

Abstract
We have investigated the possible roles of sulphonylurea receptor (SUR) type 1 and 2B in the activity of pig urethral ATP-sensitive K+ channels (KATP channels) by use of patch-clamp and reverse transcriptase–polymerase chain reaction (RT–PCR) techniques.
In voltage-clamp experiments, not only diazoxide, a SUR1 and weak SUR2B activator, but also pinacidil, a selective SUR2 activator, caused an inward current at a holding potential of −50 mV in symmetrical 140 mM K+ conditions.
Gliclazide (⩽1 μM), a selective SUR1 blocker, inhibited the 10 μM pinacidil-induced currents (Ki=177 μM) and the 500 μM diazoxide-induced currents (high-affinity site, Ki1=5 nM; low-affinity site, Ki2=108 μM) at −50 mV.
Application of tolbutamide (⩽100 μM) reversibly caused an inhibition of the 500 μM diazoxide-induced current at –50 mV.
MCC-134, a SUR type-specific KATP channel regulator (1–100 μM), produced a concentration-dependent inward K+ current, which was suppressed by the application of glibenclamide at −50 mV. The amplitude of the MCC-134 (100 μM)-induced current was approximately 50% of that of the 100 μM pinacidil-induced currents.
Using cell-attached configuration, MCC-134 activated a glibenclamide-sensitive KATP channel which was also activated by pinacidil.
RT–PCR analysis demonstrated the presence of SUR1 and SUR2B transcripts in pig urethra.
These results indicate that both SUR1 and SUR2B subunits play a functional role in regulating the activity of pig urethral KATP channels and that SUR1 contributes less than 25% to total KATP currents.
Keywords: ATP-sensitive K+ channels, diazoxide, gliclazide, MCC-134, pinacidil, tolbutamide, smooth muscle, sulphonyl-urea receptor 1, sulphonylurea receptor 2B
Introduction
Recent studies have revealed that ATP-sensitive K+ channels (KATP channels) are heteromeric complexes composed of at least two subunits; inwardly rectifying K+ channels (Kir6.x) and sulphonylurea receptors (SURs) (Aguilar-Bryan et al., 1995; Inagaki et al., 1995). The SURs are responsible not only for sulphonylurea-sensitivity (such as glibenclamide, tolbutamide, etc.) but also for intracellular nucleoside diphosphate (NDP)-sensitivity. KATP channel openers interact with the SUR subunits, and not with the Kir subunits of the KATP channels (Schwanstecher et al., 1998).
It has been reported that recombinant KATP channels solely composed of homotetrameric subunits of SUR2B resemble those found in native vascular smooth muscle (Isomoto et al., 1996; Yamada et al., 1997). Although the molecular composition of SURs assessed by reverse transcriptase–polymerase chain reaction (RT–PCR) has revealed the presence of mRNA transcripts for SUR1 and SUR2B in detrusor smooth muscle (guinea-pig, Gopalakrishnan et al., 1999; pig and human, Buckner et al., 2000), it was concluded that only SUR2B but not SUR1, is expressed as a SUR protein for regulating the activity of KATP channels in the urinary bladder (Gopalakrishnan et al., 1999). We believe that the significant differences in properties of KATP channels between urinary bladder and urethra (such as different types of SUR subunits, etc.) hold out some hope for the development of tissue-selective KATP channel openers for urge urinary incontinence (Teramoto et al., 1997). To achieve this aim, it is essential to study the properties of SUR subunits in urethral KATP channels in comparison with those in the urinary bladder.
Recently, Shindo et al. (2000), using functional expressing studies, have reported that MCC-134 is a full agonist for the SUR2B/Kir6.2 channel and a partial agonist for the SUR2A/Kir6.2 channel, whereas it inhibits the SUR1/Kir6.2, concluding that MCC-134 may be the prototype for new drugs acting selectively on distinct types of SURs.
In the present experiments, the possible roles of SUR1 and SUR2B in the activity of KATP channels in pig urethra were examined by the use of patch-clamp techniques. We have utilized diazoxide as an agonist for SUR1 and weak agonist for SUR2B, pinacidil as a selective agonist for SUR2, but not for SUR1, and MCC-134 as a novel KATP channel modulator acting through distinct types of SURs. Gliclazide and tolbutamide were used as specific antagonists for SUR1. In addition, we have also performed RT–PCR analysis in order to examine whether or not mRNA for SUR1 and SUR2B are expressed in pig urethra. The present study thus provides evidence of the pharmacological and molecular identity of SUR1 and SUR2B subunits in pig urethral KATP channels.
Methods
Cell dispersion
Fresh urethra from female pigs was collected from a local abattoir. Pig urethral myocytes were freshly isolated by the gentle tapping method after treatment with papain and collagenase, as described previously (Teramoto & Brading, 1996). Relaxed spindle-shaped cells, with length varying between 400 and 500 μm, were isolated and stored at 4°C. The dispersed cells were used within 2 h for experiments.
Recording procedure
Patch-clamp experiments were performed at room temperature (21–23°C), as described previously (Teramoto et al., 2001). Junction potentials between bath and pipette solutions were measured with a 3 M KCl reference electrode and were <2 mV, so that correction for these potentials was not necessary. Capacitance noise was kept to a minimum by maintaining the test solution in the electrode as low as possible.
Drugs and solutions
For whole-cell recording (conventional whole-cell and nystatin-perforated patch), the following solutions were used: 140 mM K+ solution containing (mM): Na+ 5, K+ 140, Mg2+ 1.2, Ca2+ 2, glucose 5, Cl− 151.4, HEPES 10, titrated to pH 7.35–7.40 with Tris base; high potassium pipette solution containing (mM): K+ 140, Cl− 140, glucose 5, EGTA 5, and HEPES 10/Tris (pH 7.35–7.40). The perforated-patch technique with nystatin was also occasionally used to record whole-cell currents (Teramoto & Brading, 1996). In short, nystatin was freshly dissolved in acidified methanol (1 N HCl to about pH 2) and the pH was readjusted to 7.4 with Tris base. This stock solution (10 mg ml−1) was diluted in the pipette solution at a final concentration of 50 μg ml−1 just before use. Whole-cell recording was performed with a pipette that was first dipped in normal pipette solution (nystatin-free) and then back-filled with nystatin-containing pipette solution. This resulted in chemical perforation of the membrane. For cell-attached recordings, the pipette and bath solution were high potassium solutions (mM): K+ 140, Cl− 140, EGTA 5, glucose 5, HEPES 10/Tris (pH 7.35–7.40). Cells were allowed to settle in the small experimental chamber (80 μl in volume). The bath solution was superfused by gravity throughout the experiments at a rate of 2 ml min−1. MCC-134 (Mitsubishi Pharma Corporation, Tokyo, Japan), gliclazide (Dainippon Pharmaceuticals Co., Ltd, Osaka, Japan), and glibenclamide were prepared daily as 100 mM stock solutions in dimethylsulphoxide (DMSO). Other drugs were obtained from Sigma Chemical (Sigma Chemical K.K., Tokyo, Japan). The maximum final concentration of DMSO in the bath solution was 0.3% for patch-clamp experiments. These concentrations were shown not to affect K+ channels.
Data analysis
The whole-cell current data were low-pass filtered at 500 Hz by an eight pole Bessel filter, sampled at 25 ms intervals (continuous traces) or 1 ms (ramp current) and analysed on a computer (PowerMac G3, Tokyo, Japan) by the commercial software ‘MacLab 3.5. 6' (ADInstruments Pty Ltd, Castle Hill, Australia). For single-channel recordings, the stored data were low-pass filtered at 2 kHz (−3 dB) and sampled by a computer with a digitalized interval of 80 μs using ‘PAT' program (kindly provided by Dr J. Dempster, University of Strathclyde, U.K.); events briefer than 80 μs were not included in the evaluation. Continuous traces in the figures were obtained from records filtered at 500 Hz for presentation (digital sampling interval, 25 ms). Values for the channel open state probability (Popen) were measured at −50 mV for 1 min.
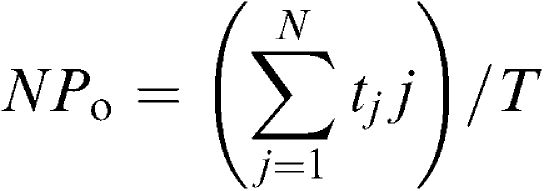 |
where tj is the time spent at each current level corresponding to j=0, 1, 2,… N, T is the duration of the recording, and N is taken as the maximum number of channels observed in the patch membrane where Popen was relatively high. Data points were fitted using the method of least-squares. Data are expressed as mean with the standard deviation (s.d.).
Equation for curve fitting of KATP channel opener-induced currents
The curve for KATP channel opener-induced current was drawn by fitting the equation using the least-squares method:
where A, K, D and nH are maximum relative current induced by KATP channel opener, apparent dissociation constant, concentration of KATP channel opener (μM) and the Hill coefficient, respectively.
Equation for curve fitting of the effects of gliclazide on KATP channel opener-induced currents
The mean amplitude of each KATP opener-induced current at the holding potential of −50 mV just before application of gliclazide (30 s duration) was normalized as 1.0. The curve was drawn by fitting the equation using the least-squares method:
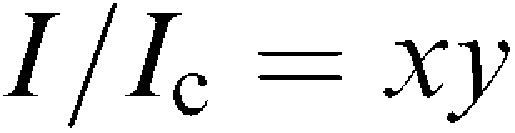 |
where I is the mean amplitude of the KATP opener-induced current in the presence of gliclazide, Ic is the mean amplitude of the KATP opener-induced current in control (in the absence of gliclazide), x is a term describing the high affinity site, and y a term describing the low affinity site.
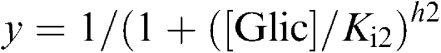 |
where [Glic] is the concentration of gliclazide (μM), Ki1, Ki2 are the gliclazide concentrations at which inhibition is half maximum at high- and low-affinity sites, respectively; h1, h2 are the Hill coefficients for the high- and low-affinity sites, respectively; and L is the fractional current amplitude remaining when the high-affinity sites are fully occupied. When only a single site is present, the equation reduces to I/Ic=x.
Statistics
Statistical analyses were performed with a two-paired t-test (two-factor with replication). Changes were considered significant at P<0.05.
RNA preparation and RT-PCR analysis
For RNA isolation, total RNA from pig urethral smooth muscle was isolated using the TRIzol reagent according to the manufacturer's instructions (RNeasy Mini Kit, QIAGEN K.K., Tokyo, Japan). First-strand synthesis of cDNA using random hexamers was carried out as follows: DNase I-treated total RNA (0.5–1.0 μg) isolated from tissues was incubated with random hexamers at 70°C for 10 min and then with PCR buffer (20 mM Tris/HCl, pH 8.4, 50 mM KCl), 2.5 mM MgCl2, 0.5 mM deoxynucleoside-5′-triphosphate, and 10 mM dithiothreitol at 25°C for 5 min. RT-PCR was initiated by the addition of Superscript II RT (200 U) at 25°C for 10 min followed by incubation at 42°C for 50 min. The reaction was terminated by incubation with RNase H at 70°C for 15 min, before chilling on ice. PCR was performed using 1 μl of cDNA in a 12.5 μl reaction containing 0.4 μM concentration of each primer, 200 μM concentration of each deoxynucleoside-5′-triphosphate, and 0.5 U of Taq polymerase (Takara Co. Ltd, Kyoto, Japan). The cycling conditions were 94°C for 25 s, 54°C for 25 s, and 72°C for 25 s for 40 cycles. An aliquot (10 μl) of the RT–PCR product was analysed on a 3% Tris-borate-EDTA polyacrylamide gel. Since no sequence information is available for KATP channel subunits in pig, generic subunit-specific primers were designed based on information from rat, mouse, and human sequences. The locations of the primers indicated are based on the subunit sequence information obtained from GenBank: SUR1 (human, L78207), and SUR2 (rat 2A, D83598; mouse 2A, D86037; mouse 2B, D86038; human 2A, AF061323; human 2B, AF061324). The sequences of the primers were as follows: for SUR1 (forward) 5′-GCGTGCAAAAGCTAAGCGAG-3′ and (reverse) 5′-GAC-GCTTGCGGTTCACAAC-3′ (corresponding to nucleotide positions 1817–1836 and 1951–1933, respectively); for SUR2A/B (forward) 5′-GCTGAAGAATATGGTCAAATC-TC-3′ and (reverse) 5′-CGGAGTGTCGTATTCCAAAATA-3′ (corresponding to nucleotide positions, 4278–4300 and 4590–4569, respectively). Control reactions were carried out where samples in the absence of reverse transcriptase were amplified to ensure that the detected product was not the result of possible DNA contamination and by use of corresponding templates as positive controls to ensure that the primers were annealing successfully. These primers gave products of the expected sizes that were confirmed by DNA sequence analysis.
Results
Comparison between the diazoxide-induced inward current and the pinacidil-induced inward current in pig urethral myocytes by use of nystatin-perforated patch
In order to investigate pharmacological properties of KATP channels in pig urethra, two distinct types of well-known KATP channel openers (diazoxide, a SUR1 activator; pinacidil, a selective SUR2 activator; reviewed by Babenko et al., 1998; Fujita & Kurachi, 2000) were utilized by use of the nystatin-perforated patch. After the establishment of the nystatin-perforated patch, approximately 15 min later, cumulative application of diazoxide (100–500 μM) caused an inward current in a concentration-dependent manner at a holding potential of −50 mV (bath solution, 140 mM K+ solution; pipette solution, 140 mM KCl solution containing 5 mM EGTA; that is, symmetrical 140 mM K+ conditions, Figure 1). After diazoxide was removed, the current recovered to the control level. Subsequently, application of pinacidil (100 μM) caused an inward current. Figure 1b shows the relation between the concentrations of each KATP channel opener (diazoxide and pinacidil) and the peak amplitude of each KATP channel opener-induced inward currents when the peak amplitude of the 100 μM pinacidil-induced current was taken as one. As shown in Figure 1c, additional application of diazoxide (500 μM) further enhanced the 10 μM pinacidil-induced currents. Similarly, pinacidil caused an additional effect on the diazoxide-induced current (Figure 1d).
Figure 1.

Comparison of the potency of diazoxide and pinacidil in activation of KATP membrane currents at −50 mV in pig urethra. Nystatin-perforated patch configuration; the bath contained a 140 mM K+ solution and pipette 140 mM KCl containing 5 mM EGTA (i.e., symmetrical 140 mM K+ conditions). (a) Diazoxide (100 – 500 μM) caused an inward membrane current in a concentration-dependent manner. On removal of diazoxide, the current recovered to the control level. Subsequent application of pinacidil (100 μM) caused an inward current. The dashed line indicates zero current level. (b) The relation between the concentrations of KATP channel openers and the relative peak amplitude of KATP channel opener-induced membrane currents in pig urethra. Note that the peak current amplitude was measured from the level before application of KATP channel openers and normalized with respect to the 100 μM pinacidil-induced current from the same dispersed myocytes. The following values were used for the curve fitting: A=1.1, K=15 μM, nH=1.2. The line for the diazoxide-induced current was drawn by eye. Each symbol indicates the mean of 3–10 observations with ±s.d. shown by vertical lines. (c) Diazoxide (500 μM) enhanced the 10 μM pinacidil-induced current. The dashed line indicates zero current level. (d) The effects of diazoxide and pinacidil were additive. The different symbols represent four separated experiments. The arrow between points indicates the order of application of the KATP channel openers (diazoxide and pinacidil) onto the same dispersed cell. Asterisks indicate a statistically significant difference, demonstrated using a paired t-test (P<0.05).
Differential sensitivity of KATP channel opener-induced membrane currents to gliclazide in pig urethra
In order to further investigate whether the SUR1 subunit may regulate the activity of KATP channels in pig urethra, gliclazide, a specific SUR1 blocker (Gribble & Ashcroft, 1999), was used. The concentration of each KATP channel opener, which caused a similar potency for the current activation on the membrane currents was chosen (10 μM pinacidil and 500 μM diazoxide). As shown in Figure 2a, application of 10 μM gliclazide, a selective SUR1 blocker, caused a small inhibitory effect on the pinacidil (10 μM)-induced current (0.94±0.07, n=4). Further application of gliclazide (100–1000 μM) inhibited the pinacidil-induced current at −50 mV (Figure 2c, 10 μM pinacidil, Ki=177 μM; 100 μM pinacidil, Ki=285 μM). In contrast, 10 nM gliclazide reduced the 500 μM diazoxide-induced current amplitude by approximately 18% (0.82±0.09, n=5, P<0.01) and higher concentrations of gliclazide inhibited the diazoxide-induced current in a concentration-dependent manner, demonstrating two inhibitory sites: a high-affinity site (Ki1=5 nM) and a low-affinity site (Ki2=108 μM) in Figure 2c. Furthermore, as shown in Figure 3a, the application of tolbutamide, a SUR1 inhibitor, caused an inhibition of the 500 μM diazoxide-induced current at –50 mV, demonstrating a concentration-dependent manner (Figure 3c). However, lower concentrations of tolbutamide (30–100 μM) had little effect on the 10 μM pinacidil-induced current. As shown in Figure 2 and Figure 3, occasionally, a small amount of spontaneous transient inward currents were observed at −50 mV. The spontaneous transient inward currents were selectively inhibited by additional application of 100 nM iberiotoxin (data not shown).
Figure 2.

Inhibitory effects of gliclazide on (a) 10 μM pinacidil- and (b) 500 μM diazoxide- induced inward membrane currents at −50 mV in symmetrical 140 mM K+ conditions by use of nystatin-perforated patch configuration. The dashed line indicates zero current. (c) Relation between relative inhibition of the peak amplitude of each KATP channel opener (pinacidil (10 and 100 μM) and 500 μM diazoxide)-induced current and the concentration of gliclazide. The following values were used for the curve fitting: 10 μM pinacidil, Ki=177 μM, h=0.8; 100 μM pinacidil, Ki=285 μM, h=0.8; diazoxide, Ki1=5 nM, h1=1.2; Ki2=108 μM, h2=0.7, L=0.74. Each symbol indicates the mean of 3–8 observations with s.d. shown by vertical lines.
Figure 3.

The effects of tolbutamide (30, 100 μM) on (a) 500 μM diazoxide- and (b) 10 μM pinacidil- induced inward membrane currents at −50 mV in symmetrical 140 mM K+ conditions by use of nystatin-perforated patch configuration. The dashed line indicates zero current. (c) Tolbutamide (30–500 μM) inhibited the 500 μM diazoxide- and the 10 μM pinacidil-induced inward membrane current at −50 mV in a concentration-dependent manner. Each column indicates the mean 4–5 observations with +s.d.
Relations between peak amplitude of MCC-134-induced currents and concentrations of MCC-134
In order to further investigate whether the SUR1 subunit may regulate the activity of KATP channels in pig urethra, MCC-134, a novel KATP channel modulator acting through distinct types of SURs (Shindo et al., 2000), was used. When the concentration of MCC-134 was increased cumulatively (from 10 to 100 μM), the peak amplitude of the MCC-134-induced basal membrane current at −50 mV increased in a concentration-dependent manner (Figure 4a). On removal of MCC-134, subsequent application of 100 μM pinacidil caused a larger amplitude inward current, which was suppressed by 5 μM glibenclamide. Further studies were carried out on the effects of the two different types of drugs (MCC-134 and pinacidil) using triangular ramp potential pulses (see inset in Figure 4a). Three of these were applied from −120 to +80 mV, obtaining the current–voltage relations in the absence and presence of each drug. Figure 4b shows the averaged membrane currents during the falling phase of the ramp pulses under the various experimental conditions. The MCC-134-induced membrane current was obtained by subtracting the averaged control current from the membrane current in the presence of 100 μM MCC-134 and demonstrated an inwardly rectifying property at positive potentials (Figure 4c). The pinacidil-induced membrane current was obtained by subtracting the averaged control current from the membrane current in the presence of pinacidil, and showed a similar inwardly rectifying property at positive potentials (Figure 4c). Figure 4d shows the relation between the concentrations of MCC-134 and the peak amplitude of the MCC-134-induced inward currents when the peak amplitude of the 100 μM pinacidil-induced current was taken as one. The MCC-134-induced inward current was suppressed by additional application of 5 μM glibenclamide (Figure 5a). However, a low concentration of gliclazide (1 μM), a selective SUR1 blocker, had no effect on the 100 μM MCC-134-induced inward current (0.99±0.03, n=4) when the peak amplitude of the MCC-134-induced current was normalized as one just before application of 100 μM MCC-134 (i.e., control). In each condition, current–voltage relations were obtained by applying four ramp pulses (from −120 to +80 mV for 1 s duration, see inset in Figure 5a) every 15 s at −50 mV (Figure 5b). Figure 5c demonstrates the net membrane current obtained by subtraction of the two mean ramp currents recorded before and during application of 5 μM glibenclamide in the presence of 100 μM MCC-134, demonstrating an inward rectification at positive potentials.
Figure 4.

Concentration-dependent effects of MCC-134 on the membrane currents at −50 mV in pig urethra. Whole-cell recording, the bath contained a 140 mM K+ solution and pipette 140 mM KCl containing 5 mM EGTA (i.e., symmetrical 140 mM K+ conditions). (a) MCC-134 caused an inward membrane current in a concentration-dependent manner. After MCC-134 was removed, the current recovered to the control level. Pinacidil induced an inward current, which was suppressed by 5 μM glibenclamide. The dashed line indicates zero current level. (b) The current–voltage curves measured from the negative-going limb (the falling phase) of the ramp pulse. Each symbol is the same as in the current trace. The lines are mean membrane currents from the three ramps in each condition. (c) The MCC-134-induced membrane current was obtained by subtraction of the two mean ramp currents recorded before and during application of 100 μM MCC-134. The pinacidil-induced current was obtained from the membrane currents in the absence and presence of 100 μM pinacidil. (d) The relation between the concentrations of MCC-134 and the relative peak amplitude of the MCC-134-induced membrane currents. The peak current amplitude was measured from the level in the presence of 5 μM glibenclamide and normalized with respect to the 100 μM pinacidil-induced current from the same dispersed cells. The curve was drawn by fitting the equation using the least-squares method at concentrations between 1 and 100 μM. The following values were used for the curve fitting: A=0.57, K=23 μM, nH=1.6. Each symbol indicates the relative amplitude at each concentration of MCC-134 (1 μM, 0.04±0.03, n=5; 10 μM, 0.11±0.07, n=6; 30 μM, 0.34±0.20, n=3; 100 μM, 0.52±0.27, n=10).
Figure 5.

Effects of glibenclamide on the 100 μM MCC-134-induced membrane current at −50 mV in a conventional whole-cell recording (the bath contained a 140 mM K+ solution and pipette 140 mM KCl containing 5 mM EGTA, i.e., symmetrical 140 mM K+ conditions). (a) Inhibitory effects of 5 μM glibenclamide on the 100 μM MCC-134-induced inward current. The vertical deflections indicate triangle ramp potential pulses (see inset) applied every 15 s from -120 to 80 mV for 1 s after a 300 ms conditioning pulse. The dashed line indicates the zero current level. (b) The mean ramp membrane currents (the negative-going limb of the ramp pulse) on an expanded time scale in each condition. Symbols as in (a). (c) Difference between the mean MCC-134-induced membrane current in the presence and absence of 5 μM glibenclamide. Net membrane current (glibenclamide-sensitive current) was obtained by subtraction of the two ramp membrane currents recorded before and during application of 5 μM glibenclamide when 100 μM MCC-134 was present in the bath.
MCC-134 activates a glibenclamide-sensitive KATP channel in pig urethra
Single-channel recordings were performed by use of cell-attached configuration.. To minimize the activity of maxi K+ channels, experiments were performed at −50 mV. In Figure 6a, application of 1 μM MCC-134 evoked opening of a 2.1 pA K+ channel. In the presence of 100 μM MCC-134, current–voltage relations were obtained by changing the holding membrane potential from −120 to −10 mV in 10 mV increments. The results indicate that the conductance of the channel is approximately 43 pS (43±1 pS, n=8) with an inwardly rectifying property at positive membrane potentials (data not shown). Subsequent application of pinacidil (100 μM) caused opening of the same amplitude K+ channel (Figure 6a). When the NPo value in the presence of 100 μM pinacidil was normalized as 1.0, the relative NPo value in the presence of MCC-134 in the same membrane patches was 0.03±0.01 (10 μM, n=4) and 0.28±0.16 (100 μM, n=4) (Figure 6c). The channel opening was reversibly inhibited by 10 μM glibenclamide (data not shown).
Figure 6.

The MCC-134-induced opening of K+ channels recorded with cell-attached configuration at a holding membrane potential of −50 mV. (a) Comparison of the properties of the MCC-134 (1–100 μM) activated K+ channels and the 100 μM pinacidil induced K+ channels in the same membrane patch at −50 mV. The dashed line indicates the current when the channel is not open. (b) All-point amplitude histograms in the presence of 100 μM MCC-134 and 100 μM pinacidil (obtained during 1 min). Continuous lines in the histograms are theoretical curves fitted with the Gaussian distribution, by the least-squares method. The abscissa scale is the amplitude of the current (pA) and the ordinate scale is the percentage value of the probability density function (%) for the recording period (1 min). (c) Each column shows the relative level of the activity of the MCC-134-induced KATP channel (mean value with +s.d.) when the mean amplitude of the pinacidil-induced channel activity was normalized as one (n=4).
Molecular expression of SURs in pig urethra
In order to investigate the molecular component of SURs in pig urethral KATP channels, RT–PCR studies were performed. The specific primers for amplification of each SUR mRNA were designed to produce a cDNA fragment. As shown in Figure 7, both SUR1 (134 bp) and SUR2B (312 bp) mRNA were expressed in rat cDNA and pig urethra with the expected fragment size for SUR1 or SUR2B.
Figure 7.

RT–PCR detection of SUR1 and SUR2B. RT–PCR was performed as described in Methods and size markers were used to indicate the size of the amplified fragments. RT-PCR yielded visible amounts of SUR1 (134 bp fragment) and SUR2B (312 bp fragment) in mRNAs from rat cDNA and pig urethra.
Discussion
It is commonly assumed that SURs are responsible for differential pharmacological effects, depending on either the types of KATP channel openers used or three subtypes of SURs (SUR1, SUR2A, and SUR2B) present. In functional expression experiments, pharmacological and electrophysiological studies have reported that SUR1/Kir6.2 represents the pancreatic ß-cell KATP channel, that SUR2A/Kir6.2 is thought to represent the cardiac KATP channel, whereas SUR2B/Kir6.1 represents the smooth muscle-type KATP channel (reviewed by Babenko et al., 1998; Fujita & Kurachi, 2000). Previous work has shown that the SUR1/Kir6.2 channel is activated by diazoxide (EC50, 50 μM) but not by pinacidil (⩽1 mM); the SUR2A/Kir6.2 channel is strongly activated by pinacidil (EC50, 10 μM) but not responsive to diazoxide (⩽200 μM); and the SUR2B/Kir6.2 channel is activated by both agents (Babenko et al., 1998; Fujita & Kurachi, 2000).
Our present observations regarding the actions of diazoxide, pinacidil, gliclazide, tolbutamide, and MCC-134 on the pig urethral KATP channels can be summarized as follows: (1) Diazoxide, a SUR1 activator, and pinacidil, a selective SUR2 activator, induce glibenclamide-sensitive inward K+ currents. Similarly, by use of tension measurements, we have demonstrated that cumulative application of pinacidil and diazoxide produced a concentration-dependent relaxation of the resting urethral tone, which was suppressed by additional application of 1 μM glibenclamide (pinacidil, EC50=1.6 μM; diazoxide, EC50=71 μM; Teramoto & Ito, 1999). (2) Gliclazide, a selective antagonist for SUR1, reversibly inhibits the diazoxide- but not the pinacidil-induced currents at low concentrations (between 10 nM and 1 μM). At higher concentrations (⩾10 μM), this agent also suppressed pinacidil-induced currents. (3) Low concentrations of tolbutamide (30–100 μM), a selective SUR1 inhibitor, inhibited the 500 μM diazoxide-induced current and the 10 μM pinacidil-induced current with a different potency. (4) The inhibitory effects on not only the 500 μM diazoxide-induced current but also the 10 μM pinacidil-induced current were reversible. Low concentrations of tolbutamide (⩽100 μM) inhibit the activity of SUR1/Kir6.2 not but SUR2A/Kir6.2 and inhibition of SUR1/Kir6.2 by tolbutamide (⩽100 μM) is readily reversible on washout, indicating that the reversibility of the inhibition by tolbutamide is a good clue to identify SUR subtypes (Gribble et al., 1998). However, application of 500 μM tolbutamide also caused a reversible inhibition of the SUR2B/Kir6.2 current activated by 200 μM diazoxide (Isomoto et al., 1996). Thus, we can only exclude the possibility that SUR2A subunits are in pig urethral KATP channels. (5) MCC-134 induces a concentration-dependent glibenclamide-sensitive inward K+ current and activates glibenclamide-sensitive KATP channels, as assessed by single-channel recordings. These results strongly suggest that SUR1 as well as SUR2B play physiological roles in the regulation of the muscle tone in pig urethra.
In the present RT–PCR studies, we have been able to demonstrate the presence of SUR1 as well as SUR2B in pig urethral smooth muscle cells although these studies have addressed only the expression of mRNA transcripts. Similarly, both SUR1 and SUR2B are widely expressed in the detrusor smooth muscle of several species (guinea-pig, Gopalakrishnan et al., 1999; pig and human, Buckner et al., 2000) as well as rat pulmonary vascular smooth muscle (Cui et al., 2002) as determined by RT–PCR analysis. However, in urinary bladder, it is concluded that SUR1 protein is not expressed at significant levels, since there was much less sensitivity to diazoxide in comparison to that of pancreatic β-cells and no high-affinity binding site for [3H]glyburide (Gopalakrishnan et al., 1999; Buckner et al., 2000). Thus, until our study, no functional study has yet been undertaken in smooth muscle KATP channels to establish the involvement of SUR1 subunits.
In the present experiments, gliclazide (⩽1 μM), a selective sulphonylurea drug for SUR1, reversibly inhibited the diazoxide-induced KATP current, but not the pinacidil- or the MCC-134-induced urethral KATP current. When the concentration of gliclazide is less than 1 μM, it is believed that gliclazide possesses selective inhibitory effects on SUR1, but not SUR2A or SUR2B, in recombinant KATP channels and that only much higher concentrations of gliclazide (⩾10 μM) directly block the channel pore region in KATP channels (Gribble & Ashcroft, 2000). Therefore, it is reasonable to assume that, in pig urethral KATP channels, gliclazide specifically inhibits SUR1 at relatively lower concentrations (⩽1 μM) and interacts with the channel pore at higher concentrations (⩾10 μM), demonstrating two affinity sites (the high- and low-affinity sites). However, gliclazide (⩾10 μM) suppressed pinacidil (10 and 100 μM)-induced current by an interaction with a single affinity site. Similarly, in rat mesenteric artery, the pinacidil-induced current was also blocked by gliclazide at a single site (Lawrence et al., 2001). These results suggest that pinacidil and MCC-134 may selectively activate SUR2B, but not SUR1 in pig urethra, coinciding with observations in recombinant KATP channels (pinacidil, Fujita & Kurachi, 2001; MCC-134, Shindo et al., 2000). In pig urethra, the peak amplitude of the 100 μM MCC-134-induced K+ currents at −50 mV was significantly smaller than that induced by pinacidil (100 μM) in the same dispersed cells, demonstrating that MCC-134 is less potent than pinacidil in activating KATP channels. Similarly, using the same membrane patches, the activity of the MCC-134-induced KATP channels was much less than that of the pinacidil-induced KATP channels in single-channel recordings. In contrast, the peak amplitude of the MCC-134 (100 μM)-induced channel current at recombinant SUR2B/Kir6.2 channels was much larger than that of the 100 μM pinacidil-induced K+ current (Shindo et al., 2000). We are not certain why these significant discrepancies exist between the native urethral KATP channels and the recombinant KATP channels regarding the effects of MCC-134. However, we suggest the possibility that MCC-134 may bind to both SUR1 (antagonistic action) and SUR2B (agonistic action) in urethral KATP channels, since the peak amplitude of the MCC-134-induced current was much smaller than that of the pinacidil-induced current because of its dual action.
In the present experiments, application of diazoxide enhanced the amplitude of the pinacidil-induced current at –50 mV, demonstrating an additional effect. Similarly, pinacidil caused a further increase in the diazoxide-induced current. These results also suggest the coexistence of SUR1 and SUR2B in pig urethral KATP channels. Yokoshiki et al. (1999) have reported that a small population of KATP channels in neonatal rat ventricular myocytes is constructed by a combination of SUR1 and SUR2A in antisense studies, suggesting the existence of heterotetrameric combinations. However, Giblin et al. (2002) have recently demonstrated biochemical and electrophysiological evidence that SUR1 and SUR2 do not interact to form heteromultimers in KATP channels. Since the sensitivity of gliclazide in pig urethral KATP channels reflected the reported pharmacological properties of SUR1, we suggest that SUR1 and SUR2B do not combine, but rather form homomultimeric subunits in pig urethral KATP channels. However, since the gliclazide-sensitive component was less than approximately 25% in the diazoxide-induced inward current, we suggest that SUR subunits in pig urethral KATP channels may be composed mainly of SUR2B and that those containing SUR1 play a minor but important role in regulating channel activity of urethral KATP channels.
In the present experiments, MCC-134 activated a 2.1 pA glibenclamide-sensitive K+ channel (i.e. KATP channel) at –50 mV, demonstrating the size of the channel conductance is 43 pS at negative potentials. The same KATP channels were activated by other types of KATP channel openers (levcromakalim, Teramoto & Brading, 1996; nicorandil, Teramoto & Brading, 1997; pinacidil, Teramoto et al., 2000; ZD6169, Teramoto et al., 2001) in pig urethra. The channel conductance of this KATP channel is intermediate between those of Kir6.1 and Kir6.2 (Babenko et al., 1998). In functional expression experiments, Cui et al. (2001) have recently reported that heteromultimerization readily occurs between Kir6.1 and Kir6.2, producing functional glibenclamide-sensitive KATP channels. Cui et al. (2001) also suggest a possibility that KATP channels in pig urethra may possess a heteromeric Kir6.1/Kir6.2 structure. Further study is certainly necessary in order to elucidate the interaction of Kir subunits in pig urethral KATP channels.
In conclusion, we have been able to demonstrate that SUR1 and SUR2B play a functional role in the activity of KATP channels in pig urethral myocytes using several pharmacological and molecular approaches.
Acknowledgments
We thank Professor A.F. Brading (University Department of Pharmacology, Oxford, U.K.) and Dr N.J. Bramich (University of Melbourne, Department of Zoology, Melbourne, Australia) for their critical reading of the manuscript. We are grateful to the Mitsubishi Pharma Corporation (Tokyo, Japan) for the generous gift of MCC-134. Gliclazide was kindly provided by the Dai-Nippon Pharmaceuticals Co. Ltd. (Osaka, Japan). We thank Professor J. Bryan (Department of Medicine, Baylor College of Medicine, Houston, U.S.A.) for generously providing the SUR1 clone. We also thank Dr M. Takano (Department of Physiology and Biophysics, Graduate School of Medicine, Kyoto University, Japan) for generously providing the SUR2B clone. This work was supported by a grant from the Kaibara Morikazu Medical Science Promotion Foundation (Fukuoka, Japan) and the AstraZeneca Research Grant 2001 (Osaka, Japan), awarded to Noriyoshi Teramoto and from a Grant-in-Aid for Scientific Research by the Ministry of Education, Science, Sports and Culture of Japan (Grant number 14540080). Takakazu Yunoki is a research fellow of the Japanese Society for the Promotion of Science (Grant number 14DC2-08992).
Abbreviations
- DMSO
dimethylsulphoxide
- Kir
inwardly-rectifying K+ channel
- KATP channels
ATP-sensitive K+ channels
- MCC-134
1-[4-(1H-imidazol-1-yl)benzoyl]-N-methyl-cyclobutanecarbothioamide
- NDP
nucleoside diphos-phates
- RT–PCR
reverse transcriptase–polymerase chain reaction
- SUR
sulphonylurea receptor
References
- AGUILAR-BRYAN L., NICHOLS C.G., WECHSLER S.W., CLEMENT J.P., IV, BOYD A.E., GONZÁLEZ G., HERRERA-SOSA H., NGUY K., BRYAN J., NELSON D.A. Cloning of the β cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–425. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- BABENKO A.P., AGUILAR-BRYAN L., BRYAN J. A view of SUR/KIR6.x, KATP channels. Annu. Rev. Physiol. 1998;60:667–687. doi: 10.1146/annurev.physiol.60.1.667. [DOI] [PubMed] [Google Scholar]
- BUCKNER S.A., MILICIC I., DAZA A., DAVIS-TABER R., SCOTT V.E., SULLIVAN J.P., BRIONI J.D. Pharmacological and molecular analysis of ATP-sensitive K+ channels in the pig and human detrusor. Eur. J. Pharmacol. 2000;400:287–295. doi: 10.1016/s0014-2999(00)00388-5. [DOI] [PubMed] [Google Scholar]
- CUI Y., GIBLIN J.P., CLAPP L.H., TINKER A. A mechanism for ATP-sensitive potassium channel diversity: Functional coassembly of two pore-forming subunits. Proc. Natl. Acad. Sci. U.S.A. 2001;98:729–734. doi: 10.1073/pnas.011370498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUI Y., TRAN S., TINKER A., CLAPP L.H. The molecular composition of KATP channels in human pulmonary artery smooth muscle cells and their modulation by growth. Am. J. Respir. Cell Mol. Biol. 2002;26:135–143. doi: 10.1165/ajrcmb.26.1.4622. [DOI] [PubMed] [Google Scholar]
- FUJITA A., KURACHI Y. Molecular aspects of ATP-sensitive K+ channels in the cardiovascular system and K+ channel openers. Pharmacol. Ther. 2000;85:39–53. doi: 10.1016/s0163-7258(99)00050-9. [DOI] [PubMed] [Google Scholar]
- GIBLIN J.P., CUI Y., CLAPP L.H., TINKER A. Assembly limits the pharmacological complexity of ATP-sensitive potassium channels. J. Biol. Chem. 2002;277:13717–13723. doi: 10.1074/jbc.M112209200. [DOI] [PubMed] [Google Scholar]
- GOPALAKRISHNAN M., WHITEAKER K.L., MOLINARI E.J., DAVIS-TABER R., SCOTT V.E., SHIEH C.C., BUCKNER S.A., MILICIC I., CAIN J.C., POSTL S., SULLIVAN J.P., BRIONI J.D. Characterization of the ATP-sensitive potassium channels (KATP) expressed in guinea pig bladder smooth muscle cells. J. Pharmacol. Exp. Ther. 1999;289:551–558. [PubMed] [Google Scholar]
- GRIBBLE F.M., ASHCROFT F.M. Differential sensitivity of beta cells and extrapancreatic KATP channels to gliclazide. Diabetologia. 1999;42:845–848. doi: 10.1007/s001250051236. [DOI] [PubMed] [Google Scholar]
- GRIBBLE F.M., TUCKER S.J., SEINO S., ASHCROFT F.M. Tissue specificity of sulfonylureas: studies on cloned cardiac and beta-cell KATP channels. Diabetes. 1998;47:1412–1418. doi: 10.2337/diabetes.47.9.1412. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT IV J.P., NAMBA N., INAZAWA J., GONZALEZ G., AGUILAR-BRYAN L., SEINO S., BRYAN J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- ISOMOTO S., KONDO C., YAMADA M., MATSUMOTO S., HIGASHIGUCHI O., HORIO Y., MATSUZAWA Y., KURACHI Y. A novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+ channel. J. Biol. Chem. 1996;271:24321–24324. doi: 10.1074/jbc.271.40.24321. [DOI] [PubMed] [Google Scholar]
- LAWRENCE C.L., PROKS P., RODRIGO G.C., JONES P., HAYABUCHI Y., STANDEN N.B., ASHCROFT F.M. Gliclazide produces high-affinity block of KATP channels in mouse isolated pancreatic beta cells but not rat heart or arterial smooth muscle cells. Diabetologia. 2001;44:1019–1025. doi: 10.1007/s001250100595. [DOI] [PubMed] [Google Scholar]
- SCHWANSTECHER M., SIEVERDING C., DÖRSCHNER H., GROSS I., AGUILAR-BRYAN L., SCHWANSTECHER C., BRYAN J. Potassium channel openers require ATP to bind and act through sulfonylurea receptors. EMBO J. 1998;17:5529–5535. doi: 10.1093/emboj/17.19.5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHINDO T., KATAYAMA Y., HORIO Y., KURACHI Y. MCC-134, a novel vascular relaxing agent, is an inverse agonist for the pancreatic-type ATP-sensitive K+ channel. J. Pharmacol. Exp. Ther. 2000;292:131–135. [PubMed] [Google Scholar]
- TERAMOTO N., BRADING A.F. Activation by levcromakalim and metabolic inhibition of glibenclamide-sensitive K channels in smooth muscle cells of pig proximal urethra. Br. J. Pharmacol. 1996;118:635–642. doi: 10.1111/j.1476-5381.1996.tb15448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERAMOTO N., BRADING A.F. Nicorandil activates glibenclamide-sensitive K+ channels in smooth muscle cells of pig proximal urethra. J Pharmacol. Exp. Ther. 1997;280:483–491. [PubMed] [Google Scholar]
- TERAMOTO N., BRADING A.F., ITO Y. Possible underestimation of the channel conductance underlying pinacidil-induced K+ currents using noise analysis in pig urethral myocytes. J. Pharm. Pharmacol. 2000;52:1395–1403. doi: 10.1211/0022357001777397. [DOI] [PubMed] [Google Scholar]
- TERAMOTO N., CREED K.E., BRADING A.F. Activity of glibenclamide-sensitive K+ channels under unstimulated conditions in smooth muscle cells of pig proximal urethra. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:418–424. doi: 10.1007/pl00005071. [DOI] [PubMed] [Google Scholar]
- TERAMOTO N., ITO Y. Comparative studies on the relaxing action of several adenosine 5′-triphosphate-sensitive K+ channel openers in pig urethra. J. Smooth Muscle Res. 1999;35:11–22. doi: 10.1540/jsmr.35.11. [DOI] [PubMed] [Google Scholar]
- TERAMOTO N., YUNOKI T., TAKANO M., YONEMITSU Y., MASAKI I., SUEISHI K., BRADING A.F., ITO Y. Dual action of ZD6169, a novel K+ channel opener, on ATP-sensitive K+ channels in pig urethral myocytes. Br. J. Pharmacol. 2001;133:154–164. doi: 10.1038/sj.bjp.0704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMADA M., ISOMOTO S., MATSUMOTO S., KONDO C., SHINDO T., HORIO Y., KURACHI Y. Sulphonylurea receptor 2B and Kir6.1 form a sulphonylurea-sensitive but ATP-insensitive K+ channel. J. Physiol. 1997;499:715–720. doi: 10.1113/jphysiol.1997.sp021963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOKOSHIKI H., SUNAGAWA M., SEKI T., SPERELAKIS N. Antisense oligodeoxynucleotides of sulfonylurea receptors inhibit ATP-sensitive K+ channels in cultured neonatal rat ventricular cells. Pflügers Arch. 1999;437:400–408. doi: 10.1007/s004240050794. [DOI] [PubMed] [Google Scholar]