

Abstract
Porous polymeric media (polymer foams) are utilized in a wide range of applications, such as thermal and mechanical insulators, solid supports for catalysis, and medical devices. A process for the production of polymer foams has been developed. This process, which is applicable to a wide range of polymers, uses a hydrocarbon particulate phase as a template for the precipitation of the polymer phase and subsequent pore formation. The use of a hydrocarbon template allows for enhanced control over pore structure, porosity, and other structural and bulk characteristics of the polymer foam. Polymer foams with densities as low as 120 mg/cc, porosity as high as 87%, and high surface areas (20 m2/g) have been produced. Foams of poly(l-lactic acid), a biodegradable polymer, produced by this process have been used to engineer a variety of different structures, including tissues with complex geometries such as in the likeness of a human nose.
Keywords: hydrocarbon porogen, tissue engineering, drug delivery
Polymeric foams are utilized in a range of applications such as mechanical dampeners, thermal, acoustic, and electrical insulators, solid supports for catalysis and separations, and medical devices (1–9). Macroporous polymeric foams have been produced by dispersion of a gaseous phase in a fluid polymer phase, leaching of a water-soluble inorganic fugitive phase, phase separation, polymer precipitation, particle sintering, extrusion, and injection molding (1, 2, 10). However, these processes do not generally offer optimal control over pore structure (cell diameter and pore interconnectivity) and bulk characteristics (density, void volume, mechanical and electrical properties) (11).
It occurred to us that, by combining two distinct foaming processes, (i) leaching of a fugitive phase with (ii) polymer precipitation, one could attain enhanced control over both porosity and bulk properties of the polymer foam. This was achieved by using a non-water-soluble particulate hydrocarbon fugitive phase derived from waxes, which allowed for the formation of pores with concomitant precipitation of the polymer phase. The macroporosity of the polymer foam was determined by the hydrocarbon fugitive phase (porogen), which also functioned as a template for the rapid precipitation of the polymer. Bulk properties of the foam could be manipulated independently of the macroporosity and pore size by incorporation of inorganic and organic fillers into the highly viscous polymer phase.
The process is applicable to a wide range of polymer systems including water-soluble polymers, as long as the following conditions are satisfied: (i) the hydrocarbon porogen is extracted below the melting temperature of the polymer, to ensure isotropy in the properties of the resulting foam; (ii) the polymer has good solubility (at least 100 mg/ml) in a solvent that is a poor solvent for the porogen, to obtain a viscous polymer solution wherein the porogen can be distributed uniformly; and (iii) the polymer has a molecular weight of at least 40,000, to ensure structural stability of the resulting foam.
Materials and Methods
Chemicals.
Poly(l-lactic acid) (PLLA) (70 kDa) and poly(l-lactic-co-glycolic acid) (PLGA) (108 kDa) were obtained from Birmingham Polymers (Birmingham, AL), poly(ethylene oxide) (5, 10, 20, 40, and 100 kDa) was purchased from Polysciences or Fluka AG, and poly(methyl methacrylate) (PMMA) (70 kDa) was purchased from Aldrich. All polymers were ground and sieved to <500 μm particles before use. Paraffin and beeswax were purchased from Fisher Scientific. All other solvents and chemicals were obtained from EM Science.
Preparation of Polymer Foams.
In the first step, a multicomponent system consisting of a viscous polymer solution and a particulate hydrocarbon porogen is compacted in a Teflon mold. In the subsequent step, the polymer/solvent/porogen phase is subjected to extraction in a hydrocarbon solvent, such as pentane or hexane, that is a nonsolvent for the polymer but miscible with the polymer solvent. As a result, the porogen is extracted with a simultaneous rapid precipitation of the polymer phase and the formation of a network of the same (Fig. 1; Table 1).
Figure 1.

A schematic representation of the steps involved in the preparation of polymeric foams (the composition details provided as an example yield a foam 1 cm3 in volume with ≈87% porosity). Step 1: The polymer (200 mg) is dissolved in a suitable solvent (e.g., methylene chloride, chloroform) and then mixed with the hydrocarbon porogen (e.g., paraffin, beeswax, bone wax) (800 mg of 300- to 500-μm particles) to yield a moldable mixture. Step 2: This mixture is then compacted in a Teflon mold (1 cm3). Step 3: The polymer/porogen mixture in the mold is then immersed in an aliphatic hydrocarbon solvent (e.g., pentane, hexane), which is a nonsolvent for the polymer, for 20 min at 45–50°C. During this step, the porogen and polymer solvent are extracted with concurrent precipitation of the polymer phase. To improve the efficiency of solvent penetration, the mold is equipped with small (1 mm in diameter) openings on all faces. Residual porogen is removed by repeating the last step three more times. The foam obtained is then dried under vacuum to remove any trace of solvents.
Table 1.
Composition and structural specifications of polymer foams
| Foam composition, ratios are wt %*† | Polymer/porogen, wt/wt | Porogen size distribution | Porogen morphology | Additives, wt % of total polymer | Total percent porosity (P), n = 3§¶ | Foam dimensions obtained, cm |
|---|---|---|---|---|---|---|
| PLLA | 0.25 | 300–500 μm | Irr | None | 88 | 1 × 1 × 1; 2.5 × 2.5 × 2.5; and 1 × 2.5 × 2.5 |
| PLLA | 0.29 | 700–800 μm | Sp | None | 86 | 1.2 × 1 × 1 |
| PLLA | 0.29 | ≈1,000 μm (1 mm) | Sp | None | 88 | 1.2 × 1 × 1 |
| PLLA | 0.5 | ≈1,000 μm (1 mm) | Sp | None | 75 | 1.2 × 1 × 1 |
| PLGA | 0.25 | 300–500 μm | Irr | None | 86 | 1 × 1 × 1; 2.5 × 2.5 × 2.5; and 1 × 2.5 × 2.5 |
| PEO | 0.43 | 300–500 μm | Irr | None | 80 | 1 × 1 × 1 |
| PMMA | 0.29 | 300–500 μm | Irr | None | 77 | 1 × 1 × 1 |
| PLLA + PEG (80:20) | 0.25 | 300–500 μm | Irr | None | 87 | 1 × 1 × 1; 2.5 × 2.5 × 2.5 |
| PLLA + PEG (60:40) | 0.25 | 300–500 μm | Irr | None | 88 | 1 × 1 × 1; 2.5 × 2.5 × 2.5 |
| PLGA + PEO (80:20) | 0.25 | 300–500 μm | Irr | None | 86 | 1 × 1 × 1; 2.5 × 2.5 × 2.5 |
| PLGA + PEG (80:20) | 0.25 | 300–500 μm | Irr | None | 88 | 1 × 1 × 1; 2.5 × 2.5 × 2.5 |
| PLLA + PEG (80:20) | 0.25 | 300–500 μm | Irr | 2.5%, Rh-B | 86 | 1 × 1 × 1 |
| PLGA + PEG (80:20) | 0.25 | 300–500 μm | Irr | 2.5%, Rh-B | 85 | 1 × 1 × 1 |
| PLLA + PEG (80:20) | 0.25 | 300–500 μm | Irr | 50%, CC†‡ | 87 | 1 × 1 × 1 |
| PLGA + PEG (80:20) | 0.25 | 300–500 μm | Irr | 50%, CC†‡ | 85 | 1 × 1 × 1; 2.5 × 2.5 × 2.5 |
| PLLA | 0.25 | 300–500 μm | Irr | 50%, Unfired HA†‡ | 85 | 1 × 1 × 1 |
| PLLA | 0.25 | 300–500 μm | Irr | 50%, Sintered HA†‡ | 85 | 1 × 1 × 1 |
| PLLA | 0.25 | 300–500 μm | Irr | 100%, Sintered HA†‡ | 80 | 1 × 1 × 1 |
| PMMA | 0.43 | 300–500 μm | Irr | 10%, CG† | 80 | 1 × 1 × 1 |
| PMMA | 0.36 | 300–500 μm | Irr | 40%, CG† | 77 | 1 × 1 × 1 |
| PMMA | 0.36 | 300–500 μm | Irr | 80%, CG† | 75 | 1 × 1 × 1 |
Spherical hydrocarbon phase resulted in pores with spherical morphology (Fig. 2 C–E) whereas polyhedral hydrocarbon phase resulted in pores with irregular morphology (Fig. 2A). PEO, polyethylene oxide; Rh-β, rhodamine-β base; CC, calcium carbonate; HA, hydroxyapatite; CG, colloidal graphite; Sp, spherical; Irr, irregular.
*Molecular weight: PLLA, 100,000; PLLGA, 108,000; PEO, 100,000; PMMA, 100,000; PEG, 20,000.
†Density (ρ) (g/ml): PLLA, 1.24; PLGA, 1.27; PEO, 1.13; PMMA, 1.18, PEG, 1.13; CC, 2.71; HA, 2.5; CG, 2.09.
‡Particle size (μm): CC, < 53; unfired HA, 45–70; sintered HA, 212–500; CG, <0.5.
§Porosity was measured as follows: (1 − ρrel) × 100; where, ρrel = (foam density)/(polymer or material density).
¶The standard deviation from the mean was ≤±3.
Thermogravimetric Analyses.
Thermogravimetric analyses were carried out on 5–10 mg of sample under dynamic nitrogen atmosphere by using a Perkin–Elmer Series 7 thermogravimetric analyzer interfaced to a PC. The curves were analyzed by using Perkin–Elmer thermogravimetric analyses software.
Surface Area and Porosity Determination.
Surface area measurements of the foams were obtained by the Brunauer-Emmett-Teller method using Argon and Krypton adsorption (15) and were performed by Porous Materials (Ithaca, NY). Porosity of the foam was characterized by using apparent density, helium pycnometry, and mercury intrusion (1).
Scanning Electron Microscopy.
For scanning electron microscopy, samples were coated with 100 Å thick layer of gold by using a Denton II (Denton Vacuum, Moorestown, NJ) vacuum sputter coater to minimize charging of the sample and then mounted onto aluminum stubs using conductive carbon tape and conductive paint to ensure efficient charge dissipation. Scanning electron microscopy images were obtained using a Hitachi (Tokyo) Scanning Electron Microscope set at 4 keV accelerating voltage at 1 × 10−9 torr vacuum.
Compressive Modulus Measurements.
The stress–strain curves of the polymeric foams were obtained by testing circular specimens (5–6 mm in diameter × 2–3 mm thick) with a dynamic mechanical analyzer (Perkin–Elmer, DMA-7), in a parallel plate configuration, at a creep rate of 2.9 kPa/min. Compressive modulus of the sample was determined as the slope of the best linear fit of the stress–strain curves in the range of 70–140 kPa stress, which for all samples was within the linear region corresponding to the densification process (1).
Resistivity Measurements.
PMMA foams incorporated with colloidal graphite were cut into strips 1 cm in length (l) × 2 mm in width (w) × 2 mm in thickness (t). The resistance (R) of the foam across its length was determined by using a Micronta (Radio Shack) multimeter. The resistivity (s) was then calculated by using the following equation: s(Ω − cm) = R ⋅ A/l, where A is the cross-sectional area (t ⋅ w), and l is the length of the foam.
Incorporation and Release of Alkaline Phosphatase (ALP).
ALP was dissolved in a solution of polyethylene glycol (PEG) (MW = 20 kDa) in phosphate buffer and subsequently was lyophilized to yield a powder of PEG containing the enzyme. A foam of PLLA containing 20% by weight of the PEG powder was then prepared. The release of ALP from the PLLA-PEG foam was measured by incubating samples of the foam in 1.0 ml of PBS at 4°C for over 2 months. At timed intervals, the PBS was replaced and the activity of the released ALP was assessed by using a standard assay (Sigma ALP assay kit and procedure No. 245).
Cell Isolation and Culture.
Chondrocytes were first isolated from the femoropatellar grooves of 2- to 3-week-old bovine calves by using type II collagenase as described (20) and were seeded on the foam in magnetically stirred spinner flasks (21). The construct shaped like a human nose was cultured in the same flasks, whereas the other cell–polymer constructs were subsequently transferred into agarose-coated Petri dishes and were placed on an orbital shaker (75 rpm) for further cultivation. All constructs were cultured for up to 4 weeks in DMEM supplemented with 10% fetal bovine serum and ascorbic acid.
Results
Polymer Foam Structure and Properties.
Polymer foams were produced by using hydrocarbon particulate phases of both spherical and polyhedral morphologies (Table 1). Scanning electron microscopy of the foams revealed the presence of a bicontinuous network of the polymer and void with two distinct pore architectures (Fig. 2). The geometry and size of larger pores (macropores) in the foam were nearly identical to those of the particulate hydrocarbon phase (Fig. 2; Table 1). Spherical hydrocarbon particles resulted in pores with spherical morphology (Fig. 2 C–E) whereas polyhedral hydrocarbon particles resulted in pores with irregular morphology (Fig. 2A). The size of the pores correlated with the size distribution of the hydrocarbon particles, with a variance of <10%. This observation confirmed our premise that the hydrocarbon particulate phase would serve as template for the formation of pores and the precipitation of the polymer phase. The smaller pores [micropores, 0.5–40 μm in size (Fig. 2B)], residing in the polymer phase, were mostly spherical and could be derived from (i) the nucleation and growth of solvent bubbles before evaporation (1) and/or (ii) phase separation. Foams of polymers ranging from amorphous/semicrystalline such as PMMA, PLLA, and PLGA to water-soluble polymer such as poly(ethylene oxide) have been prepared (Table 1). By using this process, foams with porosity in the range of 70–90% with density as low as 120 mg/cc have been obtained. Thermogravimetric analysis revealed that <0.2% of the total hydrocarbon porogen (which corresponds to <1% by weight of the foam) was typically left behind after three extractions.
Figure 2.

Scanning electron microscopic characterization of the foams. PLGA and PLLA foams were prepared by using particulate hydrocarbon porogen (fugitive phase) differing in morphology, mean diameter (size distribution), and relative ratio (wt/wt) with respect to the polymer phase. Fugitive phase in: A and B (PLGA) polyhedral, 300–500 μm, and 80 wt/wt%; C (PLLA) spherical, 700–800 mm, and 78 wt/wt/%; D (PLLA) spherical, ≈1 mm (1,000 μm), and 78 wt/wt%; and E (PLLA) spherical, ≈1 mm (1,000 μm), and 50 wt/wt %. Scanning electron micrographs showed that in general the foam structure was characterized by a continuous polymer phase with interconnected macropores (A and C–E). Analysis of the cross-sections of the polymer phase revealed the presence of a porous microarchitecture (B). Similar characteristics were observed in foams of different compositions. [Bars = 1 mm (A and C–E) and 10 μm (B).] Typical stress–strain curves of foams of different compositions are shown in F [1, PMMA (5.0); 2, PLGA (5.1); 3, PLLA (4.6); 4, PLLA-PEG-CaCO3 (3.0); 5, PLGA-PEG (1.3); 6, PLLA-PEG (0.4)]. The curves are typical of foams, wherein a densification process (linear region) follows an initial elastic deformation (toe region), due to buckling of the polymer phase (1). Foams made of PLA-PEG-CaCO3 displayed a brittle fracture at 5% strain, which is typical for materials containing minerals (1) whereas the densification process in foams made of PLLA-PEG was not uniformly linear. The numbers in parenthesis for each composition are the calculated compressive moduli (MPa) in the stress range of 70–140 kPa.
Modification of Foam Properties.
Incorporation of solids into the polymer phase is not easily achieved in the gas foaming of polymer melt because of the disruptive effect of such phases on the stability and growth of gas bubbles (1). One feature of this hydrocarbon templating process is that the rapid precipitation of the viscous polymer phase during the porogen extraction enables the incorporation of organic and inorganic fillers into the polymer phase of highly porous foams without compromising their structural integrity. Furthermore, by keeping constant the volume fraction of the porogen with respect to the foam volume (which is determined by the volume of the Teflon mold), one can tailor bulk properties such as electrical, magnetic, and mechanical properties of the foam independently of its macroporosity. As an example, we produced conductive foams of PMMA with overall porosities higher than 70% and resistivities from 600 to 7000 Ω-cm, by incorporating colloidal graphite (20–50% by polymer weight) into the PMMA phase (Table 1). In another example, we have modified mechanical properties of PLLA and PLGA foams by blending the primary polymer phase, i.e., PLLA or PLGA, with immiscible inorganic and organic phases. The blending of PLLA or PLGA with an immiscible, water-soluble polymer such as PEG (20% wt/wt) resulted in a foam with a lower compressive modulus than PLLA or PLGA alone (Fig. 2F, curves 2, 3, 5, and 6; Table 1). However, incorporation of an inorganic filler such as calcium carbonate to the PLLA-PEG (80–20) blend (Fig. 2F, curve 4) almost restored the modulus to that of 100% PLLA (Fig. 2F, curve 2), without diminution in the macroporosity. Therefore, the tradeoff between pore size and mechanical stability, which is a limitation in gas foaming process because of the capillary forces and interfacial tension during the removal of the gas phase (14), appears to be virtually absent in the present system.
Potential Biomedical Applications of Polymer Foams.
Porous polymeric structures are currently being explored as templates for the ex vivo engineering of tissue equivalents from mammalian cells (17). Control over the porosity, degradation, and mechanical properties of the templates is crucial for optimal tissue development. In particular, to obtain tissue equivalents with uniform extracellular matrix distribution, it is desirable to have scaffold erosion rates that match the rate of tissue ingrowth.
Using the hydrocarbon templating approach, one can alter the degradation of polymer foams by blending water-soluble immiscible phases into the parent polymer phase. Such blending of immiscible phases would be difficult using processes like melt foaming, because of changes in viscosity and interfacial dynamics, which would alter the evolution of gas bubbles (1, 2). Scaffolds of PLLA blended with PEG (up to 40% wt/wt), of ≈85% porosity, were seeded with bovine chondrocytes. It was observed that the amount of nonresorbed polymer in the constructs after 4 weeks in culture was significantly lower with increasing concentrations of PEG (22.7 ± 3.1, 12.0 ± 1.3, and 4.7 ± 0.9% of the total cross-sectional area for 0, 20, and 40% PEG wt/wt, respectively). Correspondingly, blending of PLLA with PEG allowed for the accumulation of an increasingly more uniform extracellular matrix, rich in cartilage-specific glycosaminoglycans, as assessed by Safranin-O stain (Fig. 3 A–C). It is plausible that degradation of the matrices occurred as a result of both dissolution (PEG phase) and hydrolysis (PLLA phase), thus presenting a potential approach to altering bioerosion.
Figure 3.
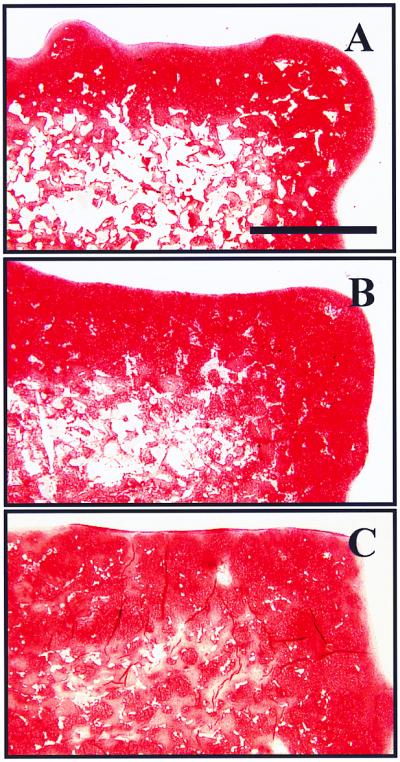
Representative histological cross-sections of engineered cartilage samples after 4 weeks in culture stained with Safranin O. Bovine chondrocytes were seeded onto foams of different compositions: PLA 100% (A), PLLA-PEG 80–20 (B), and PLLA-PEG 60–40 (C). (Bar = 1 mm.)
Current techniques for the production of porous polymeric structures for tissue engineering are generally limited to the production of membranes <4 mm in thickness (12). This limits their utility in the engineering of structurally complex tissues such as for musculoskeletal reconstruction. Because the process described herein allows for the production of large monolithic foams, porous polymeric structures of various geometries can be easily fabricated by a reductionist approach using well established computer-aided design/computer-aided machining (CAD/CAM) techniques. As an example, a PLLA foam in the likeness of a human nose was carved out of a 1-inch cube by using a surgical blade (Fig. 4A). The PLLA foam scaffold was then seeded with bovine chondrocytes and was cultured in vitro for 2 weeks in a bioreactor, to generate a structurally stable biological composite consisting of cartilaginous tissue mass and biodegradable polymer (Fig. 4B).
Figure 4.

Polymeric foams of complex shapes for guided tissue regeneration. Shown is a porous scaffold of PLLA in the likeness of a human nose (A) and a cartilaginous tissue-polymer composite produced by seeding the scaffold with bovine chondrocytes (B).
Porous polymeric templates that can deliver bioactive molecules such as growth factors have the potential to induce specific biological responses while potentially guiding tissue regeneration. A preliminary study has shown that proteins such as ALP can be incorporated into the polymer phase with minimal loss of activity and subsequently can be released for extended lengths of time (Fig. 5).
Figure 5.

Release of active ALP as a function of time.
Discussion
We have shown that a hydrocarbon particulate phase can serve both as a template and pore-forming agent in the foaming of polymers. The process described in this study offers several potential advantages over existing approaches: namely, mild and rapid processing conditions, improved control over pore structure and porosity, and versatility in the composition of the polymer phase.
Unlike conventional leaching of a water-soluble fugitive phase, in the present process, the porogen is actively extracted in an organic solvent, which can quickly penetrate the polymer–porogen mixture and promote the rapid precipitation of the polymer phase. As a result, limitations on foam thickness are virtually absent. In contrast, sintering of polymer particles and salt extraction are generally limited to the production of membranes (<4 mm), as larger structures result in anisotropy (12). Large monolithic pieces of foam as thick as 1 inch, with highly regular and interconnected pore structure, possessing relative densities (ρrel) in the range of 0.1–0.30, densities as low as 120 mg/cc [which is similar to aerogels (13)], and surface areas as high as 20 m2/g were produced by using this process (Table 1). Because porogen extraction can occur by penetration of the solvent through the micropores, foams with closed cell morphology were also obtained by decreasing the porogen/polymer ratio (Fig. 2E). In contrast, leaching of a water-soluble fugitive phase such as salt can only yield foams with a continuous open cell morphology (10).
Foams of PMMA, PLLA, and PLGA produced by this process had compressive moduli up to 5 MPa at 5% strain (Fig. 2F, curves 1–3). The good mechanical properties of the foams may be attributed to the (i) presence of a continuous polymer phase, (ii) high degree of polymer chain entanglement that can occur during the precipitation process, and (iii) increased crystallinity of the polymer phase (14) after processing, as assessed by differential scanning calorimetric analysis (data not shown). Because of the enhanced control over foam properties, this process maybe well suited for the production of low-density microcellular plastics as well (14).
The hydrocarbon templating process also allows for the incorporation of particulate phases into the primary polymer phase of the foam. As a consequence, bulk characteristics of the foam such as electromagnetic properties can be altered easily. Porous conductive foams with high surface area could have utility in the fabrication of lightweight components for batteries and fuel cells (15), or to dissipate static electricity (1). One could also envisage the production of porous structures for catalyst beds and enzyme-immobilized bioreactors, by using polymers containing derivatizable functionalities (1). Similarly, blending of different polymers (including water-soluble ones) and incorporation of inorganic fillers can provide a simple and versatile tool to control the mechanical properties and degradation characteristics of foams. Control over these properties is of particular significance when they are used as degradable templates for tissue regeneration (16–18). With further study, polymer foams produced by this approach can be potentially coupled with imaging and computer-aided design/computer-aided machining techniques to produce scaffolds with complex topographies for tissue engineering.
Acknowledgments
The authors thank Ruben Dario Sinisterra for his help with the thermogravimetric analyses of the foams and Robert Padera for the histology. This work was partly supported by a grant from the National Institutes of Health (Grant 1RO1-HL60435-01).
Abbreviations
- PLLA
poly(l-lactic acid)
- PLGA
poly(l-lactic-co-glycolic acid)
- PMMA
poly(methyl methacrylate)
- ALP
alkaline phosphatase
- PEG
polyethylene glycol
References
- 1.Gibson L, Ashby M. Cellular Solids: Structure and Properties. Oxford: Pergamon; 1997. [Google Scholar]
- 2.Suh K, Webb D. In: Encyclopedia of Polymer Science and Engineering. Kroschwitz J, editor. Vol. 3. New York: Wiley; 1985. pp. 1–59. [Google Scholar]
- 3.Yoda R. J Biomater Sci Polym Ed. 1998;9:561–626. doi: 10.1163/156856298x00046. [DOI] [PubMed] [Google Scholar]
- 4.Szycher M, Lee S. J Biomater Appl. 1992;7:142–213. doi: 10.1177/088532829200700204. [DOI] [PubMed] [Google Scholar]
- 5.Pruitt B, Levine N. Arch Surg. 1984;119:312–322. doi: 10.1001/archsurg.1984.01390150050013. [DOI] [PubMed] [Google Scholar]
- 6.Alsbjorn B. Scand J Plast Reconstr Surg. 1984;18:127–133. doi: 10.3109/02844318409057414. [DOI] [PubMed] [Google Scholar]
- 7.Bruck S. Med Prog Technol. 1982;9:1–16. [PubMed] [Google Scholar]
- 8.Guidoin R, Couture J, Assayed F, Gosselin C. Int Surg. 1988;73:241–249. [PubMed] [Google Scholar]
- 9.Lindenauer S, Weber T R, Miller T A, Ramsburgh S R, Salles C A, Kahn S P, Wojtalik R S. Biomed Eng. 1976;11:301–306. [PubMed] [Google Scholar]
- 10.Saunders J, Hansen R. In: Plastic Foams. Frisch K, Saunders J, editors. New York: Dekker; 1972. , Part I, 23–108. [Google Scholar]
- 11.Even W, Gregory D. Mater Res Soc Bull. 1994;19(6):29–33. [Google Scholar]
- 12.Lu L, Mikos A. Mater Res Soc Bull. 1996;21(11):28–32. doi: 10.1557/s088376940003181x. [DOI] [PubMed] [Google Scholar]
- 13.Heinrich T, Klett U, Fricke J. J Porous Mater. 1995;1:7–17. [Google Scholar]
- 14.LeMay J, Hopper R, Hrubesh L, Pekala R. Mater Res Soc Bull. 1990;15(12):19–45. [Google Scholar]
- 15.Fischer U, Saliger R, Bock V, Petricevic R, Fricke J. J Porous Mater. 1997;4:281–285. [Google Scholar]
- 16.James K, Kohn J. Mater Res Soc Bull. 1996;21(11):22–26. [Google Scholar]
- 17.Langer R, Vacanti J P. Science. 1993;260:920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 18.Martin I, Shastri V P, Langer R, Vunjak-Norakovic G, Freed L E. Trans Orthop Res Soc. 1999;24:57. [Google Scholar]
- 19.Bock V, Emmerling A, Saliger R, Fricke J. J Porous Mat. 1997;4:287–294. [Google Scholar]
- 20.Martin I, Vunjak-Novakovic G, Yang J, Langer R, Freed L E. Exp Cell Res. 1999;253:681–688. doi: 10.1006/excr.1999.4708. [DOI] [PubMed] [Google Scholar]
- 21.Vunjak-Novakovic, Obradovic B, Martin I, Bursac P M, Langer R, Freed L E. Biotechnol Prog. 1998;14:193–202. doi: 10.1021/bp970120j. [DOI] [PubMed] [Google Scholar]