

Abstract
Pharmacological blockade of the Human ether-a-go-go related gene (HERG) potassium channel is commonly linked with acquired long QT syndrome and associated proarrhythmia. The objectives of this study were (i) to identify and characterise any inhibitory action on HERG of the selective-serotonin re-uptake inhibitor fluvoxamine, (ii) to then determine whether fluvoxamine shared the consensus molecular determinants of HERG blockade of those drugs so far tested.
Heterologous HERG potassium current (IHERG) was measured at 37°C, using the whole-cell patch-clamp technique, from a mammalian cell line (Human embryonic kidney 293) expressing HERG channels. IHERG tails, following repolarisation from +20 to −40 mV, were blocked by fluvoxamine with an IC50 of 3.8 μM.
Blockade of wild-type HERG was of extremely rapid onset (within 10 ms) and showed voltage dependence, with fluvoxamine also inducing a leftward shift in voltage-dependent activation of IHERG. Characteristics of block were consistent with a component of closed channel (or extremely rapidly developing open channel) blockade and dependence on open and inactivated channel states. The attenuated-inactivation mutation S631A partially reduced the blocking effect of fluvoxamine.
The S6 mutations, Y652A and F656A, and the pore helix mutant S631A only partially attenuated blockade by fluvoxamine at concentrations causing profound blockade of wild-type HERG.
All HERG-blocking pharmaceuticals studied to date have been shown to block F656 mutant channels with over 100-fold reduced potency compared to their blockade of the wild-type channel. Fluvoxamine is therefore quite distinct in this regard from previously studied agents.
Keywords: Acquired long QT syndrome, arrhythmia, HERG, fluvoxamine, IKr, QT interval, ‘rapid' delayed rectifier, selective-serotonin re-uptake inhibitor, SSRI
Introduction
In ventricular myocytes of the heart, pharmacological blockade of the ‘rapid' delayed rectifier potassium (K+) current (IKr) and current carried by its cloned equivalent, Human ether-a-go-go related gene (HERG) (Sanguinetti et al., 1995; Trudeau et al., 1995), can lead to impaired membrane potential repolarisation and consequently to excessive ventricular action potential prolongation (Sanguinetti & Keating, 1997; Crumb & Cavero, 1999; Haverkamp et al., 2000). Often, the clinical result of this delayed repolarisation is acquired long QT syndrome, which is associated with a risk of the polymorphic ventricular tachyarrhythmia Torsades de pointes and potentially with sudden death (Viskin, 1999). The number of pharmaceutical agents with diverse chemical structures and pharmacological actions found to block HERG is growing, so much so that it is now a major consideration in drug design generally (Committee for Proprietary Medicinal Products, 1997; Taglialatela et al., 1998; Crumb & Cavero, 1999; Gralinski, 2000; Witchel & Hancox, 2000; Vandenberg et al., 2001).
One possible explanation for this pharmacological ‘promiscuity' is that the HERG channel has a larger vestibule within the conducting pore itself than other 6-transmembrane domain (6-TM) K+ channels (i.e. Shaker-like channels). Structure-function studies (Lees-Miller et al., 2000; Mitcheson et al., 2000) as well as analogy with the known crystal structure of the bacterial K+ channel KcsA (Doyle et al., 1998) have suggested that the high-affinity drug-binding sites are within the vestibule. The absence of highly conserved prolines (normally present in 6-TM K+ channels) at HERG amino-acid positions 655 and 657 may suggest that a ‘kink', which is present in most 6-TM K+ channels, is absent from this part of the HERG subunit. This absence may result in an increase in space within the vestibule. A potential effect of this putative increase in space inside the vestibule of HERG would be the ability of a wide variety of drugs to gain access to binding sites inside the pore.
Using alanine scanning mutagenesis of the pore-S6 region of HERG (i.e. the inner lining of the pore and vestibule), the molecular determinants of blockade of HERG have been determined for a variety of drugs, including the high-potency blocking methanesulphonanilides and the low-potency blockers, vesnarinone and chloroquine (Lees-Miller et al., 2000; Mitcheson et al., 2000; Kamiya et al., 2001; Sanchez-Chapula et al., 2002). All such studies have shown that mutation of the phenylalanine amino-acid residue at position 656 (F656) in the S6 region to either valine or alanine dramatically attenuates drug-mediated block of HERG, increasing the IC50 for these drugs by over 100-fold. Furthermore, mutation of Y652 to an alanine, in all but one case (Kamiya et al., 2001), leads to an equally dramatic attenuation of drug blockade of HERG.
Understanding the relative cardiotoxic risks associated with different antidepressants is important because the patients prescribed these drugs can be subject to overdose and self-poisoning (Henry, 1997). Despite the fact that selective-serotonin re-uptake inhibitors (SSRIs) such as fluvoxamine have been reported to compare favourably with tricyclic antidepressants (TCAs) in terms of risk of cardiotoxicity (Roos, 1983; Wouters & Deiman, 1983; Feighner et al., 1989; Laird et al., 1993; Hewer et al., 1995), recent reports have shown that the SSRIs, fluoxetine and citalopram, are low-potency blockers (half-maximal inhibitory drug concentration (IC50) values in μM range) of HERG channels (Thomas et al., 2002; Witchel et al., 2002). In fact, there are also a few observations associating fluvoxamine with the rate-corrected QT interval of the electrocardiogram (QTc) interval prolongation (Ohtani et al., 2001; Rodriguez de la Torre et al., 2001) and ventricular arrhythmia (Manet et al., 1993), particularly at excessively high drug concentrations. These rare observations might be explained if fluvoxamine can also block HERG. Fluvoxamine (see Figure 1), the first SSRI of the class of 2-aminoethyloximethers of aralkylketones, is structurally distinct from both fluoxetine and citalopram in that it lacks an asymmetric carbon in its structure and possesses an internal plane of symmetry (in contrast to fluoxetine and citalopram, it lacks enantiomers). In a preliminary report, we found that fluvoxamine, like citalopram and fluoxetine, is also a low-potency blocker of HERG (Milnes et al., 2002). In the present study, we investigated the effects of fluvoxamine on current mediated by HERG channels (IHERG) expressed in mammalian cell lines, with the aims: (a) to characterise the nature of the inhibitory action on IHERG by fluvoxamine and (b) to determine whether or not key molecular determinants of HERG blockade for previously investigated drugs (Mitcheson et al., 2000) are also important for inhibition of IHERG by fluvoxamine.
Figure 1.

Chemical structure of the SSRI, fluvoxamine.
Methods
Creation and maintenance of mammalian cell lines stably expressing HERG and HERG mutants
Experiments on wild-type HERG were performed on a cell line (Human embryonic kidney; HEK 293) stably expressing HERG (generously donated by Dr Craig January, University of Wisconsin (Zhou et al., 1998)), or another line stably expressing lower levels of HERG developed in this lab. HEK 293 cell lines stably expressing HERG and its mutants, F656A and Y652A, were created using standard techniques by subcloning appropriately mutated HERG sequences into a HERG expression vector (based on pIRES1hyg) into the BstEII/Sse8387I sites of HERG; the expression constructs were transfected using Fugene (Roche Diagnostics) into HEK 293 cells, selected, subcloned, and assayed for HERG expression by immunofluorescence (using Alomone APC-016) followed by electrophysiological validation. Treatment of these mutant cell lines was identical to treatment of the wild-type cell line except that cultures were maintained with 100 μg ml−1 of hygromycin instead of 400 μg ml−1 G418. Cells transiently transfected with S631A were made as previously described (Paul et al., 2001). Cells were passaged using a non-enzymatic agent (Splittix, AutogenBioclear) and plated out onto small sterilised glass coverslips in 30 mm petri dishes containing a modification of Dulbecco's modified Eagle's medium with Glutamax-1 (DMEM; Gibco, Gibco/Invitrogen, Paisley, U.K.), supplemented with 10% foetal bovine serum (Gibco), 400 μg ml−1 gentamycin (Gibco) and 400 μg ml−1 geneticin (G418; Gibco). The cells were incubated at 37°C for a minimum of 2 days prior to any electrophysiological study.
Electrophysiological recordings
The glass coverslips were placed in a bath (0.5 ml volume) mounted on an inverted microscope (Nikon Diaphot) and the cells were superfused with Normal Tyrode's solution which contained (in mM): 140 NaCl, 4 KCl, 2.5 CaCl2, 1 MgCl2, 10 Glucose and 5 HEPES (titrated to pH 7.45 with NaOH). In Tyrode's solutions containing elevated external potassium ([K+]o), elevated K+ was compensated for by a corresponding reduction in external Na+. Drugs were also added to this to make up test solutions at the final concentrations mentioned in the ‘Results' text. Patch pipettes (Corning 7052 glass, AM Systems Inc.) were pulled to resistances of 1–2 MΩ (Narishige PP83) and fire-polished to 3–5 MΩ (Narishige, MF83). The internal dialysis solution contained (in mM): 130 KCl, 1 MgCl2, 5 EGTA, 5 MgATP, 10 HEPES (titrated to pH 7.2 with KOH). The ‘pipette-to-bath' liquid junction potential was measured for this filling solution and was −3.2 mV. Since this value was small, no corrections of membrane potential were made. Whole-cell patch-clamp recordings of membrane currents were made using an Axopatch 1D amplifier (Axon Instruments) and a CV-4 1/100 headstage. Between 75 and 90% of the electrode series resistance could be compensated. Voltage-clamp commands were generated using ‘WinWCP', a program written and supplied free of charge by John Dempster of Strathclyde University or Clampex 8 (Axon Instruments). Data were recorded via a Digidata interface (Axon Instruments) and stored on the hard disk of a Viglen computer prior to analysis. Data digitization rates were 2 kHz during all protocols with the exception of inactivation protocols (not shown) that were digitized at 12 kHz (the maximum rate possible for these protocols with the Strathclyde software) and those described in Figure 5. Bandwidths of 2–10 kHz were set on the amplifier.
Figure 5.

State-dependence of block. Panel (a) shows representative IHERG traces recorded prior to and the first record following equilibration in 3 μM fluvoxamine, during which period membrane potential was held at −100 mV to maintain all channels in the closed state. Membrane potential was then briefly stepped to +40 mV for 10 ms prior to repolarisation to −40 mV to evoke IHERG tails. Fractional block of IHERG tails is summarised in panel (aii) (10 ms, n=4) and for comparison is shown alongside fractional block data taken from the concentration–response (unpaired t-test, P>0.15, NS denotes non-significance). Panel (b) To further investigate closed vs rapid open-state block, fluvoxamine concentration was reduced by 10-fold in order to reduce the rate of drug-channel association to the open state. Experiments were performed as in (ai) with the addition of a further trace in which membrane potential was stepped to +40 mV for 200 ms to investigate time-dependent changes in block. Data are summarised in panel (bii) showing fractional block following 10 and 200 ms activating steps. For comparison, fractional block data for 0.3 μM fluvoxamine is shown from the concentration–response relation. (ANOVA, P>0.6; NS denotes non-significance). Data from panels (a) and (b) are concordant with a component of closed state, although a component of very rapid open-state block cannot be excluded. Panel (c) shows a protocol similar to that described in Figure 4a with an additional 4 s step to +80 mV to produce profound IHERG inactivation. Representative IHERG traces are shown in the absence and presence of 3 μM fluvoxamine; plotted below is fractional block for that cell. Mean fractional block data are shown in 100 ms intervals in (cii) (n=4 cells). Bar indicates step to +80 mV. No significant change in block was observed following profound inactivation (ANOVA, P>0.9), suggesting that fluvoxamine's interaction with HERG is not hindered by strong inactivation. Panel (di) shows summarised data from experiments in which the fraction of inactive channels (Fi) following a 150 ms activating step from −80 to 0 mV was altered by elevating [K+]o from 4 to 30 mM (n=5 and 4, respectively, unpaired t-test) (see ‘sampling protocol' ‘Methods'; this time point was chosen as by 150 ms Fi had reached steady-state). Panel (dii) summarises the effect of modulating Fi on fractional block of the inactivation relieved current (I2, see ‘Methods') using an identical ‘sampling protocol' with an activating step to 0 mV for 150 ms. Reducing Fi by elevating [K+]o resulted in a significant reduction in fractional block of IHERG by 3 μM fluvoxamine (unpaired t-test, P<0.05).
On achieving the whole-cell configuration, pulses were applied until current amplitudes stabilised; experiments then commenced. The actions of fluvoxamine on HERG were rapid, with steady-state effects observed within four to five pulses (i.e. ∼1 min). Following the initial period of stabilisation, typical experimental recording periods were no longer than 5 min. Consequently, rundown of IHERG was small (∼10–15%) over the experimental period and was not corrected for. All experiments were carried out at 37±1°C.
Voltage command protocols are as described in the ‘Results' section, with the additional details as follows. Rapid time course of recovery from inactivation/deactivation: hold −80 mV, step to +40 mV for 500 ms, step to −100 mV for time durations between 1 and 10 ms, and step to +40 mV for 500 ms (during this step, peak tail current is measured). Voltage dependence of inactivation: holding potential of −80 mV, step to +40 mV for 500 ms, brief repolarisation to −100 mV for 4 ms followed by a second depolarising pulse to a range of potentials from −40 to +30 mV for a further 300 ms (during this step, peak tail current is measured).
Modulating the fraction of inactivated channels, by elevating external [K+], and studying the effect of this on fluvoxamine, an approach recently described by Weerapura et al. (2002) was employed to study the inactivated state as a target for fluvoxamine action. A modified envelope of tails (‘sampling') protocol was used, in which resurgent currents after rapid recovery from inactivation were elicited at the same potential as channel activation. Membrane potential was stepped from a holding potential of −80 to 0 mV for progressively greater durations (0–150 ms in 10 ms steps) to allow incremental channel activation/inactivation. It was then stepped to −100 mV (2 and 4 ms in 4 and 30 mM external K+, respectively) to fully relieve inactivation, while only nominal deactivation occurred, prior to depolarisation to 0 mV for 350 ms to elicit large, inactivation-relieved currents prior to a return to −80 mV. With this protocol, data were acquired at a sampling rate of 50 kHz, with the bandwidth set at 10 kHz on the amplifier. Currents were analysed to determine (1) the fraction of inactivated channels, (2) the effect of modulating this fraction by raising [K+]o on the fluvoxamine block as described by Weerapura et al. (2002). Briefly, measurement of IHERG at 0 mV prior to repolarisation to −100 mV (I1) allowed measurement of the IHERG component in the presence of inactivation. Following the step back to 0 from −100 mV, large resurgent tail currents were fitted with a mono-exponential function and extrapolated back to the beginning of the depolarisation step. This extrapolated current (I2) represents the current passing through inactivation-free channels, where I1 corresponds to current in the presence of inactivation. At 0 mV, the fraction of inactivated channels (Fi) can be calculated as
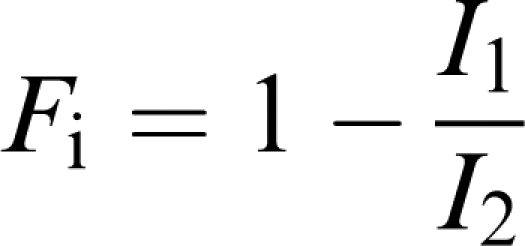 |
Having established stable currents in Tyrode's solution containing either 4 or 30 mM [K+]o, cells were superfused with fluvoxamine. An identical three pulse sampling protocol in which cells were stepped from −80 to 0 mV for 150 ms (at which time Fi was at steady-state) was then applied in the presence of fluvoxamine, enabling investigation of the effect of reducing the component of inactivated channels on HERG block of I2 by fluvoxamine as per Weerapura et al. (2002).
F656A-containing cells were tested in a superfusate containing 96 mM K+, and the protocol for testing HERG currents in these cells was to hold at −80 mV, step to +20 mV for 2000 ms and then to observe tails at −120 mV for 500 ms.
Data analysis and presentation
Data were analysed using Clampfit 8 (Axon Instruments), Excel 5.0 and Prism 3 (Graphpad Inc.) software. Data are presented as mean±standard error of the mean (s.e.m.) with the exception of IC50 and h, which are given as mean with 95% confidence intervals (CIs). Statistical comparisons were made using a two-tailed Student's t-test (paired or unpaired) or one-way analysis of variance (ANOVA) with a Bonferroni post-test using Prism 3 software (Graphpad Inc.). P-values of less than 0.05 were taken as statistically significant.
For the purposes of comparison, HERG currents were measured in two manners: peak tail current after the beginning of repolarisation (‘tail' current) and isochronal currents at the end of the activating, depolarising step (‘end-pulse' current). Throughout the text, unless otherwise stated, IHERG refers to peak tail currents. A significant time-dependent leftward shift of ∼−6 mV (P<0.01, ANOVA, n=5 cells) of the half-maximal activation of IHERG was observed during the first 6 min of achieving whole-cell access. Control experiments revealed no further significant time-dependent shift in activation (P>0.05, ANOVA, n=5 cells). Consequently, sufficient time (∼6 min) was allowed to elapse to ensure stable current–voltage relations to be recorded, thus permitting the study of possible effects of fluvoxamine on activation, independent of time-dependent changes.
Fractional block of IHERG was calculated using the following equation:
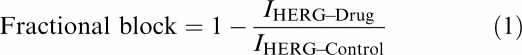 |
The relation between drug concentration and current block by fluvoxamine was determined by fitting data with a Hill equation of the form:
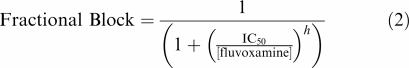 |
where IC50 is the concentration of fluvoxamine producing half-maximal inhibition of the IHERG and h is the Hill coefficient for the fit.
The voltage dependence of current activation was determined by fitting the values of the normalised tail currents with a modified Boltzmann equation of the form
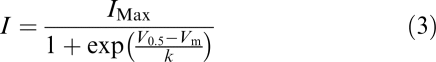 |
where I=IHERG tail amplitude following test potential Vm, IMax is the maximal IHERG tail observed, V0.5=potential at which IHERG was half-maximally activated and k is the slope factor describing IHERG activation.
The voltage dependent activation curves for IHERG tails in Figure 3c were obtained by calculating activation variables at 1 mV intervals between −50 and +40 mV. To do this, mean values for V0.5 and k derived from fits to experimental I–V data with Equation (3) were inserted into the following equation:
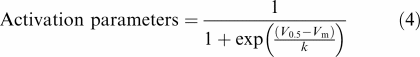 |
where ‘activation parameters' at any potential, Vm occurs within the range 0–1 and V0.5 and k have similar meanings to those in Equation (3).
Figure 3.

Voltage-dependent modulation of IHERG by 3 μM fluvoxamine. Panel (a) shows representative IHERG traces recorded in the absence (ai) and presence (aii) of 3 μM fluvoxamine. Currents (upper traces) were evoked in response to a series of incremental step depolarisations between −40 and +40 mV from a holding potential of −80 mV (lower traces). For clarity, not all steps are shown. Panel (b) shows the complete current–voltage relations for the IHERG tail current (bi) and the end-pulse current (bii) from the cell shown in panel (a). Data are shown in control and in the presence of 3 μM fluvoxamine. IHERG tails were measured as the peak current evoked on repolarisation to −40 mV relative to the holding current at −80 mV. The isochronal end-pulse current was measured as the current amplitude at the end of the conditioning voltage step prior to repolarisation to −40 mV. Tail currents were fitted with a Boltzmann sigmoidal function (see Equation (3), Methods). The values for V0.5 in control and drug for this cell were −15.4±0.2 and −24.0±0.3 mV, respectively. Values for k in control and drug were 5.1±0.1 and 4.5±0.3 mV, respectively. Panel (c) shows mean data of the voltage dependence of fractional block of IHERG tails (filled bar) and end-pulse current (hatched bars) (ANOVA with a Bonferroni post-test, # denotes P<0.001 for post-test pairwise comparison with steps to ⩾−10 mV, * denotes P<0.05 for post-test pairwise comparison with voltage steps ⩾0). Superimposed are the normalised Boltzmann functions describing voltage-dependent activation of the IHERG tails in control (solid line) and in the presence of 3 μM fluvoxamine (dashed line). Application of fluvoxamine shifted the mean half-maximal activation voltage (V0.5) by ≈−10 mV (see also ‘Results' text).
Deactivation of IHERG tails were fitted with a bi-exponential equation of the form
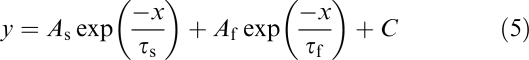 |
where τs and τf represent the time constants for the slow and fast components of the deactivating tail current and As and Af represent the total current fitted by each component, respectively. C is the residual current component not fitted by either τs or τf. This value for C was at or close to zero for fits to IHERG tail deactivation.
Drugs
Fluvoxamine maleate (Tocris) was dissolved in deionized water (Milli-Q, Millipore) to produce a stock solution of 30 mM. This stock solution was aliquoted and stored at −20°C until use. Stock solution was added to Tyrode's solution to make up the concentrations mentioned in the ‘Results' text. Solutions containing the drug were made up fresh on each experimental day. During recordings, all solutions were applied to the cells under study using a home built, warmed, multi-barrelled solution application device (Levi et al., 1996) capable of changing the bathing solution surrounding a cell in <1 s.
Results
Fluvoxamine produces a concentration-dependent inhibition of IHERG
The basic protocol used to study the effects of fluvoxamine on IHERG is shown in Figure 2a (lower trace), together with representative currents (upper traces). From a holding potential of −80 mV, the membrane potential was stepped to +20 mV for 2 s, and then repolarised to −40 mV for 4 s, before it was returned to −80 mV. The standard protocol was applied with a start-to-start interval of 12 s. Figure 2a shows representative traces recorded from a single cell in an experiment in which a range of concentrations of fluvoxamine were applied sequentially, in order to determine the concentration dependence of inhibition of the IHERG. During superfusion of each drug concentration, the cell was continually stimulated in order to ascertain steady-state inhibition. A total of six different concentrations of fluvoxamine between 10 nM and 30 μM were tested (using a minimum of six replicates for each concentration). For each drug concentration and for each cell tested, IHERG blockade was quantified by measuring IHERG tail amplitudes and end-pulse currents in control and fluvoxamine-containing solutions relative to the holding current at –80 mV. Fractional block of the IHERG tail and end-pulse current was determined using Equation (1) (see ‘Methods').
Figure 2.

Concentration–response relation for block of HERG-mediated current (IHERG) by fluvoxamine. Panel (a) shows the steady-state effect of a range of concentrations of fluvoxamine on IHERG evoked by the protocol shown. Superimposed traces were recorded from the same cell. The intermittent line marks zero current. Panel (b) shows mean data (±s.e.m.) from a series of experiments similar to that shown in panel (a). Fractional block of IHERG tails and end-pulse current are plotted as a function of fluvoxamine concentration. Data were fitted with the Hill equation yielding an IC50 and a Hill coefficient (h) (mean values shown in figure). Numbers of cells (n) are given in parentheses at each concentration.
Data from individual cells were pooled to obtain mean (±s.e.m.) fractional block values, which were then plotted against the corresponding fluvoxamine concentration as shown in Figure 2b; values were calculated for both peak tail currents and for end-pulse currents; these were not found to be significantly different (t-test, P>0.05). The mean data points were then fitted with a Hill equation (see Equation (2), ‘Methods' section). For IHERG tails, the observed IC50 was 3.8 μM (95% confidence interval (CI): 2.7 to 5.4 μM) and the Hill coefficient for the fit was close to 1 (0.9; 95% CI: 0.6–1.2); for end-pulse currents, the IC50 was 2.6 μM (95% CI: 1.8–3.8 μM) and the Hill coefficient for the fit was 0.7 (95% CI: 0.5–1.0).
Voltage dependence of IHERG inhibition by fluvoxamine
The effects of voltage on fluvoxamine's blockade of IHERG were investigated by applying voltage commands of 5 s duration from a holding potential of −80 mV to a range of test potentials. Measurements of end-pulse current and of the magnitude of IHERG tails on repolarisation to −40 mV were made in control and in 3 μM fluvoxamine-containing solutions. Currents were measured relative to the holding current at −80 mV. Representative current traces are shown in the upper panel of Figure 3a, with the corresponding voltage protocols in the lower panel. The traces shown were recorded from the same cell in control (Figure 3ai) and in the presence of 3 μM fluvoxamine (Figure 3aii). At the more negative test potentials in the range tested, 3 μM fluvoxamine exerted comparatively little effect on IHERG, and in some experiments the tail current magnitude was greater in the presence of drug than in control solution (cf. Figure 3ai and aii). At test potentials more positive than −20 mV, a marked inhibitory effect was observed in all cells.
Figure 3b shows a representative current–voltage relation for IHERG tails (Figure 3bi) and end-pulse current (Figure 3bii) in control and drug; at test potentials of −30 and −20 mV, end-pulse current and IHERG tails were increased by the drug, which is not dissimilar to the effects previously observed for azimilide on HERG (Jiang et al., 1999). In a total of seven cells, both end-pulse current and IHERG tail amplitude were significantly increased at −30 mV (P<0.05 and P<0.005, respectively, using a paired t-test) in the presence of 3 μM fluvoxamine, while at more positive potentials the blocking effect was evident. To understand this apparent dual effect of fluvoxamine in more detail, individual I–V relations were fitted with a modified Boltzmann equation (see Equation (3), ‘Methods' section). The mean V0.5 values derived from fits with Equation (3) to data from individual cells (n=7) were −18.3±2.7 mV in control solution and −28.1±1.3 mV in fluvoxamine (P<0.001 using a paired t-test), while the k values were 4.7±2.4 and 2.5±0.7 mV, respectively (P<0.05 using a paired t-test). Thus, the dual effect of fluvoxamine on IHERG tail magnitude may be explained by the drug both inhibiting IHERG and producing a leftward shift in voltage-dependent activation of the current.
The voltage–dependent activation curves for IHERG in control (solid line) and fluvoxamine (dashed line), are plotted in Figure 3c (constructed using the experimentally derived V0.5 and k values, see Equation (4), ‘Methods' section). Voltage dependence of the drug action coincided with the voltage range over the rising phase of the activation curves (i.e. the membrane potential range over which channel opening occurs). The voltage dependence of IHERG blockade by fluvoxamine was quantified by plotting mean fractional block (calculated using Equation (1)) against test potential, as shown in Figure 3c. The negative fractional block values at −30 and −20 mV reflect the alteration to voltage-dependent activation of the IHERG tails, as described above. The level of IHERG blockade produced by 3 μM fluvoxamine increased progressively up to −10 mV and levelled out between 0 and +40 mV; the dependence of fractional block on test potential was found to be statistically significant for both tail and end-pulse IHERG (P<0.001; ANOVA over the range −30 to +40 mV, see Figure 3 and legend).
The deactivation time course of IHERG tails was not significantly altered by fluvoxamine. IHERG tails recorded on repolarisation to −40 mV from a test potential of +40 mV were fitted with a bi-exponential equation (see Equation (5), ‘Methods' section). The mean values for the deactivation time constants in control solution and the presence of 3 μM fluvoxamine were 3737±256 and 3610±301 ms (τs) along with 545±51 and 507±86 ms (τf, n=7), respectively, and these were not significantly different (paired t-test: P=0.69 for τs, P=0.48 for τf,). The relative proportion of current deactivation described by the slow time constant in control and drug (As/(As+Af)) were 0.55±0.03 and 0.53±0.03, respectively, and these were also not significantly different (paired t-test, P=0.53).
Time-dependence of fluvoxamine-mediated blockade of HERG
IHERG blockade by Class III methanesulphonanilide agents during a sustained depolarisation shows a characteristic profile; it begins with little blockade initially and this progressively develops and increases over seconds during the maintained depolarisation (e.g. Spector et al., 1996; Snyders & Chaudhary, 1996). Some other HERG-blocking agents, including the TCA imipramine (Teschemacher et al., 1999) and the SSRIs citalopram and fluoxetine (Thomas et al., 2002; Witchel et al., 2002), show a different profile, with blockade evident early during the depolarising pulse. In order to study the development of IHERG blockade by fluvoxamine during a sustained pulse, long-duration (10 s) depolarising pulses were applied from a holding potential of −80 to 0 mV to elicit a stable IHERG in control solution, as shown in the representative current records in Figure 4a. Following repolarisation to −80 mV, the external solution surrounding the cell under study was then rapidly exchanged for one containing 3 μM fluvoxamine. The cell was then equilibrated (>3 min) at a holding potential of −80 mV in the fluvoxamine-containing solution in the absence of applied pulses, before an identical long-duration pulse was applied. Figure 4b shows the mean level of fractional block of current by 3 μM fluvoxamine plotted at 1 s intervals during the applied voltage pulse. Between 1 and 10 s, there was no statistically significant difference in the level of current blockade (P>0.05; one-way ANOVA with a Bonferroni post-test). The results with this experimental protocol suggest that IHERG blockade by fluvoxamine occurs rapidly (i.e. <1 s during depolarisation to 0 mV), as has been seen with other low-potency psychotropic agents (Teschemacher et al., 1999; Witchel et al., 2002).
Figure 4.

Time-dependence of IHERG blockade by fluvoxamine. Panel (a) shows typical long pulse current traces recorded in a cell expressing HERG. The cell was repeatedly stepped from a holding potential of −80 to 0 mV for 10 s (lower trace) to evoke stable steady-state currents (upper trace). Stimulation was then terminated and the cell was allowed to equilibrate in 3 μM fluvoxamine for >3 min, while being held at −80 mV. The cell was then subjected to the same voltage command. Traces immediately prior to and the first step following the application of 3 μM fluvoxamine are shown. Panel (b) shows the mean data (±s.e.m.) for the experiment in panel (a) using six cells. Time zero coincides with the voltage step from −80 to 0 mV. Following development of full block (which occurs within 1 s), no time-dependent change was observed. The intermittent straight line at 0.5 is shown for comparison. Panel (c) shows representative current traces evoked by an envelope of tails protocol. Cells were held at −80 mV and stepped to +40 mV for 25–600 ms and then tail currents were evoked on repolarisation to −40 mV. Peak tail currents at each time point were measured. Representative current traces were recorded in the same cell under control conditions and in the presence of 3 μM fluvoxamine. Panel (d) shows mean data (±s.e.m.) from seven cells for the envelope of tails experiments. Fractional blockade of the HERG tail current was plotted as a function of the depolarising pulse duration. Block developed mono-exponentially with a t1/2 of 7.5 ms.
The time course of development of IHERG blockade by fluvoxamine was further investigated using an ‘envelope of tails' protocol (Trudeau et al., 1995) that was similar to that used in a recent investigation of pharmacological blockade of IHERG in this laboratory (Paul et al., 2001). Membrane potential was voltage-clamped at a holding potential of −80 mV and stepped to +40 mV for increasing durations between 25 and 600 ms, after which IHERG tails were evoked on repolarisation to −40 mV. This protocol permitted the investigation of the effect of progressive IHERG activation on inhibition by fluvoxamine. Original current traces (upper panel) obtained using this protocol are shown in control (Figure 4c). The cells were then held at −80 mV, while the external solution was exchanged for one containing 3 μM fluvoxamine for >3 min. The protocol was then repeated (Figure 4c). Figure 4d shows the time dependence of fractional block of the evoked IHERG tails (after test potentials of +40 mV) fitted with a mono-exponential association, yielding a t1/2 of 7.5 ms for the onset of post-depolarisation channel block. Standard-error bars for fractional block for the shortest pulse durations were large, however, and inhibition levels were not statistically different from those at longer pulse durations (ANOVA, P=0.4).
Investigating further the state dependence of wild-type IHERG blockade by fluvoxamine
The data gathered using the ‘envelope of tails' protocols showed that blockade by 3 μM fluvoxamine was present even at the shortest test pulse examined. As previously reported for imipramine (Teschemacher et al., 1999) and canrenoic acid (Caballero et al., 2003), this might reflect a component of closed channel inhibition, or alternatively result from extremely rapid development of blockade on channel gating (as suggested for fluoxetine and its binding to activated IHERG; Thomas et al., 2002). For fluvoxamine, this issue was investigated further as follows: cell membrane potential was held at −100 mV (which greatly favours the closed-channel state(s)), and from this potential a brief 10 ms depolarisation to +40 mV was applied and IHERG observed on repolarisation to −40 mV. The protocol was applied first in control and then after 3 min equilibration in fluvoxamine at −100 mV. Consequently, all HERG channels were closed during the addition and equilibration of the drug-containing superfusate; thus, binding sites dependent upon channel activation would be expected to be inaccessible to the fluvoxamine until the 10 ms step to +40 mV. As shown in Figure 5a, 3 μM fluvoxamine produced an attenuation of IHERG even after only a 10 ms activating pulse. The level of inhibition observed was similar to that seen with 2000 ms duration command pulses used to obtain concentration–response data (Figure 5aii).
Since IHERG block can only be assessed following channel opening (i.e. when current is flowing through open channels), it can be difficult to distinguish between closed- and extremely rapid open-state-dependent channel block (Dumaine et al., 1998; Caballero et al., 2003). The rate of development of a rapid open-channel blockade should be dependent upon drug concentration. Thus, a similar experiment was performed using a 10-fold lower concentration of fluvoxamine (Figure 5b). Here again, inhibition of IHERG was observed after only a brief 10 ms depolarisation from −100 to +40 mV. The level of inhibition observed was similar to that produced by a 200 ms pulse to +40 from −100 mV and to that seen with the 2000 ms pulses used to obtain concentration–response data (Figure 5bii). The fact that drug concentrations differing 10-fold both produced such rapid IHERG inhibition is concordant with the drug being able to bind to the closed-channel state.
Pure closed-channel-state-dependent drug binding might be expected to result in a decrease in channel inhibition with time during sustained depolarisation or in an inverse dependence of inhibition on test voltage. Since IHERG inhibition by fluvoxamine showed neither of these characteristics, it seemed likely that the drug interacted also with one or both of the activated and inactivated channel states. Indeed, the data in Figure 3 show that fluvoxamine modified the voltage dependence of current activation.
For fluoxetine, depolarisation beyond +40 mV was reported to decrease the level of observed inhibition suggesting that inactivation reduced drug block (Thomas et al., 2002). We therefore employed a modified version of the protocol used in Figure 4a to investigate whether or not this also occurred with fluvoxamine. In this protocol, a step to +80 mV was imposed from and back to 0 mV. The reduction in IHERG during the phase at +80 mV (Figure 5c) reflects the increased number of channels in the inactive state at this potential. The protocol was applied in normal Tyrode's solution and in 3 μM fluvoxamine and fractional block during each phase of the voltage command was monitored. Figures 5ci and cii show that the level of IHERG inhibition was unaltered on stepping from 0 to +80 mV and was also unaltered on stepping back to 0 from +80 mV. This shows clearly that, in contrast to fluoxetine (Thomas et al., 2002), increasing inactivation did not decrease IHERG block and this is consistent with both activated and inactivated channels interacting with the drug.
A method recently published by Weerapura et al. (2002) was used to quantify and determine the effect of altering the fraction of inactivated channels (Fi) upon the action of fluvoxamine (test potential 0 mV; see ‘Methods' for details). The bar charts in Figure 5d summarise the data obtained. The value for Fi that we obtained (∼0.86) was comparable to that reported by Weerapura et al. (2002; ∼0.92 in 2 mM [K+]o at ambient temperature). Increasing external K+ from 4 to 30 mM decreased significantly the value of Fi by 25% (unpaired t-test, P<0.005) (Figure 5di) and also reduced the level of fluvoxamine inhibition of activation (inactivation-free) current (I2 as described by Weerapura et al. (2002); Figure 5dii). This indicates that interaction with the inactivated state is involved in the overall inhibitory action of fluvoxamine.
In further experiments, we did not detect any significant effects of fluvoxamine on the time-dependent recovery from inactivation or on the voltage dependence of IHERG inactivation. In a three-pulse protocol with varying durations (1–10 ms) of recovery from inactivation (accompanied by lesser amounts of deactivation) at −100 mV (see ‘Methods' and Yang et al., 1997), normalised current amplitude on the third (depolarising, +40 mV) pulse (Itail(time)/Itail(maximum)) at each time point failed to show any statistically significant difference between control and 3 μM fluvoxamine (P>0.1, paired t-test). Furthermore, in a three-pulse protocol in which the third pulse was at varying voltages (see ‘Methods' and Snyders & Chaudhary, 1996), fitting the rapidly diminishing current due to inactivation to a mono-exponential equation, resulted in τ values that were not significantly different at any voltage in the presence or absence of 3 μM fluvoxamine (P>0.05, one-way ANOVA with Bonferroni post-test).
Alanine substitution of F656, Y652 or S631 results in partial attenuation of blockade
Our data from wild-type HERG suggests that fluvoxamine exhibited mixed state-dependent channel blockade. Its interactions with HERG were further investigated by examining the effects of S6 or H5 mutations upon observed levels of inhibition. Cell lines expressing the mutant HERG channels F656A, Y652A and S631A were tested for the ability of fluvoxamine to block them using concentrations of fluvoxamine that cause profound block of the wild-type channel. Owing to poor expression associated with F656A, as has been previously reported (Mitcheson et al., 2000), that mutant was tested using a superfusate containing 96 mM K+ with a voltage command protocol testing tail currents at −120 mV; the procedure of using raised external K+ and observing tails at hyperpolarising potentials in wild-type HERG-based currents is well established (Faravelli et al., 1996). The other mutants were tested using a standard HERG-activating protocol and observing tails at −40 mV (Figure 6). Under these conditions, all the three mutant channels had significantly attenuated levels of block compared with that of wild-type HERG, but currents from mutant channels were still blocked by at least 50%. By contrast, using 120 nM E-4031 (∼10-fold the IC50 under these conditions) on F656A resulted in 14.7±2.8% (n=3) block in 96 mM K+, and Y652A was blocked 8.3±7.1% (n=5) in 4 mM K+ superfusate. Similar experiments applying E-4031 to cells expressing wild-type HERG yielded current block in excess of 93±1% in both 4 mM (n=3) and 96 mM (n=3) external K+.
Figure 6.

Fluvoxamine block of F656A, Y652A and S631A. Panels (a) and (b) show representative current records from cells expressing wild-type and mutant HERG channels. Panel (a) shows the effect of application of 30 μM fluvoxamine on wild-type HERG (ai) and Y652A mutant HERG (aii). Panel (b) shows the effect of 90 μM fluvoxamine on HERG wild-type (bi) and F656A (bii); tails were measured on repolarisation to −120 mV. Experiments in panel (a) are performed in the presence of 4 mM external K+, experiments in panel (b) are performed in the presence of 96 mM external K+ to maximise the low expression of the F656A mutant (hence, inward tail currents). Panel (c) is a summary showing mean fractional block data for the mutants tested, compared to the effects on the wild-type channel. Statistical comparisons were made using an unpaired t-test (#P<0.0001, *P<0.001).
Discussion
The data presented in this study show that the SSRI fluvoxamine blocks HERG, and that this blockade is only partially attenuated by the mutations F656A, Y652A and S631A. IHERG inhibition by fluvoxamine was very rapidly developing and is mechanistically distinct from that of the SSRI fluoxetine, for which inactivation was reported to decrease, as opposed to contribute to, blockade (Thomas et al., 2002). To the best of our knowledge, fluvoxamine is the first drug for which mutation of F656 has been observed to produce only a partial (as opposed to 100-fold) attenuation of block and it is only the second agent (vesnarinone aside; Kamiya et al., 2001) for which the mutation Y652A does not profoundly attenuate blockade (Lees-Miller et al., 2000; Mitcheson et al., 2000; Sanchez-Chapula et al., 2002). This is also the first study in which the F656A and Y652A mutants have been tested in a mammalian cell line.
With the possible exception of scorpion toxin blockers of HERG (which have molecular determinants of HERG blockade in HERG's S5-pore extracellular linker region (i.e. the ‘turret'; Pardo-Lopez et al., 2002), all HERG-blocking drugs that have been examined for their molecular determinants of blockade have been shown to be dependent on a variety of amino-acid residues in the pore-S6 region (the inner lining of the pore). In a number of previous studies, alanine-scanning mutagenesis has been used to test the amino acids in this region, and, of those mutants that were successfully expressed and conducted current, only a small number of amino-acid residues have been found to be important determinants of HERG blockade by highly diverse drugs ranging from high-affinity methanesulphonanilides to low-affinity vesnarinone: T623, S624, V625, G648, Y652, F656 and V659 (Lees-Miller et al., 2000; Mitcheson et al., 2000; Kamiya et al., 2001; Sanchez-Chapula et al., 2002). Presumably, this list represents the residues of S6 that face into the vestibule (∼ every third amino acid of the putative α helix) and those residues deep in the selectivity filter that might make contact with a drug deep within the vestibule. Of the drugs tested, only vesnarinone's blockade was not at least 100-fold attenuated in Y652A, and inhibition of HERG by absolutely all previously tested drugs was attenuated dramatically by F656A. Interestingly, although fluvoxamine's blockade was significantly attenuated by F656A and Y652A, this was not nearly to the level of virtually all other high-affinity or low-affinity HERG blockers that have been tested to date against these mutations.
Our experiments with wild-type HERG suggest that fluvoxamine exhibits mixed channel-state dependence of inhibition. The profile of rapid development of blockade during a sustained depolarisation (see Figure 4) differs markedly from the more slowly developing blockade exhibited by the higher affinity methanesulphonanilides (Snyders & Chaudhary, 1996; Spector et al., 1996). The ‘envelope of tails' data (see Figure 4) also indicate that IHERG block by fluvoxamine achieves a maximum level very rapidly on depolarisation to positive potentials. The lack of reduction of blockade by strong depolarisation to +80 mV, together with the reduced blockade seen when the fraction of inactivated IHERG was decreased (by raising [K+]o; Figure 5d), indicate that fluvoxamine interacts with both activated and inactivated channels. The extremely rapid blockade (within 10 ms) of current produced by both 3 and 0.3 μM fluvoxamine is consistent with a role also for closed-channel blockade. It must be noted though that, since IHERG block can only be observed by measuring elicited current, it can be difficult to distinguish between closed- and extremely rapid open-channel block. Also, the lack of IHERG blockade at −30 and −20 mV would, on initial inspection, seem difficult to reconcile with a role for closed-channel inhibition. However, the leftward shift in voltage-dependent activation observed with fluvoxamine means that at these potentials, at which IHERG would usually be only small, significantly more current would be seen on depolarisation than in the absence of drug. This net increase in current could conceivably mask the effects of closed-channel block at these potentials. Voltage-dependent IHERG blockade by an agent with mixed dependence on closed- and open-channel states would not be unprecedented (cf. amiodarone; Kiehn et al., 1999). Like fluvoxamine, azimilide has also been reported to exert an agonist action at negative voltages, while inhibiting current at more positive potentials (Jiang et al., 1999; Walker et al., 2000). Azimilide also produces a leftward shift in voltage-dependent activation (Walker et al., 2000). Analysis of the actions of azimilide using mutant HERG channels has implicated the S5-pore linker region in mediating the positive effects of the drug (Jiang et al., 1999), which acts rapidly on the channel from the outside of the cell; in that study the molecular determinants of the blocking effects were not described, and presumably involve another region of the channel (i.e. not the turret).
We used the attenuated-inactivation mutant S631A to further investigate a role for inactivation in the effect of fluvoxamine. This mutation partially attenuated the inhibitory effect of fluvoxamine, an observation consistent with a proposal that the process of inactivation is not obligatory for binding to occur, but influences the potency of drug binding. It has recently been reported for cisapride's high-affinity blockade from within the HERG vestibule that reduced drug affinity of non-inactivating HERG mutant channels is not due to changes in inactivation per se, but to changes in inactivation gating-associated reorientation of the residues in the S6 domain that comprise the high-affinity drug-binding site (Chen et al., 2002). In the case of fluvoxamine, however, when the S631A data are considered alongside our observations with wild-type HERG, they support a role for channel inactivation in influencing the potency of blockade.
On the basis of our data, it is not possible to conclude completely where in the HERG channel fluvoxamine binds. Rapid open- or inactivated-state channel block and partial attenuation by the mutations F656A, Y652A and S631A suggest some binding to a drug receptor site within the pore-S6 region (as has been established for MK-499). However, the lack of profound attenuation of block among the mutant channels suggests that the obligatory determinants of fluvoxamine's blockade may exhibit at least some differences from those reported for methanesulphonanilides, cisapride and terfenadine (Mitcheson et al., 2000). The possible contribution of closed-channel blockade to the overall action of fluvoxamine may also be of relevance here, as the mutations F656A, Y652A and S631A would only be anticipated to influence blockade contingent upon channel gating and binding within the pore.
Overall, the lack of profound attenuation of fluvoxamine's blockade of HERG by mutation of F656 is unique among all drugs in the range of drug classes tested so far (i.e. dofetilide, MK-499, cisapride, terfenadine, vesnarinone and chloroquine; Mitcheson et al., 2000; Lees-Miller et al., 2000; Kamiya et al., 2001; Sanchez-Chapula et al., 2002), and is therefore an observation of particular note. It suggests that at least part of fluvoxamine's action on HERG involves a mechanism that is distinct from that previously described.
Acknowledgments
This work was supported by project grants from the British Heart Foundation to HJW and JCH (PG/2001104 and PG/2000123) and by a Wellcome Trust fellowship to JCH. We are grateful to Lesley Arberry, Terri Harding for technical assistance, and to Shaun Ryder for guidance during this study.
Abbreviations
- ANOVA
analysis of variance
- CI
confidence interval
- h
Hill coefficient
- HEK
Human embryonic kidney
- HERG
Human ether-a-go-go related gene
- IC50
half-maximal inhibitory drug concentration
- IHERG
current mediated by the HERG channel
- IK
delayed rectifier potassium current
- IKr
‘rapid' delayed rectifier potassium current
- IKs
‘slow' delayed rectifier potassium current
- k
slope factor describing voltage-dependent activation
- [K+]o
extracellular potassium concentration
- QT interval
QT interval of the electrocardiogram
- QTc
rate-corrected QT interval of the electrocardiogram
- SSRI
selective-serotonin re-uptake inhibitor
- TCA
tricyclic antidepressant
- 6-TM
6-Transmembrane spanning domains
- t1/2
time point of half-maximal change
- τf
fast time constant of deactivation
- τs
slow time constant of deactivation
- s.e.m.
standard error of the mean
- V0.5
half-maximal activation voltage
References
- CABALLERO R., MORENO I., GONZALEZ T., ARIAS C., VALENZUELA C., DELPON E., TAMARGO J. Spironolactone and its main metabolite, canrenoic acid, block human ether-a-go-go-related gene channels. Circulation. 2003;107:889–895. doi: 10.1161/01.cir.0000048189.58449.f7. [DOI] [PubMed] [Google Scholar]
- CHEN J., SEEBOHM G., SANGUINETTI M.C. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12461–12466. doi: 10.1073/pnas.192367299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS Points to Consider: The Assessment for the Potential for QT Prolongation by Non-Cardiovascular Medicinal Products 1997London: European Agency for the Evaluation of Medicinal Products; CPMP/986/96. 1997 [Google Scholar]
- CRUMB W., CAVERO I. QT interval prolongation by non-cardiovascular drugs: issues and solutions for novel drug development. Pharm. Sci. Technol. Today. 1999;2:270–280. doi: 10.1016/s1461-5347(99)00172-8. [DOI] [PubMed] [Google Scholar]
- DOYLE D.A., MORAIS CABRAL J.H., PFUETZNER R.A., KUO A., GULBIS J.M., COHEN S.L., CHAIT B.T., MACKINNON R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- DUMAINE R., ROY M.L., BROWN A.M. Blockade of HERG and Kv1.5 by ketoconazole. J. Pharmacol. Exp. Ther. 1998;286:727–735. [PubMed] [Google Scholar]
- FARAVELLI L., ARCANGELI A., OLIVOTTO M., WANKE E. A HERG-like K+ channel in rat F-11 DRG cell line: pharmacological identification and biophysical characterization. J. Physiol. (Lond.) 1996;496:13–23. doi: 10.1113/jphysiol.1996.sp021661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEIGHNER J.P., BOYER W.F., MEREDITH C.H., HENDRICKSON G.G. A placebo-controlled inpatient comparison of fluvoxamine maleate and imipramine in major depression. Int. Clin. Psychopharmacol. 1989;4:239–244. doi: 10.1097/00004850-198907000-00006. [DOI] [PubMed] [Google Scholar]
- GRALINSKI M.R. The assessment of potential for QT interval prolongation with new pharmaceuticals: impact on drug development. J. Pharmacol. Toxicol. Methods. 2000;43:91–99. doi: 10.1016/s1056-8719(00)00100-3. [DOI] [PubMed] [Google Scholar]
- HAVERKAMP W., BREITHARDT G., CAMM A.J., JANSE M.J., ROSEN M.R., ANTZELEVITCH C., ESCANDE D., FRANZ M., MALIK M., MOSS A., SHAH R. The potential for QT prolongation and pro-arrhythmia by non-anti-arrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Cardiovasc. Res. 2000;47:219–233. doi: 10.1016/s0008-6363(00)00119-x. [DOI] [PubMed] [Google Scholar]
- HENRY J.A. Epidemiology and relative toxicity of antidepressant drugs in overdose. Drug Saf. 1997;16:374–390. doi: 10.2165/00002018-199716060-00004. [DOI] [PubMed] [Google Scholar]
- HEWER W., ROST W., GATTAZ W.F. Cardiovascular effects of fluvoxamine and maprotiline in depressed patients. Eur. Arch. Psychiatry Clin. Neurosci. 1995;246:1–6. doi: 10.1007/BF02191808. [DOI] [PubMed] [Google Scholar]
- JIANG M., DUN W., FAN J.S., TSENG G.N. Use-dependent ‘agonist' effect of azimilide on the HERG channel. J. Pharmacol. Exp. Ther. 1999;291:1324–1336. [PubMed] [Google Scholar]
- KAMIYA K., MITCHESON J.S., YASUI K., KODAMA I., SANGUINETTI M.C. Open channel block of HERG K+ channels by vesnarinone. Mol. Pharmacol. 2001;60:244–253. doi: 10.1124/mol.60.2.244. [DOI] [PubMed] [Google Scholar]
- KIEHN J., THOMAS D., KARLE C.A., SCHOLS W., KUBLER W. Inhibitory effects of the class III antiarrhythmic drug amiodarone on cloned HERG potassium channels. Naunyn Schmiedeberg's Arch. Pharmacol. 1999;359:212–219. doi: 10.1007/pl00005344. [DOI] [PubMed] [Google Scholar]
- LAIRD L.K., LYDIARD R.B., MORTON W.A., STEELE T.E., KELLNER C., THOMPSON N.M., BALLENGER J.C. Cardiovascular effects of imipramine, fluvoxamine, and placebo in depressed outpatients. J. Clin. Psychiatry. 1993;54:224–228. [PubMed] [Google Scholar]
- LEES-MILLER J.P., DUAN Y., TENG G.Q., DUFF H.J. Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol. Pharmacol. 2000;57:367–374. [PubMed] [Google Scholar]
- LEVI A.J., HANCOX J.C., HOWARTH F.C., CROKER J., VINNICOMBE J. A method for making rapid changes of superfusate whilst maintaining temperature at 37 degrees C. Pflügers Arch. 1996;432:930–937. doi: 10.1007/s004240050217. [DOI] [PubMed] [Google Scholar]
- MANET P., HILPERT F., FOUET P., TOLEDANO D. Ventricular arrhythmia during fluvoxamine poisoning. Therapie. 1993;48:62–63. [PubMed] [Google Scholar]
- MILNES J.T., HANCOX J.C., WITCHEL H.J. Actions of the selective serotonin re-uptake inhibitor fluvoxamine on herg-mediated potassium current in a mammalian cell line. J. Physiol. 2002;544P:25. [Google Scholar]
- MITCHESON J.S., CHEN J., LIN M., CULBERSON C., SANGUINETTI M.C. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U.S.A. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHTANI H., ODAGIRI Y., SATO H., SAWADA Y., IGA T. A comparative pharmacodynamic study of the arrhythmogenicity of antidepressants, fluvoxamine and imipramine, in guinea pigs. Biol. Pharm. Bull. 2001;24:550–554. doi: 10.1248/bpb.24.550. [DOI] [PubMed] [Google Scholar]
- PARDO-LOPEZ L., ZHANG M., LIU J., JIANG M., POSSANI L.D., TSENG G.N. Mapping the binding site of a human ether-a-go-go-related gene-specific peptide toxin (ErgTx) to the channel's outer vestibule. J. Biol. Chem. 2002;277:16403–16411. doi: 10.1074/jbc.M200460200. [DOI] [PubMed] [Google Scholar]
- PAUL A., WITCHEL H.J., HANCOX J.C. Inhibition of HERG potassium channel current by the class 1a antiarrhythmic agent disopyramide. Biochem. Biophys. Res. Commun. 2001;280:1243–1250. doi: 10.1006/bbrc.2001.4269. [DOI] [PubMed] [Google Scholar]
- RODRIGUEZ DE LA TORRE B., DREHER J., MALEVANY I., BAGLI M., KOLBINGER M., OMRAN H., LUDERITZ B., RAO M.L. Serum levels and cardiovascular effects of tricyclic antidepressants and selective serotonin reuptake inhibitors in depressed patients. Ther. Drug Monit. 2001;23:435–440. doi: 10.1097/00007691-200108000-00019. [DOI] [PubMed] [Google Scholar]
- ROOS J.C. Cardiac effects of antidepressant drugs. A comparison of the tricyclic antidepressants and fluvoxamine. Br. J. Clin. Pharmacol. 1983;15 Suppl 3:439S–445S. doi: 10.1111/j.1365-2125.1983.tb02135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCHEZ-CHAPULA J.A., NAVARRO-POLANCO R.A., CULBERSON C., CHEN J., SANGUINETTI M.C. Molecular determinants of voltage-dependent human ether-a-go-go related gene (HERG) K+ channel block. J. Biol. Chem. 2002;277:23587–23595. doi: 10.1074/jbc.M200448200. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JIANG C., CURRAN M.E., KEATING M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., KEATING M.T. Role of delayed rectifier potassium channels in cardiac repolarization and arrhythmias. News Physiol. Sci. 1997;12:152–157. [Google Scholar]
- SNYDERS D.J., CHAUDHARY A. High affinity open channel block by dofetilide of HERG expressed in a human cell line. Mol. Pharm. 1996;49:949–955. [PubMed] [Google Scholar]
- SPECTOR P.S., CURRAN M.E., KEATING M.T., SANGUINETTI M.C. Class III antiarrhythmic drugs block HERG, a human cardiac delayed rectifier K+ channel. Open-channel block by methanesulfonanilides. Circ. Res. 1996;78:499–503. doi: 10.1161/01.res.78.3.499. [DOI] [PubMed] [Google Scholar]
- TAGLIALATELA M., CASTALDO P., PANNACCIONE A., GIORGIO G., ANNUNZIATO L. Human ether-a-go-go related gene (HERG) K+ channels as pharmacological targets: present and future implications. Biochem. Pharmacol. 1998;55:1741–1746. doi: 10.1016/s0006-2952(98)00002-1. [DOI] [PubMed] [Google Scholar]
- TESCHEMACHER A.G., SEWARD E.P., HANCOX J.C., WITCHEL H.J. Inhibition of the current of heterologously expressed HERG potassium channels by imipramine and amitriptyline. Br. J. Pharmacol. 1999;128:479–485. doi: 10.1038/sj.bjp.0702800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS D., GUT B., WENDT-NORDAHL G., KIEHN J. The antidepressant drug fluoxetine is an inhibitor of human ether-a-go-go-related gene (HERG) potassium channels. J. Pharmacol. Exp. Ther. 2002;300:543–548. doi: 10.1124/jpet.300.2.543. [DOI] [PubMed] [Google Scholar]
- TRUDEAU M.C., WARMKE J.W., GANETZKY B., ROBERTSON G.A. HERG, a human inward rectifier in the voltage gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- VANDENBERG J.I., WALKER B.D., CAMPBELL T.J. HERG K+ channels: friend and foe. Trends Pharmacol. Sci. 2001;22:240–246. doi: 10.1016/s0165-6147(00)01662-x. [DOI] [PubMed] [Google Scholar]
- VISKIN S. Long QT syndromes and torsade de pointes. Lancet. 1999;354:1625–1633. doi: 10.1016/S0140-6736(99)02107-8. [DOI] [PubMed] [Google Scholar]
- WALKER B.D., SINGLETON C.B., TIE H., BURSILL J.A., WYSE K.R., VALENZUELA S.M., BREIT S.N., CAMPBELL T.J. Comparative effects of azimilide and ambasilide on the human ether-a-go-go-related gene (HERG) potassium channel. Cardiovasc. Res. 2000;48:44–58. doi: 10.1016/s0008-6363(00)00155-3. [DOI] [PubMed] [Google Scholar]
- WEERAPURA M., HEBERT T.E., NATTEL S. Dofetilide block involves interactions with open and inactivated states of HERG channels. Pflügers Arch. 2002;443:520–531. doi: 10.1007/s004240100720. [DOI] [PubMed] [Google Scholar]
- WITCHEL H.J., HANCOX J.C. Familial and acquired Long QT Syndrome and the cardiac rapid delayed rectifier potassium current. Clin. Exp. Pharm. Physiol. 2000;27:753–766. doi: 10.1046/j.1440-1681.2000.03337.x. [DOI] [PubMed] [Google Scholar]
- WITCHEL H.J., PABBATHI V.K., HOFMANN G., PAUL A.A., HANCOX J.C. Inhibitory actions of the selective serotonin re-uptake inhibitor citalopram on HERG and ventricular L-type calcium currents. FEBS Lett. 2002;512:59–66. doi: 10.1016/s0014-5793(01)03320-8. [DOI] [PubMed] [Google Scholar]
- WOUTERS W., DEIMAN W. Acute cardiac effects of fluvoxamine and other antidepressants in conscious rabbits. Arch. Int. Pharmacodyn. Ther. 1983;263:197–207. [PubMed] [Google Scholar]
- YANG T., SNYDERS D.J., RODEN D.M. Rapid inactivation determines the rectification and [K+]o dependence of the rapid component of the delayed rectifier K+ current in cardiac cells. Circ. Res. 1997;80:782–789. doi: 10.1161/01.res.80.6.782. [DOI] [PubMed] [Google Scholar]
- ZHOU Z., GONG Q., YE B., MAKIELSKI J.C., ROBERTSON G.A., JANUARY C.T. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys. J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]