh in CHO-m2, -m3 and -m2m3 cells
The robust ERK activation stimulated by incubation of CHO-m2 cells with MCh (100 μM) for 5 min was completely abolished by PTx pretreatment (Figure 9a). Therefore, it was anticipated that PTx pretreatment would result in a purely M3 mACh receptor–Gq/11-mediated ERK activation in CHO-m2m3 cells. As can be seen in Figure 9b, PTx treatment caused a marked decrease in the maximal ERK activation elicited by MCh, and a leftward shift in the concentration-dependency (pEC50 values: −PTx, 7.04±0.13; +PTx, 7.68±0.18; P<0.05). In the light of this unexpected result, the effect of PTx pretreatment on the agonist-stimulated ERK response in CHO-m3 cells was also investigated. As can be seen in Figure 9c, PTx pretreatment of CHO-m3 cells caused a marked (>50-fold) leftward shift in the concentration-dependency of ERK activation by MCh (pEC50 values: −PTx, 6.00±0.16; +PTx, 7.88±0.29; P<0.05).
Figure 9.

Effects of PTx pretreatment on the concentration-dependencies of ERK activation by MCh in CHO-m2, -m3 and -m2m3 cells. (a) CHO-m2, (b) CHO-m2m3 and (c) CHO-m3 cell monolayers were pretreated with PTx (100 ng ml−1, 24 h) or vehicle before challenge with the indicated concentrations of MCh (panel a; 100 μM). Incubations were terminated, cell lysates prepared and kinase assays performed as described in the Methods section. Data are expressed either as a ‘fold' increase over basal ERK activity (panels a and b), or as a percentage of the response to 100 μM MCh in vehicle-treated CHO-m3 cells (panel c). In all cases, data are shown as means±s.e. mean for at least three separate experiments.
Discussion
Although it is clear that in some cases signal transduction can be viewed as a series of parallel signalling pathways, activated by specific subsets of receptors that bring about distinct cellular responses, there is also considerable evidence for ‘cross-talk' between signalling pathways (Gudermann et al., 1996; Selbie & Hill, 1998; Schaefer & Weber, 1999). Indeed, the apparent overlap between the signalling pathways activated by different extracellular stimuli can sometimes be perplexing (see Pawson & Saxton, 1999). Nevertheless, it is becoming increasingly clear that the intracellular milieu is a highly structured environment and unwanted cross-talk is efficiently eliminated in vivo by preventing the components of the different pathways from coming into intimate contact through compartmentation and/or the ‘scaffolding' of signalling complexes (Gudermann et al., 1996; Burack & Shaw, 2000; Dumont et al., 2002). However, a number of physiologically important processes have been shown to depend upon cross-talk between pathways activated by distinct receptors to bring about phenomena such as coincidence detection (Sunahara et al., 1996; Batchelor & Garthwaite, 1997; Sweatt, 2001).
In the present study, we have investigated whether cross-talk occurs between the pathways that lie downstream of M2 and M3 mACh receptors stably coexpressed in CHO cell-lines. The clones isolated in this study, which demonstrated a coexpression of these receptor populations, both exhibited M3>M2 mACh receptor densities (see Table 1). While this differs from the situation in a number of tissues, for example, the majority of smooth muscle types where the M2 mACh receptor predominates, it does reflect the receptor distributions in some tissues (e.g. rat uterine smooth muscle; Choppin et al., 1999), and we considered that the CHO-m2m3 cell-lines we had isolated and characterized should provide useful information on mACh receptor cross-talk at multiple loci downstream of receptor activation.
Cross-talk at the level of second messenger generation
In both the CHO-m2m3 and -m3 cell-lines, but not in CHO-m2 cells, MCh addition caused a rapid and large increase in Ins(1,4,5)P3. In the coexpressing cell-line, the initial peak increase in Ins(1,4,5)P3 was greater, and the concentration-dependency was approx three-fold to the left of the responses seen in CHO-m3 cells expressing a comparable M3 mACh receptor density. Furthermore, elimination of productive coupling to cellular Gi/o proteins in CHO-m2m3 cells decreased both the peak response and shifted the concentration-dependency to that seen in CHO-m3 cells. These data strongly suggest that in the CHO-m2m3 cell-line, agonist stimulation of the M2 mACh receptor facilitates the M3-driven Ins(1,4,5)P3 response. Such an effect might be brought about by a number of mechanisms, including a Gi-derived βγ subunit activation of PLC-β isoenzymes that is not observed with M2 mACh receptor activation alone as the effect may be conditional upon a Gq/11α–PLC-β binding interaction (Chan et al., 2000). Alternatively, the liberation of βγ subunits through M2 mACh receptor activation may facilitate Gq/11α-mediated signalling by increasing the efficiency of Gq/11 heterotrimer presentation to the activated M3 mACh receptor (Quitterer & Lohse, 1999). A further possibility is that the enhanced response seen in CHO-m2m3 cells might be mediated through an M2-mediated enhancement of a Ca2+ entry pathway to facilitate phospholipase C isoenzymic activities (Rhee, 2001). However, our preliminary data on MCh-stimulated [Ca2+]i responses in the CHO-m3 and CHO-m2m3 cell lines have provided no evidence in support of this possibility (Hornigold, D.C., Daniels, D. & Challiss, R.A.J., unpublished data).
As well as stimulating Ins(1,4,5)P3 accumulation, M3 mACh receptor activation also increased cyclic AMP accumulation by a mechanism that was attenuated by M2 mACh receptor coactivation. Thus, PTx pretreatment of CHO-m2m3 cells resulted in a marked increase (by approx 100%) in peak cyclic AMP accumulation without affecting the concentration-dependency of this effect. Under conditions where basal cyclic AMP accumulation was increased by forskolin a rather different picture emerged. Thus, while agonist stimulation of M2 mACh receptors resulted in a monophasic inhibition of the forskolin-stimulated response, M3 receptor activation caused a biphasic response where low concentrations of MCh caused an inhibition, which was superseded by an enhancement of the forskolin-stimulated accumulation at higher agonist concentrations. In experiments not reported here, it has been shown that the inhibitory effect of MCh in CHO-m3 cells is dependent on the presence of extracellular Ca2+ and is blocked by lanthanides (La3+ and Gd3+). These and other data suggest that receptor-gated and/or store-depletion-operated Ca2+- channels cause Ca2+ entry that inhibits specific adenylyl cyclase (AC5/AC6) isoenzymes in CHO cells, a mechanism observed previously in other cell-types (Fagan et al., 1998; Wong et al., 2000). Irrespective of the mechanisms underlying the biphasic cyclic AMP response seen in CHO-m3 cells, an interaction between M2- and M3-mediated mechanisms to inhibit forskolin-stimulated adenylyl cyclase activity was clearly evident at low (⩽1 μM) MCh concentrations. This was confirmed by the demonstration that following PTx pretreatment, the magnitude and concentration-dependency of the inhibitory effects of MCh in CHO-m2m3 cells were similar to those seen in CHO-m3 cells.
Cross-talk at the level of the MAP kinases
In CHO cells, stimulation of M2 and M3 mACh receptors caused comparable activations of ERK, whereas JNK activity was activated only by M3 mACh receptors, in agreement with previous work (Wylie et al., 1999). The pathways linking M2/M3 mACh receptor activation to ERK have been investigated in CHO and other cell-types. Such studies have often shown the dependence of the M3-driven response on PKC and possibly other intermediary proteins (Kim et al., 1999; Wylie et al., 1999; Slack, 2000; Budd et al., 2001), while the M2-driven ERK response has been proposed to be Gβγ-dependent and may involve pathways distinct from, or overlapping with those seen following M3 mACh receptor activation (Winitz et al., 1993; Crespo et al., 1994; Lopez-Ilasaca et al., 1997; Wylie et al., 1999). In the CHO-m2m3 cell-lines, the concentration-dependency of ERK activation by MCh was dramatically left-shifted (by more than 25-fold), the maximal ERK response increased, and the duration of the ERK activation prolonged, compared to the MCh responses observed in either CHO-m2 or -m3 cells. The M2/M3 synergism, at least with respect to the sensitivity increase for ERK activation, is unlikely to be specific to these mACh receptor subtypes, but a more general consequence of coactivating Gq/11- and Gi/o-coupled GPCRs, as activation of endogenous P2Y receptors in CHO-m2 cells also caused a significant leftward shift in the MCh concentration–effect curve for ERK activation. However, the presence of Gq/11- and Gi/o-coupled GPCRs responsive to a common hormone/neurotransmitter may optimize the synergism. In contrast to the consequences of M2/M3 mACh receptor coexpression for the regulation of ERK activity, the profile of JNK activation in CHO-m2m3 cells showed only modest differences from the CHO-m3 cell-line. Although constitutively active Giα proteins can be shown to stimulate JNK activity (Yamauchi et al., 2000), in our hands M2 mACh receptor activation did not stimulate JNK activity in CHO cells and therefore the lack of M2/M3 interaction to promote JNK activation was anticipated.
The observation of an altered duration of ERK activation in the receptor coexpressing cell-line may be particularly significant, as it has been shown that how well receptor-mediated ERK activation is maintained markedly affects the outcome with respect to cell fate (Marshall, 1995). Thus, a number of well-characterized examples now exist to show that the longevity of ERK activation, rather than simply the magnitude of the response, determines cellular decision-making with respect to processes such as proliferation, cell growth and transformation (Tombes et al., 1998; Orsini et al., 1999; Murphy et al., 2002). Therefore, if the increased agonist sensitivity and responsiveness, together with a more sustained pattern of ERK activation, are general features of cells coexpressing M2/M3 mACh receptors this may be of particular physiological significance.
Gq/11 and Gi/o involvement in M3 mACh receptor-ERK signalling
Unexpected results were obtained with respect to ERK activation profiles generated in CHO-m2m3 cells following PTx pretreatment. It had been anticipated that toxin ablation of M2 mACh receptor-Gi/o signalling would result in agonist concentration–effect curves that overlay those obtained in CHO-m3 cells; however, while the maximum responsiveness decreased to levels typically seen in CHO-m3 cells (i.e. from approx 30-fold down to 10–15 fold), no rightward shift in the concentration-dependency was observed. Interestingly, a partial explanation for these data was provided by experiments where CHO-m3 cells were PTx-treated. Under these conditions, the concentration–effect curve for the agonist-stimulated increase in ERK activity was dramatically left-shifted. Taken together these data suggest that under normal conditions M3 mACh receptors couple to both Gq/11 and Gi/o proteins and inactivation of the Gi/o protein subpopulation appears to result in a much greater sensitivity (approx 100-fold) to agonist with respect to ERK activation. These data contrast with an elegant previous study by Blaukat et al. (2000) who demonstrated that efficient ERK activation by B2 bradykinin, and M1 and M3 mACh receptors is dependent on cooperation between Gq/11α and Gi/oα signals in HEK293T cells.
We have previously shown that M3 mACh receptors can couple to Gi/o proteins in CHO cell membrane preparations by Gα-subtype-specific immunoprecipitation of G protein-[35S]-GTPγS complexes (Akam et al., 2001). Using the same technique here, we have shown that PTx pretreatment did not affect receptor-Gq/11 coupling in CHO-m3 cells, and while MCh stimulated an increased maximal yield of Gq/11α protein-[35S]-GTPγS complexes in CHO-m2m3 cell membranes, the concentration dependency of this stimulation was unaffected by Gi/o inactivation. Thus, the observed effects of PTx on receptor-Gq/11 coupling in membranes derived from CHO-m3 and -m2m3 cells provide little insight into what may underlie the effects seen at the level of ERK regulation.
How is it possible to rationalize a situation where M2 mACh receptor stimulation per se stimulates a robust and entirely PTx-sensitive ERK activity, while M3 mACh receptor stimulation also appears to recruit a Gi/o component, but in this case to attenuate the Gq/11-driven ERK response? Furthermore, in the CHO-m2m3 cell-line costimulation of the M2-receptor must either suppress or overcome any Gi/o protein component stimulated by the M3-receptor such that M2/M3 mACh receptor coactivation results in the marked leftward curve shift and increased responsiveness. At present, we can only speculate on the mechanism(s) that underlies the data presented here. One possibility is that distinct Gi/oα protein subpopulations are activated by M2 and M3 receptors that respectively mediate stimulatory or inhibitory effects on the pathways regulating ERK activity. There is some evidence for this in the CHO cell-lines used here (Dell'Acqua et al., 1993; Akam et al., 2001) and in other systems (Migeon et al., 1995). In addition, it is possible that M2 and M3 receptors may facilitate the release of distinct Gi/o-derived βγ-subunit combinations, or there may be micro-compartmentation of mACh receptor access to Gi/o pools (Gudermann et al., 1996; Albert & Robillard, 2002).
In summary, we have demonstrated a number of interactions between M2 and M3 mACh receptor signalling pathways that are unlikely to result from simple additivity between convergent signalling events. In particular, we report that coactivation of M2 and M3 mACh receptors in a CHO cell background results in a synergistic activation of ERK where both the sensitivity and responsiveness to agonist stimulation are increased. These data raise the question of whether ERK activation is similarly facilitated in tissues coexpressing these mACh receptor populations.
Acknowledgments
We thank Roche Bioscience (Palo Alto, U.S.A.) for financial support to D.C.H. and P.D.R. We gratefully acknowledge the work of Donna K. Boxall and Barbara Keys in helping to establish the CHO-m2m3 clonal cell-lines. We thank Professor C. Melchiorre (University of Bologna, Italy) for the gift of tripitramine, Dr Noel Davies for providing expert advice on the curve-fitting analysis. Finally, we would like to express our gratitude to Professor Steve Nahorski (University of Leicester), and Drs Richard Eglen, Anthony Ford and Don Daniels (Roche Bioscience, Palo Alto, USA) for their help, advice and encouragement during this project.
Abbreviations
- CCh
carbachol
- CHO
Chinese hamster ovary
- ERK
extracellular signal-regulated kinase
- GPCR
G protein-coupled receptor
- GTPγS
guanosine 5′-[γ-thio]triphosphate
- Ins(1,4,5)P3
inositol 1,4,5-trisphosphate
- JNK
c-Jun N-terminal kinase
- mACh
muscarinic acetylcholine
- MAPK
mitogen-activated protein kinase
- MCh
methacholine
- NMS
N-methyl-scopolamine
- PLC
phosphoinositide-specific phospholipase C
- PTx
pertussis toxin
- UTP
uridine 5′-triphosphate
References
- AKAM E.C., CHALLISS R.A.J., NAHORSKI S.R. Gq/11 and Gi/o activation profiles in CHO cells expressing human muscarinic receptors: dependence on agonist as well as receptor-subtype. Br. J. Pharmacol. 2001;132:950–958. doi: 10.1038/sj.bjp.0703892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALBERT P.R., ROBILLARD L. G protein specificity: traffic direction required. Cell. Signal. 2002;14:407–418. doi: 10.1016/s0898-6568(01)00259-5. [DOI] [PubMed] [Google Scholar]
- BATCHELOR A.M., GARTHWAITE J. Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature. 1997;385:74–77. doi: 10.1038/385074a0. [DOI] [PubMed] [Google Scholar]
- BLAUKAT A., BARAC A., CROSS M.J., OFFERMANNS S., DIKIC I. G protein-coupled receptor-mediated mitogen-activated protein kinase activation through cooperation of Gαq and Gαi signals. Mol. Cell. Biol. 2000;20:6837–6848. doi: 10.1128/mcb.20.18.6837-6848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BONNER T.I., BUCKLEY N.J., YOUNG A.C., BRANN M.R. Identification of a family of muscarinic acetylcholine receptor genes. Science. 1987;237:527–532. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- BROWN B.L., ALBANO J.D.M., ELKINS R.P., SGHERZI A.M., TAMPION W. A simple and sensitive saturation assay method for the measurement of adenosine 3′,5′-cyclic monophosphate. Biochem. J. 1971;121:561–563. doi: 10.1042/bj1210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUDD D.C., WILLARS G.B., MCDONALD J.E., TOBIN A.B. Phosphorylation of the Gq/11-coupled M3-muscarinic receptor is involved in receptor activation of the ERK-1/2 mitogen-activated protein kinase pathway. J. Biol. Chem. 2001;276:4581–4587. doi: 10.1074/jbc.M008827200. [DOI] [PubMed] [Google Scholar]
- BURACK W.R., SHAW A.S. Signal transduction: hanging on a scaffold. Curr. Opin. Cell Biol. 2000;12:211–216. doi: 10.1016/s0955-0674(99)00078-2. [DOI] [PubMed] [Google Scholar]
- BURFORD N.T., NAHORSKI S.R. Muscarinic m1 receptor-stimulated adenylate cyclase activity in Chinese hamster ovary cells is mediated by Gsα and is not a consequence of phosphoinositidase C activation. Biochem. J. 1996;315:883–888. doi: 10.1042/bj3150883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAULFIELD M.P., BIRDSALL N.J.M. IUPHAR XVII. Classification of muscarinic acetylcholine receptors. Pharmacol. Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- CHALLISS R.A.J., BLANK J.L.Muscarinic acetylcholine receptor signalling pathways in smooth muscle Muscarinic Receptor Subtypes in Smooth Muscle. 1997Boca Raton: CRC Press, Inc; 39–86.ed. Eglen, R.M. [Google Scholar]
- CHALLISS R.A.J., BATTY I.H., NAHORSKI S.R. Mass measurements of inositol 1,4,5-trisphosphate in rat cerebral cortex slices using a radioreceptor assay: effects of neurotransmitters and depolarization. Biochem. Biophys. Res. Commun. 1988;157:684–691. doi: 10.1016/s0006-291x(88)80304-8. [DOI] [PubMed] [Google Scholar]
- CHAN J.S.C., LEE J.W.M., HO M.K.C., WONG Y.H. Preactivation permits subsequent stimulation of phospholipase C by Gi-coupled receptors. Mol. Pharmacol. 2000;57:700–708. doi: 10.1124/mol.57.4.700. [DOI] [PubMed] [Google Scholar]
- CHOPPIN A., STEPAN G.J., LOURY D.N., WATSON N., EGLEN R.M. Characterization of the muscarinic receptor in isolated uterus of sham-operated and ovariectomized rats. Br. J. Pharmacol. 1999;127:1551–1558. doi: 10.1038/sj.bjp.0702696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRESPO P., XU N., SIMONDS W.F., GUTKIND J.S. Ras-dependent activation of MAP kinase pathway mediated by G protein βγ-subunits. Nature. 1994;369:418–420. doi: 10.1038/369418a0. [DOI] [PubMed] [Google Scholar]
- DELL'ACQUA M.L., CARROLL R.C., PERALTA E.G. Transfected m2 muscarinic acetylcholine receptors couple to Gαi2 and Gαi3 in Chinese hamster ovary cells. J. Biol. Chem. 1993;268:5676–5685. [PubMed] [Google Scholar]
- DHANASEKARAN N., HEASLEY L.E., JOHNSON G.L. G protein-coupled receptor systems involved in cell growth and oncogenesis. Endocrine Rev. 1995;16:259–270. doi: 10.1210/edrv-16-3-259. [DOI] [PubMed] [Google Scholar]
- DICKENSON J.M., BLANK J.L., HILL S.J. Human adenosine A1 receptor and P2Y2-purinoceptor-mediated activation of the mitogen-activated protein kinase cascade in transfected CHO cells. Br. J. Pharmacol. 1998;124:1491–1499. doi: 10.1038/sj.bjp.0701977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUMONT J.E., DREMIER S., PIRSON I., MAENHAUT C. Cross signalling, cell specificity, and physiology. Am. J. Physiol. 2002;283:C2–C28. doi: 10.1152/ajpcell.00581.2001. [DOI] [PubMed] [Google Scholar]
- EGLEN R.M., HEGDE S.S., WATSON N. Muscarinic receptor subtypes and smooth muscle function. Pharmacol. Rev. 1996;48:531–565. [PubMed] [Google Scholar]
- EGLEN R.M., REDDY H., WATSON N., CHALLISS R.A.J. Muscarinic acetylcholine receptor subtypes in smooth muscle. Trends Pharmacol. Sci. 1994;15:114–119. doi: 10.1016/0165-6147(94)90047-7. [DOI] [PubMed] [Google Scholar]
- FAGAN K.A., MONS N., COOPER D.M.F. Dependence of the Ca2+-inhibitable adenylyl cyclase of C6-2B glioma cells on capacitative Ca2+ entry. J. Biol. Chem. 1998;273:9297–9305. doi: 10.1074/jbc.273.15.9297. [DOI] [PubMed] [Google Scholar]
- GUDERMANN T., GROSSE R., SCHULTZ G. Contribution of receptor/G protein signaling to cell growth and transformation. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000;361:345–362. doi: 10.1007/s002109900208. [DOI] [PubMed] [Google Scholar]
- GUDERMANN T., KALKBRENNER F., SCHULTZ G. Diversity and selectivity of receptor-G protein interaction. Annu. Rev. Pharmacol. Toxicol. 1996;36:429–459. doi: 10.1146/annurev.pa.36.040196.002241. [DOI] [PubMed] [Google Scholar]
- GUTKIND J.S. The pathways connecting G protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J. Biol. Chem. 1998;273:1839–1842. doi: 10.1074/jbc.273.4.1839. [DOI] [PubMed] [Google Scholar]
- HEGDE S.S., CHOPPIN A., BONHAUS D., BRIAUD S., LOEB M., MOY T.M., LOURY D., EGLEN R.M. Functional role of M2 and M3 muscarinic receptors in the urinary bladder of rats in vitro and in vivo. Br. J. Pharmacol. 1997;120:1409–1418. doi: 10.1038/sj.bjp.0701048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM J.Y., YANG M.S., OH C.D., KIM K.T., HA M.J., KANG S.S., CHUN J.S. Signalling pathway leading to an activation of mitogen-activated protein kinase by stimulating M3 muscarinic receptor. Biochem. J. 1999;337:275–280. [PMC free article] [PubMed] [Google Scholar]
- KURACHI Y. G protein regulation of cardiac muscarinic potassium channel. Am. J. Physiol. 1995;269:C821–C830. doi: 10.1152/ajpcell.1995.269.4.C821. [DOI] [PubMed] [Google Scholar]
- LOPEZ-ILASACA M., CRESPO P., PELLICI P.G., GUTKIND J.S., WETZKER R. Linkage of G protein-coupled receptors to the MAPK signalling pathway through PI 3-kinase γ. Science. 1997;275:394–397. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- MARSHALL C.J. Specificity of receptor tyrosine kinase signalling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- MATSUI M., MOTOMURA D., KARASAWA H., FUJIKAWA T., JIANG J., KOMIYA Y., TAKAHASHI S., TAKETO M.M. Multiple functional defects in peripheral autonomic organs in mice lacking muscarinic acetylcholine receptor gene for the M3 subtype. Proc. Natl. Acad. Sci. U.S.A. 2000;97:9579–9584. doi: 10.1073/pnas.97.17.9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELCHIORRE C., BOLOGNESI M.L., CHIARINI A., MINARINI A., SPAMPINATO S. Synthesis and biological activity of some methoctramine-related tetraamines bearing a 11-acetyl-5,11-dihydro-6H-pyrido[2,3-b][1,4]-benzodiazepine-6-one moiety as antimuscarinics: a second generation of highly selective M2 muscarinic receptor antagonists. J. Med. Chem. 1993;36:3734–3737. doi: 10.1021/jm00075a032. [DOI] [PubMed] [Google Scholar]
- MIGEON J.C., THOMAS S.L., NATHANSON N.M. Differential coupling of m2 and m4 muscarinic receptors to inhibition of adenylyl cyclase by Giα and Goα subunits. J. Biol. Chem. 1995;270:16070–16074. doi: 10.1074/jbc.270.27.16070. [DOI] [PubMed] [Google Scholar]
- MITCHELL F.M., RUSSELL M., JOHNSON G.L. Differential calcium dependence in the activation of c-jun kinase and mitogen-activated protein kinase by muscarinic acetylcholine receptors in rat 1a cells. Biochem. J. 1995;309:381–384. doi: 10.1042/bj3090381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY L.O., SMITH S., CHEN R.-H., FINGAR D.C., BLENIS J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell. Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- OFFERMANNS S., WIELAND T., HOMANN D., SANDMANN J., BOMBIEN E., SPICHER K., SCHULTZ G., JAKOBS K.-H. Transfected muscarinic acetylcholine receptors selectively couple to Gi-type proteins and Gq/11. Mol. Pharmacol. 1994;45:890–898. [PubMed] [Google Scholar]
- ORSINI M.J., KRYMSKAYA V.P., ESZTERHAS A.J., BENOVIC J.L., PANETTIERI R.A., PENN R.B. MAPK superfamily activation in human airway smooth muscle: mitogenesis requires prolonged p42/p44 activation. Am. J. Physiol. 1999;277:L479–L488. doi: 10.1152/ajplung.1999.277.3.L479. [DOI] [PubMed] [Google Scholar]
- PAWSON T., SAXTON T.M. Signaling networks–do all roads lead to the same genes. Cell. 1999;97:675–678. doi: 10.1016/s0092-8674(00)80779-5. [DOI] [PubMed] [Google Scholar]
- QUITTERER U., LOHSE M.J. Crosstalk between Gαi- and Gαq-coupled receptors is mediated by Gβγ exchange. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10626–10631. doi: 10.1073/pnas.96.19.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RHEE S.G. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAMBROOK J., FRITSCH E.F., MANIATIS T. 1989Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 16.32–16.40.Molecular Cloning, a Laboratory Manual [Google Scholar]
- SAWYER G.W., EHLERT F.J. Muscarinic M3 receptor inactivation reveals a pertussis toxin-sensitive contractile response in the guinea pig colon: evidence for M2/M3 receptor interactions. J. Pharmacol. Exp. Ther. 1999;289:464–476. [PubMed] [Google Scholar]
- SCHAEFFER H.J., WEBER M.J. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell. Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SELBIE L.A., HILL S.J. G protein-coupled receptor cross-talk: the fine-tuning of multiple receptor-signalling pathways. Trends Pharmacol. Sci. 1998;19:87–93. doi: 10.1016/s0165-6147(97)01166-8. [DOI] [PubMed] [Google Scholar]
- SLACK B.E. The m3 muscarinic acetylcholine receptor is coupled to mitogen-activated protein kinase via protein kinase C and epidermal growth factor receptor kinase. Biochem. J. 2000;348:381–387. [PMC free article] [PubMed] [Google Scholar]
- STENGEL P.W., GOMEZA J., WESS J., COHEN M.L. M2 and M4 receptor knockout mice: muscarinic receptor function in cardiac and smooth muscle in vitro. J. Pharmacol. Exp. Ther. 2000;292:877–885. [PubMed] [Google Scholar]
- STENGEL P.W., YAMADA M., WESS J., COHEN M.L. M3-receptor knockout mice: muscarinic receptor function in atria, stomach fundus, urinary bladder, and trachea. Am. J. Physiol. 2002;282:R1443–R1449. doi: 10.1152/ajpregu.00486.2001. [DOI] [PubMed] [Google Scholar]
- SUNAHARA R.K., DESSAUER C.W., GILMAN A.G. Complexity and diversity of mammalian adenylyl cyclases. Annu. Rev. Pharmacol. Toxicol. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- SWEATT J.D. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- TOBIN A.B., NAHORSKI S.R. Rapid agonist-mediated phosphorylation of m3-muscarinic receptors revealed by immunoprecipitation. J. Biol. Chem. 1993;268:9817–9823. [PubMed] [Google Scholar]
- TOMBES R.M., AUER K.L., MIKKELSEN R., VALERIE K., WYMANN M.P., MARSHALL C.J., MCMAHON M., DENT P. The mitogen-activated protein (MAP) kinase cascade can either stimulate or inhibit DNA synthesis in primary cultures of rat hepatocytes depending upon whether its activation is acute/phasic or chronic. Biochem. J. 1998;330:1451–1460. doi: 10.1042/bj3301451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Y.X., DHULIPALA D.K., LI L., BENOVIC J.L., KOTLIKOFF M.I. Coupling of M2 muscarinic receptors to membrane ion channels via phosphoinositide 3-kinase-γ and atypical protein kinase C. J. Biol. Chem. 1999;274:13859–13864. doi: 10.1074/jbc.274.20.13859. [DOI] [PubMed] [Google Scholar]
- WESS J. Molecular basis of muscarinic acetylcholine receptor function. Trends Pharmacol. Sci. 1993;14:308–313. doi: 10.1016/0165-6147(93)90049-p. [DOI] [PubMed] [Google Scholar]
- WESSLER I., KIRKPATRICK C.J., RACKE K. Non-neuronal acetylcholine, a locally acting molecule, widely distributed in biological systems: expression and function in humans. Pharmacol. Ther. 1998;77:59–79. doi: 10.1016/s0163-7258(97)00085-5. [DOI] [PubMed] [Google Scholar]
- WINITZ S., RUSSELL M., QUIAN N.X., GARDNER A., DWYER L., JOHNSON G.L. Involvement of Ras and Raf in the Gi-coupled acetylcholine muscarinic m2 receptor activation of mitogen-activated protein (MAP) kinase kinase and MAP kinase. J. Biol. Chem. 1993;268:19196–19199. [PubMed] [Google Scholar]
- WONG M.P.M., COOPER D.M.F., YOUNG K.W., YOUNG J.M. Characteristics of the Ca2+ -dependent inhibition of cyclic AMP accumulation by histamine and thapsigargin in human U373 MG astrocytoma cells. Br. J. Pharmacol. 2000;130:1021–1030. doi: 10.1038/sj.bjp.0703411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WYLIE P.G., CHALLISS R.A.J., BLANK J.L. Regulation of extracellular-signal regulated kinase and c-Jun N-terminal kinase by G-protein-linked muscarinic acetylcholine receptors. Biochem. J. 1999;338:619–628. [PMC free article] [PubMed] [Google Scholar]
- YAMAUCHI J., KAWANO T., NAGAO M., KAZIRO Y., ITOH H. Gi-dependent activation of c-Jun N-terminal kinase in human embryonal kidney 293 cells. J. Biol. Chem. 2000;275:7633–7640. doi: 10.1074/jbc.275.11.7633. [DOI] [PubMed] [Google Scholar]
- ZHOLOS A.V., BOLTON T.B. Muscarinic receptor subtypes controlling the cationic current in guinea-pig ileal smooth muscle. Br. J. Pharmacol. 1997;122:885–893. doi: 10.1038/sj.bjp.0701438. [DOI] [PMC free article] [PubMed] [Google Scholar]