

Abstract
This study investigates the influence of intestinal inflammation on: (1) the control of intestinal neurotransmission and motility by prejunctional α2-adrenoceptors and (2) the expression of intestinal α2-adrenoceptors. Experimental colitis was induced by intrarectal administration of 2,4-dinitrobenzenesulphonic acid (DNBS) to rats.
UK-14,304 inhibited atropine-sensitive electrically evoked contractions of ileal and colonic longitudinal muscle preparations. UK-14,304 acted with similar potency, but higher efficacy, on tissues from DNBS-treated animals; its effects were antagonized with greater potency by phentolamine than rauwolscine.
Electrically induced [3H]noradrenaline release from ileal preparations was reduced in the presence of colitis. Tritium outflow was decreased by UK-14,304 and stimulated by rauwolscine or phentolamine: these effects were enhanced in preparations from animals with colitis.
Reverse transcription–polymerase chain reaction and Western blot assay demonstrated the protein expression of α2A-adrenoceptors in mucosal and muscular tissues isolated from ileum and colon. The induction of colitis increased α2A-adrenoceptor expression in both ileal and colonic muscular layers, without concomitant changes in mucosal tissues.
Induction of colitis reduced gastrointestinal propulsion of a charcoal suspension in vivo. In this setting, the gastrointestinal transit was inhibited by intraperitoneal (i.p.) UK-14,304 and stimulated by i.p. rauwolscine. After pretreatment with guanethidine, the stimulant action of rauwolscine no longer occurred, and UK-14,304 exerted a more prominent inhibitory effect that was antagonized by rauwolscine.
The present results indicate that, in the presence of intestinal inflammation, prejunctional α2-adrenoceptors contribute to an enhanced inhibitory control of cholinergic and noradrenergic transmission both at inflamed and noninflamed distant sites. Evidence was obtained that such modulatory actions depend on an increased expression of α2A-adrenoceptors within the enteric nervous system.
Keywords: α2-Adrenoceptors, acetylcholine, noradrenaline, enteric nervous system, intestinal motility, intestinal inflammation
Introduction
The gastrointestinal tract is extensively innervated by extrinsic noradrenergic neurons, whose projections end in close proximity to several cellular targets (i.e. epithelial cells, neurons, blood vessels). Locally, through their interaction with α2-adrenoceptors, endogenous catecholamines modulate a variety of digestive functions, including secretions and motility (Del Tacca et al., 1982; De Ponti et al., 1996; Liu & Coupar, 1997), gastro-colonic sensation (Malcom et al., 2000), and epithelial cell proliferation (Schaak et al., 2000). Both sympathetic and cholinergic axon terminals of the enteric nervous system are equipped with prejunctional α2-adrenoceptors, the activation of which inhibits noradrenaline and acetylcholine release, respectively (Blandizzi et al., 1993; Funk et al., 1995; Colucci et al., 1998).
Inflammatory diseases of the human gut (e.g. inflammatory bowel disease, coeliac disease, infectious gastroenteritis) are commonly associated with alterations of enteric functions, including increased secretion and motor abnormalities, which are thought to contribute to the generation of digestive symptoms (Collins, 1996, Giaroni et al., 1999; Sharkey & Kroese, 2001). For this reason, there is currently great interest in understanding the pathophysiological changes occurring in the enteric nervous system as a consequence of intestinal inflammation (Sharkey & Kroese, 2001). Such alterations occur throughout the gut, at both inflamed and noninflamed sites, and they depend, at least in part, on mutual interactions between enteric nerves and the digestive immune system (Collins, 1996).
The role played by adrenoceptors in intestinal inflammation remains poorly understood. Experimental colitis in rats was associated with downregulation of β3-adrenoceptors in colonic circular smooth muscle (Zhao et al., 2001). An increased density of α2-adrenergic binding sites was detected in cell membrane preparations obtained from jejunal tissues of guinea-pigs with small bowel inflammation (Martinolle et al., 1993). However, data on the possible influence exerted by prejunctional α2-adrenoceptors on enteric neurotransmission and digestive motility in the presence of intestinal inflammation are lacking. Accordingly, the present study was designed to assess the influence of experimentally induced intestinal inflammation on: (1) the control of enteric neurotransmission and intestinal motility by prejunctional α2-adrenoceptors and (2) the expression pattern of intestinal α2-adrenoceptors.
Methods
Animals
Albino male Sprague–Dawley rats, 200–250 g body weight, were used throughout the study. The animals were fed standard laboratory chow and tap water ad libitum and were not used for at least 1 week after their delivery to the laboratory. They were housed (five in each cage) in temperature-controlled rooms on a 12-h light–dark cycle at 22–24°C and 50–60% humidity. Their care and handling were in accordance with the provisions of the European Union Council Directive 86-609, recognized and adopted by the Italian Government.
Induction of colitis
Colitis was induced in rats in accordance with the method previously described by Barbara et al. (2000). Briefly, during a short anaesthesia with diethyl ether, 30 mg of 2,4-dinitrobenzenesulphonic acid (DNBS) in 0.25 ml of 50% ethanol was administered intrarectally via a polyethylene PE-60 catheter inserted 8 cm proximal to the anus. Control rats received 0.25 ml of 50% ethanol. Animals underwent subsequent experimental procedures 6 days after DNBS administration, in order to allow a full development of histologically evident colonic inflammation.
Assessment of colitis
At 6 days after treatment with DNBS or its vehicle, animals were euthanized and the severity of intestinal inflammation was evaluated macroscopically and histologically in accordance with the criteria previously reported by Wallace & Keenan (1990), as modified by Barbara et al. (2000). Briefly, the macroscopic criteria were based on the following: the presence of adhesions between the colon and other intraabdominal organs; consistency of colonic faecal material (as an indirect marker of diarrhoea); thickening of the colonic wall; the presence and extension of hyperaemia and macroscopic mucosal damage (assessed with the aid of a ruler). Microscopic criteria for damage and inflammation were assessed by light microscopy on haematoxylin- and eosin-stained histological sections obtained from whole-gut specimens, taken from a region of the inflamed colon immediately adjacent to the gross macroscopic damage and from terminal ileum. Histological criteria included: degree of mucosal architecture changes; cellular infiltration; external muscle thickening; presence of crypt abscess and goblet cell depletion. All the parameters of macroscopic and histological damage were recorded and scored for each rat by two observers blinded to the treatment.
Recording of contractile activity from longitudinal muscle preparations of ileum
Longitudinal muscle strips of proximal ileum containing the Auerbach plexus were prepared as previously reported by Collins et al. (1989). In order to record the motor activity of longitudinal muscle, ileal preparations weighing 60–90 mg were set up in organ baths of 10 ml capacity (overflow system), containing oxygenated Krebs solution at 37°C, and connected vertically to isotonic transducers (Basile, Comerio, Italy) under a constant tension of 1 g. The Krebs solution had the following composition (in mM): NaCl 113, KCl 4.7, CaCl2 2.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25, glucose 11.5 (pH 7.4±0.1). Each preparation was allowed to equilibrate for at least 30 min, with intervening washings at intervals of 10 min. The contractile activity of preparations was recorded by the polygraph Gemini 7080 (Basile, Comerio, Italy). A pair of coaxial platinum electrodes was positioned at distance of 10 mm from the longitudinal axis of each preparation in order to deliver transmural electrical stimulation. Electrical stimuli were applied as 5-s trains of square wave pulses (0.5 ms duration, 5 Hz frequency, 30 mA intensity), applied every 60 s. In preliminary experiments, this pattern of electrical stimulation elicited motor responses consisting of relaxations followed by contractions, which were partly insensitive to atropine. Therefore, all subsequent experiments were performed after incubation of ileal preparations for 30 min with guanethidine (10 μM), N ω-nitro-L-arginine methylester (L-NAME, 100 μM) and capsaicin (10 μM), to suppress the electrically induced motor activity of longitudinal smooth muscle because of the release of noradrenaline, nitric oxide, and sensory peptides (Maggi & Giuliani, 1995). Q1Indeed, once released from intestinal afferent nerves, sensory peptides can induce noncholinergic contractions through the activation of intrinsic myenteric neurons (Barthò et al., 1999). The α2-adrenoceptor agonist UK-14,304 was added cumulatively to the bathing fluid in 0.5-log unit increments. A period of 3–5 min was allowed between subsequent increments of concentration in order to enable a full development of the effect of the agonist. In preliminary experiments, aiming to assay the sensitivity of intestinal preparations to UK-14,304, the agonist was also added to the bathing fluid in a noncumulative manner. In agonist–antagonist interaction experiments, rauwolscine (α2-adrenoceptor antagonist) or phentolamine (α1/α2-adrenoceptor antagonist) was added to the bath 20 min before the agonist. Drugs were given in volumes ⩽1% of total bath volume (10 ml). The effects of test drugs were expressed as percent changes of control cholinergic contractions elicited by electrical stimulation. The potency of UK-14,304 was expressed as EC50 (concentration of the agonist that produces 50% of the maximal response for that agonist). The percent maximum inhibition of the control cholinergic motor activity (Emax) was also evaluated. Both parameters were calculated from single concentration–response curves and then averaged. The potencies of rauwolscine and phentolamine were expressed as pKd values from the equation:
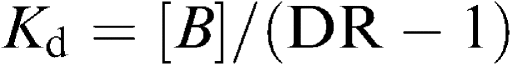 |
where B is the molar concentration of the antagonist and DR is the ratio of equally effective concentrations of the agonist (EC50) in the presence and absence of the antagonist (Furchgott, 1972).
Recording of contractile activity from longitudinal muscle preparations of colon
Specimens of distal colon were placed into cold preoxygenated Krebs solution, opened along the mesenteric insertion, and cut along the longitudinal axis into strips of approximately 3 mm width and 30 mm length. The colonic preparations were then used in order to assess the effects of test drugs on electrically induced cholinergic motor activity of longitudinal muscle. The experiments were performed as reported above for ileal preparations. Electrical stimulation was delivered to colonic strips as 5-s trains of square wave pulses (0.5 ms, 5 Hz, 30 mA), applied every 60 s. As also observed for ileal preparations, in preliminary experiments colonic strips developed electrically induced motor responses that could not be completely prevented by atropine. For this reason, all subsequent experiments were carried out in the presence of guanethidine (10 μM), L-NAME (100 μM), and capsaicin (10 μM). The effects of UK-14,304, rauwolscine and phentolamine on cholinergic contractile activity of colonic longitudinal smooth muscle were assayed as described above for the ileum.
Measurement of [3H]noradrenaline release from longitudinal muscle preparations of ileum
Longitudinal muscle strips of ileum, containing the Auerbach plexus, were prepared as reported above. The ileal preparations were then used to assess the effects of UK-14,304, rauwolscine, and phentolamine on electrically induced [3H]noradrenaline release, in accordance with the procedure previously described (Blandizzi et al., 2000), with minor modifications. Ileal strips, weighing 60–90 mg, were incubated for 30 min in Krebs solution at 37°C, oxygenated with 95% O2+5% CO2 (preincubation period), and then loaded with 1-7,8-[3H]noradrenaline (3.5 μCi ml−1) for 60 min in 2 ml of Krebs solution. Ascorbic acid 0.03 mm and disodium ethylenediamine tetraacetic acid (EDTA) 0.1 mM were included in the medium in order to prevent the oxidative breakdown of noradrenaline. At the end of loading period, the ileal strips were repeatedly washed with Krebs solution, transferred into organ baths (5-ml capacity, filled with Krebs solution), and superfused at a flow rate of 1 ml min−1 with Krebs solution bubbled with 95% O2–5% CO2 at 37°C. The superfusing Krebs solution contained cocaine 5 μM and hydrocortisone 1 μM to inhibit the reuptake of noradrenaline. The first 60-min collection of effluent was discarded (preperfusion), after which 3-min fractions were collected for 90 min. During the superfusion period, the ileal preparations underwent electrical field stimulation (BM-ST6 stimulator, Biomedica Mangoni, Pisa, Italy), delivered as square wave pulses (20 V cm−1) of 0.5 ms duration at 8 Hz (480 pulses), during the third (S1) and the 20th (S2) collection period. At the end of superfusion, the radioactivity contained in each fraction was measured by liquid scintillation counting (Betamatic, Kontron Instruments, Milan, Italy). The radioactive content of ileal strips was also measured. For this purpose, each preparation was weighed and then incubated for 30 min in 1 ml of 10% trichloroacetic acid at room temperature. An aliquot of supernatant (50 μl) was added to 5 ml of scintillator and the tritium content of the tissue was measured by liquid scintillation spectrometry. The test drugs were added to the superfusion solution in the 12th collection period, namely between S1 and S2, and exposure to each drug continued until the end of experiment. Tritium efflux into the superfusate was calculated as the fraction of tritium present in the ileal strip at the onset of the respective collection period (fractional rate; min−1). The increase in tritium outflow, evoked by electrical stimulation, was calculated as the percentage of tritium content of the tissue at the onset of electrical stimulation. The effects of test drugs on the evoked tritium outflow were expressed as ratio of the percentage release during S2 over that obtained during S1 (S2/S1), namely in the presence and absence of drug, respectively (Blandizzi et al., 2000).
Reverse transcription–polymerase chain reaction analysis of α2A-adrenoceptors
Experiments were performed on specimens of both ileum and colon, excised as reported above. In both cases, care was taken to carefully dissect the mucosa from muscular layers. Tissue specimens were then used to assess the expression of the gene encoding for α2A-adrenoceptors by reverse transcription (RT) of mRNA followed by polymerase chain reaction (PCR). For this purpose, tissue samples were immediately frozen in liquid nitrogen and stored at −80°C until use. At the time of the extraction, tissues were disrupted in mortars kept cold by filling with liquid nitrogen. Total RNA was isolated by means of Trizol® (Life Technologies, Carlsbad, CA, U.S.A.) and chloroform. Total RNA (1 μg) served as template for single-strand cDNA synthesis in a reaction using 2 μl random hexamer oligonucleotide primers (0.5 μg/μl) with 200 U of MMLV-reverse transcriptase in manufacturer's buffer containing 500 μM deoxynucleotide triphosphate mixture (dNTP) and 10 mM dithiothreitol. cDNA samples were subjected to PCR in the presence of specific oligonucleotide primers based on the nucleotide sequence of the previously cloned α2A-adrenoceptor rat gene (sense: 5′-CAGGTTCGTGCTGGCG GTGGTGATC-3′; antisense: 5′-GGTCTGTAAGCAGCACAGCCCGAG-3′; expected size 264 base pairs) (Wilborn et al., 1998). PCR, consisting of 25 μl of RT products, Taq polymerase 2.5 U, dNTP 100 μM and oligonucleotide primers 0.5 μM, was carried out by a PCR-Express thermocycler (Hybaid, Ashford, Middlesex, U.K.) at the following conditions: 35 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 2 min, and extension at 72°C for 1 min, followed by a final extension at 72°C for 10 min. Aliquots of RNA not subjected to RT were included in PCR reactions to verify the absence of genomic DNA. The efficiency of RNA extraction, RT and PCR was evaluated by specific sets of oligonucleotide primers for the constitutively expressed rat β-actin gene (sense: 5′-TCATGAAGTGTGACGTTGACATCCGT-3′; antisense: 5′-CTTAGAAGCATTTGCGGTGCACGATG-3′; expected size 286 base pairs). The amplified cDNA products were separated by 1.5% agarose gel electrophoresis in a Tris buffer 40 mM containing 2 mM EDTA, 20 mM acetic acid (pH 8), and stained with ethidium bromide. cDNA bands were then visualized by UV light and quantitated by densitometric analysis with NIH Image computer program (Scion Corporation, Frederick, MD, U.S.A.). The relative expression of α2A-adrenoceptor mRNA was normalized to that of β-actin.
Western blot analysis of α2A-adrenoceptors
Experiments were carried out on specimens of both ileum and colon, excised as reported above. In both cases, care was taken to carefully dissect the mucosa from muscular layers. Mucosal and muscular samples were then homogenized in lysis buffer containing: N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulphonic acid) 10 mM, NaCl 30 mM, EDTA 0.2 mM, phenylmethylsulphonyl fluoride 2 mM, leupeptin 10 μg ml−1, aprotinin 10 μg ml−1, sodium fluoride 1 mM, sodium orthovanadate 1 mM, glycerol 2%, MgCl2 0.3 mM, Triton X-100 1%, using the homogenizer Powergen 35 (Fisherbrand, Middleton, Manchester, U.K.). Homogenates were centrifuged at 20,000 × g for 15 min at 4°C. The supernatants were separated from pellets and stored at −80°C. Protein concentration was determined by the Bradford method (Bio-Rad protein assay reagent, Hercules, CA, U.S.A.).
Equivalent amounts of protein lysates (50 μg) were separated by electrophoresis on a sodium dodecylsulphate–polyacrylamide gel (12%) and transferred onto a nitrocellulose membrane. The blots were blocked overnight with 5% nonfat driedmilk in phosphate-buffered saline and incubated for 1 h at room temperature with a polyclonal rabbit antiserum raised against a synthetic peptide corresponding to amino-acid residues 218–235 of human, mouse, rat, and pig α2A-adrenoceptor (dilution 1 : 500, PA1-048; Affinity Bioreagents, Golden, CO, U.S.A.). After repeated washings with 0.1% Tween-20 in Tris-buffered saline (TBS-T), a peroxide-conjugated goat anti-rabbit antibody (dilution 1 : 3000, SantaCruz Biotechnology, Santa Cruz, CA, U.S.A.) was added for 1 h at room temperature. After repeated washings with TBS-T, the immunoreactive bands were visualized by enhanced chemiluminescence (ECL, Amersham Biosciences Europe, Cologno Monzese, Italy). The relative expression of α2A-adrenoceptors was quantified by densitometric analysis with NIH Image computer program (Scion Corporation, Frederick, MD, U.S.A.).
In vivo evaluation of gastrointestinal transit
The quantitative evaluation of gastrointestinal transit was carried out as previously reported by Singh et al. (1996), with minor changes. For this purpose, both DNBS- and vehicle-treated rats were fasted 36 h before the beginning of experiments. Free access to water ad libitum was allowed until 2 h before the beginning of experiments. At that time, 2 ml of a charcoal suspension (10% charcoal in 12.5% arabic gum) was injected into the gastric lumen by a polyethylene orogastric catheter. After 25 min, animals were killed by cervical dislocation and the small intestine was quickly removed avoiding stretching. The gastrointestinal transit was then evaluated by comparing the distance travelled by the charcoal meal from the pyloric sphincter with the total length of the small intestine from the pyloric sphincter to the ileo-caecal junction. In experiments aiming to assay the effects of test drugs on gastrointestinal transit, rauwolscine and UK-14,304 were administered by intraperitoneal (i.p.) route 30 and 20 min before the intragastric injection of charcoal meal, respectively. Control rats received drug vehicles by i.p. route at the same times. In additional experiments, the evaluation of gastrointestinal transit was performed on rats subjected to chemical ablation of sympathetic nervous pathways, according to the procedure previously reported by Ying et al. (1998). For this purpose, animals were treated with guanethidine 70 μmol kg−1 i.p. once daily for two consecutive days. The last dose was administered 3 h before the administration of charcoal meal for the assay of gastrointestinal transit.
Drugs
The following drugs and reagents were used: diethyl ether, guanethidine, L-NAME, capsaicin, tetrodotoxin, atropine sulphate, phentolamine hydrochloride, ascorbic acid, EDTA, cocaine hydrochloride, disodium hydrocortisone 21-phosphate, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulphonic acid), phenylmethylsulphonyl fluoride, leupeptin, aprotinin, sodium orthovanadate, glycerol, Triton X-100, sodium dodecylsulphate, polyacrylamide, Tween-20 (Sigma Chemicals Co., St Louis, MO, U.S.A.); hexamethonium 2-chloride, rauwolscine hydrochloride (RBI, Natik, MA, U.S.A.); UK-14,304 [S-bromo-N(4,5- dihydro-1H-imidazol-2-yl)-6-quinoxalinamine] (Tocris, Balwin, MO, U.S.A.); DNBS dihydrate (ICN Biomedicals, OH, U.S.A.); 1-7,8-[3H]-noradrenaline (specific activity 36 Ci mmol−1; Amersham Pharmacia Biotech, Milan, Italy). Random hexamers, dithiothreitol, MMLV-reverse transcriptase, Taq polymerase, dNTP mixture, ethidium bromide (Promega, Madison, WI, U.S.A.). Other reagents were of analytical grade. Unless otherwise stated, all drugs were dissolved in distilled water or in 0.9% sterile saline for in vivo treatments. UK-14,304 was dissolved in dimethylsulphoxide and further dilutions were made with distilled water (in vitro assays) or 0.9% sterile saline (in vivo administrations). Drugs administered i.p. were injected in a volume of 0.5 ml per rat.
Statistical analysis
Results are given as mean±s.e.m. The significance of differences was evaluated by: Student's t-test for unpaired data (assessment of intestinal inflammation); one-way analysis of variance (ANOVA) for unpaired data (RT–PCR and Western blot assays); two-way ANOVA for unpaired data (noradrenaline release); two-way ANOVA for repeated measures (in vitro contractile activity); three-way ANOVA for unpaired data (in vivo gastrointestinal transit). Post hoc analysis was performed by Tukey or Dunnett test, as appropriate. P-values lower than 0.05 were considered significant; ‘n' indicates the number of experiments. In the case of in vitro experiments, the ileal and colonic preparations included in each test group were obtained from distinct animals, and therefore, in the present study, ‘n' refers also to the number of animals used per experimental group. EC50 values were interpolated from concentration–response curves. All statistical procedures, curve fitting, and calculations of 95% confidence intervals (95% CI) were performed by means of commercial software (GraphPad Prism™, software package version 2.01 for Windows 95, from GraphPad Software Inc., San Diego, CA, U.S.A.; SPSS™, software package version 11, from SPSS Inc., Chicago, IL, U.S.A.).
Results
Assessment of intestinal inflammation
At 6 days after DNBS administration, the distal colon was thickened and ulcerated with evident areas of transmural inflammation. Adhesions were often present and the bowel was occasionally dilated. In contrast, no changes were observed in the terminal ileum. The colitis was characterized by an intense granulocyte infiltrate extending throughout the mucosa and submucosa, and often involving the muscolaris propria that appeared thickened. There was a greater than five-fold increase in both macroscopic and microscopic damage scores over ethanol-treated control animals. By contrast, all ileal specimens obtained from animals with colitis did not show any relevant histopathological abnormality. Mean score values of both macroscopic and microscopic damage estimated for ileal and colonic samples are summarized in Table 1 .
Table 1.
Macroscopic and microscopic damage scores estimated for ileal and colonic specimens 6 days after induction of colitis with DNBS
| Ileum | Colon | |||
|---|---|---|---|---|
| Vehicle | DNBS | Vehicle | DNBS | |
| Macroscopic damage | 0.45±0.09 | 0.52±0.15 | 1.85±0.32 | 10.92±1.71* |
| Microscopic damage | 0.22±0.05 | 0.27±0.06 | 1.27±0.23 | 8.16±1.44* |
Each value represents the mean of eight to ten experiments ± s.e.m.
Significant difference from the respective values obtained from vehicle-treated rats: P<0.05.
Contractile activity of longitudinal muscle preparations of ileum
During the resting period, ileal segments developed a pattern of spontaneous contractile activity that did not interfere with electrically induced motor responses. Under the experimental conditions adopted in this study, the application of electrical stimuli to ileal strips, obtained from vehicle-treated or inflamed animals, evoked recurrent phasic contractions that were abolished by tetrodotoxin (1 μM), or atropine (0.1 μM), but were unaffected by hexamethonium (10 μM), indicating an involvement of postganglionic cholinergic neurons. Figure 1 displays typical trace recordings showing the inhibitory effects of atropine either in the absence (Figure 1a) or in the presence (Figure 1c) of colitis.
Figure 1.

Representative trace recordings showing the effects of atropine (ATR, 1 μM) or UK-14,304 (1 μM) on the contractile activity of longitudinal smooth muscle evoked by transmural electrical stimulation (0.5 ms, 5 Hz, 30 mA, 25 pulses every 60 s) of ileal (a–d) or colonic preparations (e–h), obtained from rats either in the absence (a,b,e,f) or in the presence (c,d,g,h) of DNBS-induced colitis.
UK-14,304 (0.001–10 μM) caused a concentration-dependent inhibition of electrically evoked cholinergic contractions of ileal strips prepared from vehicle-treated rats (Figure 1b and Figure 2a). In this case, the EC50 was 50.9 nM (95% CI=28.4–91.2), and a maximal inhibition of –37.8±5.2% occurred at the concentration of 3 μM (n=7). Rauwolscine (1 μM) or phentolamine (1 μM) did not significantly affect the electrically induced motor activity when tested alone, whereas they antagonized the inhibitory actions of UK-14,304 (Table 2 ). When assayed on ileal preparations obtained from animals with colitis, UK-14,304 was more effective in decreasing the evoked contractile activity of longitudinal muscle (EC50=46.8 nM, 95% CI=31.5–69.5; Emax=–82.8±4.7% at 3 μM; n=8; P<0.05) (Figure 1d and Figure 2a). Under these conditions, the inhibitory effects of UK-14,304 were antagonized by rauwolscine (1 μM) or phentolamine (1 μM) (Table 2).
Figure 2.

Effects of increasing concentrations of UK-14,304 on cholinergic contractile activity of longitudinal smooth muscle evoked by transmural electrical stimulation of ileal (a) or colonic (b) preparations obtained from rats either in the absence or in the presence of DNBS-induced colitis. Each point represents the mean of seven to eight experiments ±s.e.m. (vertical bars). Significant difference from the respective values obtained in vehicle-treated animals: *P<0.05; significant difference from the respective control values: aP<0.05.
Table 2.
Antagonism of rauwolscine or phentolamine against the inhibitory effects of UK-14,304 on cholinergic contractions
| pKd (ileum) | pKd (colon) | |
| Vehicle | ||
| Rauwolscine 1 μM | 7.61 (7.20–8.02) | 7.73 (7.44–8.02) |
| Phentolamine 1 μM | 8.94 (8.61–9.27) | 9.06 (8.64–9.48) |
| DNBS | ||
| Rauwolscine 1 μM | 7.55 (7.23–7.87) | 7.68 (6.87–8.05) |
| Phentolamine 1 μM | 9.13 (8.75–9.51) | 9.24 (8.89–9.59) |
Ileal and colonic preparations were obtained from rats either in the absence or in the presence of DNBS-induced colitis. The cholinergic contractile activity of longitudinal muscle was evoked by electrical transmural stimulation of intestinal tissues (0.5 ms, 5 Hz, 30 mA, 25 pulses every 60 s). Values reported for pKd, with 95% CI in brackets, were determined from the rauwolscine- or phentolamine-induced shift of the concentration-response curves (n=6 for each point) of UK-14,304 at the level of EC50.
Contractile activity of longitudinal muscle preparations of colon
During the equilibration period, the longitudinal smooth muscle of colonic preparations, obtained from either vehicle- or DNBS-treated rats, developed a spontaneous contractile activity, which remained stable throughout the experiment without interfering with the motor responses elicited by application of electrical stimulation. As already observed for ileal preparations, following incubation with guanethidine, L-NAME and capsaicin, the application of electrical stimuli elicited recurrent phasic contractions, which were sensitive to tetrodotoxin (1 μM) or atropine (0.1 μM), but insensitive to hexamethonium (10 μM). Figure 1 displays typical trace recordings showing the inhibitory effects of atropine either in the absence (Figure 1e) or in the presence (Figure 1g) of colitis.
In the absence of colitis, UK-14,304 (0.001–10 μM) decreased the cholinergic contractions evoked by electrical stimulation in a moderate, but concentration-dependent manner (Figure 1f and Figure 2b). An EC50 value of 54.2 nM (95% CI=43.4–67.5) was estimated for this agonist, with a maximal inhibition of –36.7±6.1% at the concentration of 3 μM (n=8). Rauwolscine (1 μM) or phentolamine (1 μM) alone did not modify the pattern of evoked motor responses. However, both these antagonists counteracted the inhibitory effects of UK-14,304 (Table 2).
In the presence of colitis, UK-14,304 caused a marked and concentration-dependent inhibition of electrically evoked cholinergic contractions (Figure 1h and Figure 2b). In this case, the EC50 and Emax values were 47.9 nM (95% CI=30.9–74.1) and –75.3±4.7% at 3 μM (n=8; P<0.05), respectively. As already observed for vehicle-treated animals, rauwolscine and phentolamine did not interfere with the electrically induced contractile activity, whereas both drugs antagonized the inhibitory actions of UK-14,304 (Table 2).
[3H]noradrenaline release from longitudinal muscle preparations of ileum
In control experiments, performed on ileal preparations from vehicle-treated rats (n=6), after a 60-min initial preperfusion, the spontaneous tritium overflow approached a rate of 0.0015±0.00012 min−1 and did not vary significantly throughout the experiment. When the superfused ileal strips were exposed to electrical stimulation, the tritium efflux increased significantly from 0.0014±0.00010 to 0.0034±0.00016 min−1 (P<0.05). The increase in tritium outflow was observed usually in four 3-min fractions; the release peaked during this period and then declined exponentially to the prestimulation value. The evoked tritium efflux was 1.72±0.08% for S1 and 1.65±0.06% for S2, not significantly different from each other; the S2/S1 calculated ratio was 0.96±0.04% (Figure 3a; Table 3 ).
Figure 3.

Tritium efflux from longitudinal muscle preparations of rat ileum preincubated with [3H]noradrenaline. (Abscissa): Time of superfusate collection. (Ordinate): Efflux of tritium per min, expressed as fraction of tissue tritium at the onset of the respective collection period. Electrical field stimulation during S1 and S2 consisted of 480 pulses (20 V cm−1, 0.5 ms) at 8 Hz. Ileal preparations were obtained from rats either in the absence (a) or in the presence (b) of colitis. Each point represents the mean of six experiments ± s.e.m. (vertical bars).
Table 3.
Effects of UK-14,304, rauwolscine or phentolamine on tritium overflow
| Evoked tritium overflow | ||
|---|---|---|
| S1 | S2/S1 | |
| Vehicle control | 1.72±0.08 | 0.96±0.04 |
| UK-14,304 1 μM | 1.68±0.06 | 0.52±0.03a |
| Rauwolscine 1 μM | 1.65±0.09 | 1.68±0.18a |
| Rauwolscine 10 μM | 1.76±0.09 | 2.21±0.23a |
| Phentolamine 1 μM | 1.73±0.07 | 1.97±0.15a |
| Phentolamine 10 μM | 1.70±0.05 | 2.84±0.17a |
| DNBS control | 1.07±0.09* | 0.99±0.03 |
| UK-14,304 1 μM | 1.03±0.07* | 0.35±0.06*,a |
| Rauwolscine 1 μM | 1.02±0.05* | 2.06±0.11*,a |
| Rauwolscine 10 μM | 1.05±0.05* | 2.73±0.13*,a |
| Phentolamine 1 μM | 1.03±0.08* | 2.92±0.15*,a |
| Phentolamine 10 μM | 1.11±0.06* | 3.66±0.11*,a |
Longitudinal muscle strips of ileum were obtained from rats either in the absence or in the presence of DNBS-induced colitis. Each preparation was preincubated with [3H]noradrenaline, superfused with Krebs solution, and subjected twice to electrical stimulation (0.5 ms, 20 V cm−1, 8 Hz, 480 pulses). The effects of test drugs on tritium outflow are expressed as S1 (percentage of the tritium content of the tissue at the onset of the first electrical stimulation), or S2/S1 (ratio of the percentage release during the second stimulation over that obtained during the first stimulation). Each value represents the mean of five to six experiments±s.e.m.
Significant difference from the respective values obtained from vehicle-treated rats: *P<0.05
significant difference from the respective control values: aP<0.05.
In ileal preparations obtained from animals with DNBS-induced colitis (n=6), the basal tritium overflow was 0.0017±0.00014 min−1, and this value did not vary significantly until the end of the experiments. The first electrical stimulation (S1) caused a significant increase in tritium efflux, from 0.0017±0.00013 to 0.0029±0.00024 min−1 (P<0.05). The electrically stimulated increment of tritium outflow was 1.07±0.09% for S1 and 1.05±0.11% for S2 with a S2/S1 ratio of 0.99±0.03. (Figure 3b; Table 3). The values observed for tritium efflux elicited by S1 and S2 were significantly lower than those estimated for ileal preparations obtained from vehicle-treated animals, whereas the S2/S1 ratios did not differ (Table 3).
In experiments performed on ileal strips from vehicle-treated animals, UK-14,304 significantly inhibited the electrically induced outflow of tritium in a concentration-dependent manner, with apparent EC50 and Emax values of 115.4 nM and –52.5±3.6% (at 3 μM), respectively (Figure 4; Table 3). When tested on ileal preparations from rats with colitis, UK-14,304 caused a more prominent inhibition of electrically stimulated tritium efflux (Figure 4; Table 3). In this case, EC50 and Emax were 97.7 nM and –70.1±4.8% (at 3 μM), respectively. The latter value was significantly higher than that estimated for intestinal tissue isolated from rats in the absence of colitis (P<0.05). Additional experiments were carried out to assay the effects of α2-adrenoceptor antagonists on [3H]noradrenaline release. Rauwolscine (1–10 μM) or phentolamine (1–10 μM) significantly increased the tritium outflow evoked by electrical stimulation of ileal strips prepared from vehicle-treated rats. The stimulant actions exerted by both antagonists were enhanced when these drugs were assayed on ileal preparations isolated from animals with experimental colitis (Table 3).
Figure 4.

Effects of increasing concentrations of UK-14,304 on tritium outflow evoked by electrical field stimulation of longitudinal muscle preparations of rat ileum preincubated with [3H] noradrenaline. Electrical stimulation during S1 and S2 consisted of 480 pulses (20 V cm−1, 0.5 ms) at 8 Hz. Ileal preparations were obtained from rats either in the absence or in the presence of DNBS-induced colitis. Each point represents the mean of five to six experiments±s.e.m. (vertical bars). Significant difference from the respective values obtained in vehicle-treated animals: *P<0.05; significant difference from the respective control values: aP<0.05.
RT-PCR analysis of α2A-adrenoceptors
RT–PCR assay of intestinal tissues revealed the expression of α2A-adrenoceptor mRNA at the level of mucosal and muscular layers of both ileum and colon retrieved from vehicle- or DNBS-treated animals (Figure 5). The densitometric analysis of the amplified cDNA bands showed a significant enhancement of α2A-adrenoceptor expression in ileal and colonic muscular layers obtained from animals with colitis (+54.7 and +59.2%, respectively). However, the induction of colitis did not significantly alter the expression of α2A-adrenoceptor mRNA in the mucosa of ileum or colon (+12.9 and +9.8%, respectively) (Figure 5).
Figure 5.

RT–PCR analysis of α2Aadrenoceptor (α2A-AR) and β-actin mRNA expression in mucosal (Muc) and muscular (Musc) layers of ileum or colon isolated from animals in the absence (vehicle) or in the presence of DNBS-induced colitis (DNBS). Each panel displays two representative agarose gels, referring to the amplification of α2A-adrenoceptor and β-actin cDNAs, and a column graph referring to the densitometric analysis of α2A-adrenoceptor cDNA bands normalized to the expression of β-actin. M=size markers. Each column represents the mean of five separate experiments±s.e.m. (vertical bars). Significant difference from the respective values obtained in vehicle-treated animals: *P<0.05.
Western blot analysis of α2A-adrenoceptors
Western blot analysis of intestinal tissues, excised from vehicle- or DNBS-treated animals, showed that the expression of α2A-adrenoceptors (45 kDa) could be detected at the level of mucosal and muscular layers of both ileum and colon (Figure 6). The densitometric analysis of immunoreactive bands revealed a significant increase of α2A-adrenoceptor expression in both ileal and colonic muscular layers retrieved from animals subjected to DNBS-induced colitis (+67.8 and +50.8%, respectively). By contrast, treatment with DNBS did not significantly affect the expression of α2A-adrenoceptors in the mucosal layer of both ileum and colon (+12.8 and +16.5%, respectively) (Figure 6).
Figure 6.

Western blot analysis of α2A-adrenoceptor (α2A-AR) expression in mucosal (Muc) and muscular (Musc) layers of ileum or colon isolated from animals in the absence (vehicle) or in the presence of DNBS-induced colitis (DNBS). Each panel displays a representative blot and a column graph referring to the densitometric analysis of immunoreactive bands. Each column represents the mean of four separate experiments±s.e.m. (vertical bars). Significant difference from the respective values obtained in vehicle-treated animals: *P<0.05.
Gastrointestinal transit
The first series of experiments was aimed to assess the gastrointestinal transit in the absence of colitis. In vehicle-treated animals, the distance travelled by the charcoal suspension accounted for 72.9±6.4% (n=10) of the total length of small intestine. UK-14,304 (0.2 μmol kg−1 i.p.) caused a significant inhibition of gastrointestinal transit, whereas rauwolscine (1 μmol kg−1 i.p.) was without effect (Figure 7a). Similar results were obtained with UK-14,304 and rauwolscine when they were assayed on animals subjected to pretreatment with guanethidine in order to ablate the sympathetic nervous pathways (Figure 7c). In both cases, the inhibitory effects of UK-14,304 on gastrointestinal transit were antagonized by rauwolscine (Figure 7a and c).
Figure 7.

Effects of i.p. administrations of UK-14,304 (0.2 μmol kg−1), rauwolscine (1 μmol kg−1), or rauwolscine plus UK-14,304 on gastrointestinal transit in rats. Data displayed in panels a and b were obtained from animals in the absence or in the presence of DNBS-induced colitis, respectively. Data displayed in panels c and d refer to experiments where both vehicle-treated and DNBS-treated animals received guanethidine 70 μmol kg−1 i.p. daily for two consecutive days, before performing the gastrointestinal transit assay. Each value represents the mean of seven to ten experiments ±s.e.m. (vertical bars). Significant difference from the respective values obtained in vehicle-treated animals: *P<0.05; significant difference from the respective control values: aP<0.05; significant difference from the respective values obtained in guanethidine-untreated animals: bP<0.05.
In the second series of experiments, the gastrointestinal transit was evaluated in rats with DNBS-induced colitis. In this case, the distance travelled by the charcoal suspension was 54.3±4.4% of the total small intestine (n=8) (Figure 7b). Such a value was significantly lower than that measured in the absence of experimental colitis (P<0.05). Under these conditions, UK-14,304 (0.2 μmol kg−1 i.p.) was less effective in reducing the gastrointestinal transit, whereas rauwolscine (1 μmol kg−1 i.p.) caused a significant enhancement (Figure 7b). When animals with colitis were subjected to pretreatment with guanethidine, the gastrointestinal transit was 67.2±2.8% (n=7), this value being not significantly different from that estimated in the absence of colitis. In this setting, the inhibitory effect of UK-14,304 on the gastrointestinal transit was significantly higher than that estimated in vehicle-treated rats, whereas the stimulant action of rauwolscine no longer occurred (Figure 7d). As observed in the absence of colitis, the inhibitory actions of UK-14,304 on both guanethidine-untreated and guanethidine-treated animals were counteracted by pretreatment with rauwolscine (Figure 7b, d).
Discussion
Intestinal inflammation is associated with alterations of gut functions (motility, absorption/secretion), which may contribute to generation of various symptoms. Diarrhoea is not the only symptom experienced by patients with inflammatory bowel disease, since they also complain of changes in bowel habit, bloating, abdominal pain/discomfort, nausea, and other irritable bowel syndrome-like symptoms (Hendrickson et al., 2002). The pathophysiological mechanisms accounting for this complex array of symptoms have not been fully clarified. Changes in both intrinsic and extrinsic enteric nerves may contribute to altered motor function in the inflamed gut (Collins, 1996; Sharkey & Kroese, 2001). The present study demonstrates that the induction of colitis is associated with significant changes in the α2-adrenoceptor-mediated control of both enteric neurotransmission and digestive motility, not only in the colon but also at distant sites.
When applied to ileal or colonic preparations from rats treated with intrarectal vehicle (uninflamed), the agonist UK-14,304 inhibited the electrically induced cholinergic contractions with high potency and moderate efficacy, suggesting that the α2-adrenoceptors play a role in the prejunctional modulation of cholinergic transmission under standard conditions. In the presence of colitis, UK-14,304 decreased with higher efficacy the evoked cholinergic contractions at both colonic and ileal level. These results provide the first demonstration that colonic inflammation enhances the α2-adrenoreceptor-mediated inhibitory control of enteric cholinergic transmission, and they further expand the available knowledge on neuroplastic changes occurring in the inflamed gut (De Giorgio et al., 2001; Sharkey & Kroese, 2001). Of interest, the present findings are in keeping with previous data showing a significant decrease in electrically induced acetylcholine release from myenteric plexus preparations obtained from the small intestine of nematode-infected rats (Collins et al., 1989).
Three genetically distinct α2-adrenoceptor subtypes, named α2A, α2B and α2C, have been identified in mammals (Hieble et al., 1997). In the present investigation, an interaction with α2A-adrenoceptor subtypes seems to account for the inhibitory actions of UK-14,304 on cholinergic motility in both uninflamed and inflamed rats. Indeed, in both ileal and colonic tissues, phentolamine was more potent than rauwolscine in antagonizing the inhibitory effects of UK-14,304. This pharmacological pattern has been previously reported for rat, guinea-pig, and mouse α2A-adrenoceptors, and is not compatible with affinity profiles of α2B- or α2C-adrenoceptors (Simonneaux et al., 1991; Trendelenburg et al., 1997). Consistent with this view, recent experiments, performed on α2A-adrenoceptor knockout mice, have provided conclusive evidence that enteric cholinergic neurons are subjected to presynaptic modulation by α2A-adrenoceptor subtypes (Scheibner et al., 2002).
Beside the participation of intrinsic cholinergic pathways in the processes of remodelling and adaptation of enteric neurons in intestinal inflammation (Collins et al., 1989; Hosseini et al., 1999), several lines of evidence suggest that the extrinsic sympathetic nerves supplying the gut are involved (Collins, 1996; Sharkey & Kroese, 2001). Substantial changes in the evoked noradrenaline release have also been shown in experimental gut inflammation (Swain et al., 1991) both at inflamed and distant noninflamed sites (Jacobson et al., 1995). In keeping with these studies, the evoked tritium outflow in the present ileal preparations, preincubated with [3H]noradrenaline, was significantly lower in rats with colitis, as compared to uninflamed animals. Furthermore, our data demonstrate that the application of UK-14,304 to ileal preparations from animals with or without colitis significantly depresses the electrically evoked tritium efflux, whereas phentolamine or rauwolscine exerts potentiating effects. The latter observation might appear inconsistent with data obtained from in vitro experiments on muscle contraction, where both phentolamine and rauwolscine per se failed to affect the electrically induced cholinergic responses. This apparent discrepancy may be explained considering that muscle contraction experiments were performed in the presence of guanethidine, which, because of its action on noradrenergic fibres, prevented the enhancing effect of α2-adrenoceptor antagonists.
The most interesting observation is that the α2-adrenoceptor ligands act on the evoked [3H]noradrenaline with different degrees of efficacy, when assayed on ileal preparations retrieved from animals with or without colitis. A comparison of the concentration–response curves, constructed for UK-14,304, clearly indicates that this agonist inhibits with greater efficacy the release of [3H]noradrenaline in ileal tissues from inflamed animals. Likewise, the stimulant actions of rauwolscine or phentolamine on the evoked tritium output are further enhanced when these antagonists are tested on ileal preparations from animals with colitis. These findings suggest that, after induction of colitis, the sympathetic pathways supplying the gut undergo an enhanced prejunctional modulation, mediated by α2-adrenoceptors located on noradrenergic nerve endings.
Three major mechanisms may be hypothesized to account for the enhanced inhibitory control mediated by α2-adrenoceptors on cholinergic and noradrenergic transmissions in animals with colitis: (1) increased receptor affinity; (2) increased receptor density and/or recruitment and (3) changes in transduction pathways. In the present study, both RT–PCR and Western blot assays provide consistent evidence that the induction of colitis is associated with a significant increase in α2-adrenoceptor mRNA and protein expression in the muscle layers. These findings suggest that colonic inflammation promotes an increment of α2-adrenoceptor density within the neuromuscular compartments of both inflamed and noninflamed gut. In support of this view, the inhibitory effects of UK-14,304 on cholinergic motility and [3H]noradrenaline release in animals with colitis are characterized by higher Emax, but similar EC50 values, than in uninflamed animals. These results expand previous radioligand binding data, showing that intestinal inflammation is associated with an upregulation of α2-adrenoceptors in gut tissues (Martinolle et al., 1993). However, the possibility that our results might be interpreted in terms of changes in the efficiency of receptor–effector coupling mechanisms cannot be ruled out, and this issue should be addressed in separate studies. Although the mechanisms underlying the enhancement of α2-adrenoceptor expression and function at noninflamed gut sites were not specifically investigated in the present study, two main hypotheses can be proposed to explain these findings: (1) changes at distant sites might depend on systemic effects elicited by inflammatory/immune mediators released from colonic inflammatory lesions; this mechanism has been previously suggested by Jacobson et al. (1995) to explain the reduced noradrenaline release from preparations of non-inflamed transverse colon and ileum in rat models of colitis induced by 2,4,6-trinitrobenzenesulphonic acid (TNBS) or nematodes; (2) neural and receptor changes at remote sites might be related to nerve reflex pathways, as proposed by Moreels et al. (2001) for rat TNBS-induced ileitis.
To verify whether changes in α2-adrenoceptor expression, evoked by gut inflammation, might reflect variations of in vivo intestinal motility, the effects of UK-14,304 and rauwolscine have been tested in a model of gastrointestinal transit. Data obtained from these experiments refer only to transit in the small intestine, and therefore a correlation between in vitro and in vivo results was not made for colonic motility. In the absence of colitis, UK-14,304 causes a rauwolscine-sensitive inhibition of small intestinal transit. This finding is in line with previous reports suggesting that the activation of α2-adrenoceptors may lead to a decreased intestinal motor activity, via the inhibition of enteric cholinergic neurons (De Ponti et al., 1996; Stebbing et al., 2001). During colitis, gastrointestinal transit was reduced and, at variance with data obtained from in vitro experiments, UK-14,304 exerted a moderate inhibitory effect. In this setting, rauwolscine completely restored the propulsory activity, indicating that, in the presence of colonic inflammation, the sympathetic nerve pathways mediate a tonic inhibitory control on bowel motility through the activation of α2-adrenoceptors. Such hypothesis was tested in animals treated with guanethidine, to induce a pharmacological ablation of sympathetic pathways. In this case, the stimulant action of rauwolscine on gastrointestinal transit of inflamed animals no longer occurred, and UK-14,304 decreased the charcoal propulsion to a greater extent than in uninflamed rats. These findings, taken together with data from the present experiments on in vitro cholinergic motility, suggest that the induction of colonic inflammation causes a tonic activation of noradrenergic sympathetic nerves and a concomitant increase of α2-adrenoceptor expression, resulting in an enhanced inhibitory control of digestive motor activity. On the other hand, the reduced noradrenaline release, observed in our in vitro experiments, might reflect the activation of counterregulatory mechanisms against the increase of sympathetic tone promoted by intestinal inflammation. In line with this view, evidence of altered sympathetic activity was previously obtained both in patients with inflammatory bowel disease (Lindgren et al., 1991) and in preclinical models of intestinal inflammation (Sharkey & Kroese, 2001), whereas in vitro experiments on intestinal tissues showed a decrease in noradrenaline release at inflamed and noninflamed sites (Collins, 1996).
The use of guanethidine as an adrenergic neuron blocker deserves some comments. We are aware that guanethidine has been suggested to act also as a nicotinic receptor blocker at the level of enteric neurons (Blommaart et al., 1999). Therefore, when interpreting the present results from guanethidine-treated animals, we took into account the possibility that nicotinic transmission might be implicated in the reduction of intestinal transit elicited by colitis. However, the putative antagonism of guanethidine on nicotinic receptors seems to occur only at high concentrations (>10 μM), and was not substantiated by radioligand binding experiments (Blommaart et al., 1999). Furthermore, the colitis-induced reduction of intestinal transit was fully reversed by rauwolscine, suggesting the presence of a sympathetic inhibitory tone on digestive motility, driven via activation of α2-adrenoceptors. On this basis, it appears unlikely that guanethidine affected the intestinal transit in inflamed animals via an interaction with nicotinic receptors.
In conclusion, the results obtained in the present study indicate that, in the presence of intestinal inflammation, prejunctional α2-adrenoceptors contribute significantly to pathophysiological changes characterized by an enhanced inhibitory control of cholinergic and noradrenergic neurotransmission both at inflamed and non-inflamed distant sites. Direct evidence was also obtained that an increased expression of α2A-adrenoceptors within the enteric nervous system may contribute to such modulatory actions.
Acknowledgments
The present work was supported in part by a grant for scientific research issued from the Italian Ministry of Education, University and Research (COFIN 1999, Project number 9905222532).
Abbreviations
- ANOVA
analysis of variance
- DNBS
2,4-dinitrobenzenesulphonic acid
- EDTA
ethylenediaminetetraacetic acid
- L-NAME
Nω-nitro-L-arginine methylester
- PCR
polymerase chain reaction
- RT
reverse transcription
- TBS-T
Tween-20 in Tris-buffered saline
- TNBS
2,4,6-trinitrobenzenesulphonic acid
References
- BARBARA G., XING Z., HOGABOAM C.M., GAULDIE J., COLLINS S.M. Interleukin 10 gene transfer prevents experimental colitis in rats. Gut. 2000;46:344–349. doi: 10.1136/gut.46.3.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARTHÒ L., LENARD L., LAZAR Z., MAGGI C.A. Connections between P2 purinoceptors and capsaicin-sensitive afferents in the intestine and other tissues. Eur. J. Pharmacol. 1999;375:203–210. doi: 10.1016/s0014-2999(99)00253-8. [DOI] [PubMed] [Google Scholar]
- BLANDIZZI C., TARKOVACS G., NATALE G., DEL TACCA M., VIZI E.S. Functional evidence that [3H]acetylcholine and [3H]noradrenaline release from guinea pig ileal myenteric plexus and noradrenergic terminals is modulated by different presynaptic alpha-2 adrenoceptor subtypes. J. Pharmacol. Exp. Ther. 1993;267:1054–1060. [PubMed] [Google Scholar]
- BLANDIZZI C., TOGNETTI M., COLUCCI R., DEL TACCA M. Histamine H3 receptors mediate inhibition of noradrenaline release from intestinal sympathetic nerves. Br. J. Pharmacol. 2000;129:1387–1396. doi: 10.1038/sj.bjp.0703194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLOMMAART P.J.E., FERWERDA G., KODDE A., TYTGAT G.N.J., BOECKXSTAENS G.E. Nicotinic acetylcholine receptor blocking effect of guanethidine in the rat gastric fundus. Br. J. Pharmacol. 1999;128:903–908. doi: 10.1038/sj.bjp.0702865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLLINS S.M. The immunomodulation of enteric neuromuscular function: implications for motility and inflammatory disorders. Gastroenterology. 1996;111:1683–1699. doi: 10.1016/s0016-5085(96)70034-3. [DOI] [PubMed] [Google Scholar]
- COLLINS S.M., BLENNERHASSETT P.A., BLENNERHASSETT M.G., VERMILLION D.L. Impaired acetylcholine release from the myenteric plexus of Trichinella-infected rats. Am. J. Physiol. 1989;257:G898–G903. doi: 10.1152/ajpgi.1989.257.6.G898. [DOI] [PubMed] [Google Scholar]
- COLUCCI R., BLANDIZZI C., CARIGNANI D., PLACANICA G., LAZZERI G., DEL TACCA M. Effects of imidazoline derivatives on cholinergic motility in guinea-pig ileum: involvement of presynaptic α2-adrenoceptors or imidazoline receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;357:682–691. doi: 10.1007/pl00005225. [DOI] [PubMed] [Google Scholar]
- DE GIORGIO R., BARBARA G., BLENNERHASSETT P., WANG L., STANGHELLINI V., CORINALDESI R., COLLINS S.M., TOUGAS G. Intestinal inflammation and activation of sensory nerve pathways: a functional and morphological study in the nematode infected rat. Gut. 2001;49:822–827. doi: 10.1136/gut.49.6.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE PONTI F., GIARONI C., COSENTINO M., LECCHINI S., FRIGO G. Adrenergic mechanisms in the control of gastrointestinal motility: from basic science to clinical applications. Pharmacol. Ther. 1996;69:59–78. doi: 10.1016/0163-7258(95)02031-4. [DOI] [PubMed] [Google Scholar]
- DEL TACCA M., SOLDANI G., BERNARDINI C., MARTINOTTI E. Pharmacological studies on the mechanisms underlying the inhibitory and excitatory effects of clonidine on gastric acid secretion. Eur. J. Pharmacol. 1982;81:255–261. doi: 10.1016/0014-2999(82)90443-5. [DOI] [PubMed] [Google Scholar]
- FUNK L., TRENDELENBURG A.U., LIMBERGER N., STARKE K. Subclassification of presynaptic α2-adrenoceptors: α2D-autoreceptors and α2D-adrenoceptors modulating release of acetylcholine in guinea-pig ileum. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995;352:58–66. doi: 10.1007/BF00169190. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F.The classification of adrenoceptors (adrenergic receptors). An evaluation from the standpoint theory Catecholamines, Handbook of Experimental Pharmacology 1972Berlin: Springer; 283–335.ed. Blascho, H. & Muscholl, E. Vol. 33, pp [Google Scholar]
- GIARONI C., DE PONTI F., COSENTINO M., LECCHINI S., FRIGO G.M. Plasticity in the enteric nervous system. Gastroenterology. 1999;117:1438–1458. doi: 10.1016/s0016-5085(99)70295-7. [DOI] [PubMed] [Google Scholar]
- HENDRICKSON B.A., GOKHALE R., CHO J.H. Clinical aspects and pathophysiology of inflammatory bowel disease. Clin. Microbiol. Rev. 2002;15:79–94. doi: 10.1128/CMR.15.1.79-94.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIEBLE J.P., RUFFOLO R.R., STARKE K.Identification, characterization and subclassification of α2-adrenoceptors: an overview α2-Adrenergic Receptors: Structure, Function and Therapeutic Implications 1997Reading, MA: Harwood Academic Publishers; 1–18.ed. Lanier, S.M. Limbird, L.E. pp [Google Scholar]
- HOSSEINI J.M., GOLDHILL J.M., BOSSONE C., PINEIRO-CARRERO V., SHEA-DONOHUE T. Progressive alterations in circular smooth muscle contractility in TNBS-induced colitis in rats. Neurogastroent. Motil. 1999;11:347–356. doi: 10.1046/j.1365-2982.1999.00165.x. [DOI] [PubMed] [Google Scholar]
- JACOBSON K., MCHUGH K., COLLINS S.M. Experimental colitis alters myenteric nerve function at inflamed and noninflamed sites in the rat. Gastroenterology. 1995;109:718–722. doi: 10.1016/0016-5085(95)90378-x. [DOI] [PubMed] [Google Scholar]
- LINDGREN B., LILJA B., ROSEN I., SUNDKVIST G. Disturbed autonomic nerve function in patients with Crohn's disease. Scand. J. Gastroenterol. 1991;26:361–366. doi: 10.3109/00365529108996495. [DOI] [PubMed] [Google Scholar]
- LIU L., COUPAR I.M. Role of α2-adrenoceptors in the regulation of intestinal water transport. Br. J. Pharmacol. 1997;120:892–898. doi: 10.1038/sj.bjp.0700958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGGI C.A., GIULIANI S. Role of tachykinins as excitatory mediators of NANC contraction in the circular muscle of rat small intestine. J. Auton. Pharmacol. 1995;15:335–350. doi: 10.1111/j.1474-8673.1995.tb00400.x. [DOI] [PubMed] [Google Scholar]
- MALCOM A., CAMILLERI M., KOST L., BURTON D.D., FETT S.L., ZINSMEISTER A.R. Towards identifying optimal doses for α2 adrenergic modulation of colonic and rectal motor and sensory function. Aliment. Pharmacol. Ther. 2000;14:783–793. doi: 10.1046/j.1365-2036.2000.00757.x. [DOI] [PubMed] [Google Scholar]
- MARTINOLLE J.P., MORE J., DUBECH N., GARCIA-VILLAR R. Inverse regulation of alpha and beta adrenoceptor during trinitrobenzenesulphonic acid (TNB)-induced inflammation in guinea-pig small intestine. Life Sci. 1993;53:1499–1508. doi: 10.1016/0024-3205(93)90112-g. [DOI] [PubMed] [Google Scholar]
- MOREELS T.G., DE MAN J.G., DE WINTER B.Y., TIMMERMANS J.-P., HERMAN A.G., PELCKMANS P.A. Effect of 2,4,6- trinitrobenzenesulfonic acid (TNBS)-induced ileitis on the motor function of non-inflamed rat gastric fundus. Neurogastroenterol. Motil. 2001;13:339–352. doi: 10.1046/j.1365-2982.2001.00273.x. [DOI] [PubMed] [Google Scholar]
- SCHAAK S., CUSSAC D., CAYLA C., DEVEDJIAN J.C., GUYOT R., PARIS H., DENIS C. Alpha-2 adrenoceptors regulate proliferation of human intestinal cells. Gut. 2000;47:242–250. doi: 10.1136/gut.47.2.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHEIBNER J., TRENDELENBURG A.U., HEIN L., STARKE K., BLANDIZZI C. α2-Adrenoceptors in the enteric nervous system: a study in α2A-adrenoceptor- deficient mice. Br. J. Pharmacol. 2002;135:697–704. doi: 10.1038/sj.bjp.0704512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARKEY K.A., KROESE A.B.A. Consequences of intestinal inflammation on the enteric nervous system: neuronal activation induced by inflammatory mediators. Anat. Rec. 2001;262:79–90. doi: 10.1002/1097-0185(20010101)262:1<79::AID-AR1013>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- SIMONNEAUX V., EBADI M., BYLUND D.B. Identification and characterization of α2D-adrenergic receptors in bovine pineal gland. Mol. Pharmacol. 1991;40:235–241. [PubMed] [Google Scholar]
- SINGH L., FIELD M.J., HUNTER J.C., OLES R.J., WOODRUFF G.N. Modulation of the in vivo actions of morphine by the mixed CCK-A/B receptor antagonist PD 142898. Eur. J. Pharmacol. 1996;307:283–289. doi: 10.1016/0014-2999(96)00281-6. [DOI] [PubMed] [Google Scholar]
- STEBBING M.J., JOHNSON P.J., VREMEC M.A., BORNSTEIN J.C.Role of α2-adrenoceptors in the sympathetic inhibition of motility reflexes of guinea-pig ileum J. Physiol. 2001534.2465–478.(London) [DOI] [PMC free article] [PubMed] [Google Scholar]
- SWAIN M.G., BLENNERHASSETT P.A., COLLINS S.M. Impaired sympathetic nerve function in the inflamed rat intestine. Gastroenterology. 1991;100:675–682. doi: 10.1016/0016-5085(91)80011-w. [DOI] [PubMed] [Google Scholar]
- TRENDELENBURG A.U., SUTEJ I., WAHL C.A., MOLDERINGS G.J., RUMP L.C., STARKE K. A re-investigation of questionable subclassifications of presynaptic α2-autoreceptors: rat vena cava, rat atria, human kidney and guinea-pig urethra. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:721–737. doi: 10.1007/pl00005111. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., KEENAN C.M. An orally active inhibitor of leukotriene synthesis accelerates healing in a rat model of colitis. Am. J. Physiol. 1990;258:G527–G534. doi: 10.1152/ajpgi.1990.258.4.G527. [DOI] [PubMed] [Google Scholar]
- WILBORN T.W., SUN D., SCHAFER J.A. Expression of multiple alpha-adrenoceptor isoforms in rat CCD. Am. J. Physiol. 1998;275:F111–F118. doi: 10.1152/ajprenal.1998.275.1.F111. [DOI] [PubMed] [Google Scholar]
- YING L., YI C.J., CHUNG O. Central CGRP inhibits pancreatic enzyme secretion by modulation of vagal parasympathetic outflow. Am. J. Physiol. 1998;275:G957–G963. doi: 10.1152/ajpgi.1998.275.5.G957. [DOI] [PubMed] [Google Scholar]
- ZHAO A., BOSSONE C., PIÑEIRO-CARRERO V., SHEA-DONOHUE T. Colitis-induced alterations in adrenergic control of circular smooth muscle in vitro in rats. J. Pharmacol. Exp. Ther. 2001;299:768–774. [PubMed] [Google Scholar]