

Abstract
This study investigated the effects of galanin (GAL) on inhibition of cholinergic (vagal) activity in the mouse heart using control galanin knockout (GAL-KO) and GAL-1R receptor knockout (GAL-1R-KO) mice.
In pentobarbitone anaesthetised mice, supramaximal stimulation every 30 s of the vagus nerve innervating the heart, increased pulse interval (PI) by approximately 50 ms or decreased heart rate by approximately 100 beats min−1. This response was attenuated by intravenous administration of GAL (dose ranged from 0.8 to 13 nmol kg−1) in a dose-dependent manner.
In GAL-KO mice, the magnitude of inhibition of the increase in PI (ΔPI) following a bolus dose of GAL was not different from the ΔPI in control mice, and neuropeptide Y (NPY), previously shown to attenuate vagal inhibitory activity in mice, evoked a comparative inhibition of ΔPI in GAL-KO mice.
In GAL-1R-KO mice, an intravenous, bolus injection of GAL had no inhibitory effect on vagal activity.
In control mice, stimulation of the sympathetic nerve at 25 V, 10 Hz for 2 min in the presence of propranolol evoked a long-lasting attenuation of ΔPI. The inhibitory effect on ΔPI was reduced in the presence of the NPY Y2 antagonist, BIIE0246.
In GAL-1R-KO mice, stimulation of the sympathetic nerve in the presence of propranolol evoked an attenuation of ΔPI not significantly different from the response in control mice in the presence of BIIE0246. Following administration of BIIE0246 in GAL-1R-KO mice, the inhibition of ΔPI that followed stimulation of the sympathetic nerve was abolished.
These findings support the view that the nerve terminals of parasympathetic neurons in the mouse heart possess both GAL-1R and NPY Y2 receptors which, when activated, reduce acetylcholine release.
Keywords: Galanin, galanin knockout, GAL-1R receptor knockout, neuropeptide Y, BIIE0246, parasympathetic, sympathetic, mouse, heart
Introduction
Galanin (GAL) is a 29/30 amino-acid neuropeptide, first isolated from porcine gut in 1983 (Tatemoto et al., 1983), and is widely distributed throughout the central and peripheral nervous systems. It has been found in numerous autonomic ganglia and has been implicated in the sympathetic control of organ function of several systems, including the cardiovascular system. It has a diverse range of biological activity which affects cognition/memory, sensory/pain processing, feeding behaviour and neurotransmitter/hormone secretion and its action is mediated via specific receptors (Crawley, 1996; Iismaa & Shine, 1999). Radioligand assays using 125I-GAL suggest the existence of pre- and postsynaptic activity mediated by GAL neurons in the nucleus tractus solitarius (nTS) and the dorsal motor nucleus of the vagus (dmnX) of the rat. High densities of GAL immunoreactive nerve terminals were found within the medial subnucleus of the nTS, in the periventricular region and within the medial part of the dmnX. High densities of 125I-GAL binding sites were also found in the entire medial and lateral part of the nTS (Harfstrand et al., 1987). The presence of GAL in both the nTS and dmnX suggests a role for GAL in vagal control/modulation.
Centrally, GAL coexists with neurotransmitters including noradrenaline (NAd) in the locus coerulus and with acetylcholine (ACh) in basal forebrain neurons which project to the hippocampus (Melander et al., 1985). In monkey hippocampus, GAL inhibits potassium-evoked release of ACh and muscarinic receptor-mediated stimulation of phosphoinositide turnover, acting as a modulator of cholinergic function (Fisone et al., 1991). In the periphery, high numbers of GAL immunoreactive neurons were found in the stellate, coeliac, lower lumbar and sacral sympathetic ganglia of the cat, many colocalised with neuropeptide Y (NPY)-containing cells (Kummer, 1987). GAL-positive cell bodies have also been detected in the parasympathetic submandibular and sphenopalatine ganglia in the cat (Lindh et al., 1989).
In the heart, GAL evokes a long-lasting attenuation of cardiac vagal activity in the cat, with no effect on blood pressure (BP), while NPY increases BP but has no inhibitory effect on cardiac vagal activity (Revington et al., 1990). In the Australian possum (Trichosurus vulpecula), GAL and not NPY is colocalised with NAd in the thoracic sympathetic ganglia (Morris et al., 1992). In this species exogenous GAL, but not NPY, attenuated cardiac vagal activity, mimicking the effect of sympathetic stimulation and increasing BP (Courtice et al., 1993). While GAL has not been detected as yet in sympathetic nerves in humans, it has been detected in the adrenal medulla and pancreatic tissue, and increased plasma levels of GAL have been reported in humans postexercise (Legakis et al., 2000). Intravenous infusion of GAL in humans increased resting heart rate (HR), possibly by decreasing sinus arrhythmia (Carey et al., 1993). GAL was also proposed to be a modulator of sympathetic transmission in humans, as it reduced the potentiating effect of pyridostigmine-induced cholinergic enhancement on release of NAd (degli Uberti et al., 1996). It was suggested that GAL achieved the reduction in NAd release by decreasing preganglionic cholinergic stimulation of sympathetic postganglionic neurons.
Three receptors have been cloned for GAL; they are GAL-1R, GAL-2R and GAL-3R (Parker et al., 1995; Howard et al., 1997; Kolakowski Jr et al., 1998). One receptor (GAL-1R) has been detected in the heart of the mouse (Wang et al., 1997), and GAL-1R has been identified in mouse whole-heart preparations (Xu et al., 1995). In humans, there is considerable peripheral expression of the GAL-1R receptor, with highest levels in the heart (Sullivan et al., 1997).
The aim of this study was to examine the effects of GAL on cardiac vagal activity in mice. The effects of exogenous GAL were observed on cardiac vagal activity in anaesthetised control, GAL-KO and GAL-1R-KO mice.
Methods
Animals
All experiments were conducted with the approval of the Animal Care and Ethics committee of UNSW, and in accordance with the code of practice for Use of Animals of the National Health and Medical Research Council of Australia.
Mice homozygous for the targeted disruption of the neurotransmitter GAL, galanin knockout (GAL-KO, 129 SvJ background; Wynick et al., 1998) and homozygous for the disruption of the galanin 1R receptor knockout (GAL-1R-KO, 129 SvJ background; Jacoby et al., 2002a, 2002b) aged between 12 and 30 weeks were bred by Dr Tiina Iismaa (Garvan Institute of Medical Research, Darlinghurst, Australia). Control mice for each of the knockout groups were of the same background.
Experimental procedure
Control, GAL-KO and Gal-R1-KO mice of each sex were anaesthetised with sodium pentabarbitone (Nembutal, Boehringer-Ingelhiem; 30 mg kg−1, i.p.). Supplementary doses of anaesthetic were given as required throughout the experimental period (1 : 10 diluted with saline). The trachea was cannulated, high in the neck of each mouse and attached to a positive pressure rodent ventilator (Ugo Basile 6025). Each mouse was artificially ventilated for the duration of the experiment. Respiratory rate and tidal volume were adjusted and blood gases were monitored via a blood gas analyser (Corning 278 blood gas analyser) to allow fine adjustment of ventilation parameters (pO2≈80 mmHg, pCO2≈40 mmHg, pH 7.4). Blood gases were checked several times throughout the experimental period and if necessary, adjustments were made to either tidal volume or frequency of respiratory pump. After any adjustment, blood gases were rechecked. The femoral artery was cannulated for continuous recording of arterial BP via a Statham physiological pressure transducer (P23XL), and the femoral vein was cannulated for administration of drugs. Temperature was monitored via a rectal probe and maintained at 37±1°C. Subcutaneous needle electrodes were used to record the electrocardiogram (ECG), which was displayed on a storage oscilloscope (Gould 1401) to monitor correct triggering of pulse interval (PI) record. The ECG was used to obtain PI (time between heart beats) following processing with Neurolog modules (Digitimer, England: NL 200, 303, 601). Both PI and BP were recorded on a Grass polygraph (Model 79, Grass Instruments) and digitally stored using a data acquisition and analysis system (Grass Instruments, Polyview). Resting PI (the time between heart beats) and mean arterial pressure (MAP) were obtained prior to and after cutting both vagus nerves high in the neck. The nerves were cut to eliminate vagally mediated reflex effects on HR, occurring when BP increases following injection of peptide.
Vagal isolation
The peripheral end of the right vagus nerve was cleared of connective tissue, placed across platinum electrodes and stimulated every 30 s at a supramaximal voltage of 7.5 V, 1 ms, 2–4 Hz for 5 s using a square wave stimulator (Grass SD9). The frequency chosen for use was that which increased PI by ∼50 ms (i.e. at a resting PI of 151 ms, an increase in PI of 50 ms is equivalent to a decrease in HR of 98 beats min−1 for control mice (HR change from 397 to 299 beats min−1); a decrease in HR of 111 beats min−1 for GAL-KO mice with a resting PI of 141 ms (HR change from 425 to 314 beats min−1: see Figure 1a top panel); in GAL-1R-KO mice with a resting PI of 130 ms, an increase in PI of 50 ms is equivalent to a decrease in HR of 128 beats min−1 (HR change from 461 to 333 beats min−1) and in the corresponding control mice to a change in HR of 145 beats min−1 (HR change from 496 to 351 beats min−1: see Figure 1a bottom panel)). The change in PI (ΔPI) evoked by vagal stimulation is linearly related to stimulation frequency and not dependent on the resting PI (see Parker et al., 1984), while the magnitude of the fall in HR in response to stimulation of the vagus nerve is dependent on the resting HR and not linearly related. We chose to use ΔPI because of its linear relationship to stimulus frequency.
Figure 1.

Control (□), GAL-KO mice (▪). Top panels, mean±s.e.m. of resting PI in control and GAL-KO mice, with intact vagus nerves (solid bars) and after the vagus nerves are cut (hatched bars) are shown in panel (a). The histogram shows no difference in PI in GAL-KO mice from controls. After the vagus nerves were cut, PI significantly decreased in both groups of mice. There was no significant difference in PI between the two groups of mice before or after the nerves were cut. There was no difference in MAP between the two groups of mice. *P<0.05. Bottom panels show mean±s.e.m. of resting PI in control and GAL-1R-KO mice with intact vagus nerves (solid bars) and after the vagus nerves are cut (hatched bars) are shown in panel (a). The histogram shows no difference in PI in GAL-1R-KO mice from controls. After the vagus nerves were cut, PI decreased significantly in both groups of mice. There was no significant difference in PI between the two groups of mice before or after the nerves are cut. There was no difference in MAP between the two groups of mice, panel (b). **P<0.01.
Sympathetic isolation
In the mouse, it was not possible to identify the cardiac nerve as done in the rat due to its size and location (Smith-White et al., 2002). Therefore, a fine nerve that lay in the neck, dorsal to the vagus and in close proximity to the carotid artery was traced to the heart and identified as a sympathetic branch by acceleration of the heart when stimulated. The nerve was stimulated towards the heart and evoked an increase in HR that could be blocked by propranolol, and a small increase in BP, consistent with sympathetic activation. The nerve was lifted clear of all tissue to avoid stimulus spread and placed across platinum electrodes, kept covered and moist when not stimulated. The nerve was stimulated with stimulus parameters: 25 V, 1 ms, for a period of 2 min at a frequency of 10 Hz. A lower voltage of 10 V, 6 Hz inhibited vagal bradycardia for a short period, and was abolished following administration of propranolol consistent with the action of NAd. The inhibition evoked by higher stimulation survived β-adrenoceptor blockade. During the period of sympathetic stimulation, the vagus nerve was not stimulated.
Recording procedures and analysis
Cardiac sympathetic nerve stimulation and bolus injection of GAL are proposed to attenuate subsequent slowing of the heart evoked by stimulation of the vagus nerve. We interpret the magnitude of the per cent inhibition of the increase in PI (ΔPI) evoked by stimulation of the vagus nerve as an indication of prejunctional activity of NPY and related analogs (Potter et al., 1989). This was calculated using the equation:
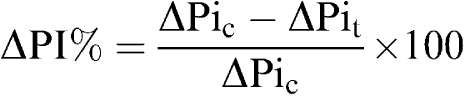 |
ΔPic is the increase in PI evoked by vagal stimulation in the control period and ΔPit is the response for the treatment period. The treatment period refers to the change in increase in PI in either the presence of the peptide, or following sympathetic stimulation. ΔPi is calculated using Polyview (Grass Instruments) from the area under curve produced by the increase in PI following vagal stimulation. T50 is the time to half recovery of the inhibitory response from maximal inhibition of the increase in PI evoked by vagal stimulation. Both resting PI and ΔPI evoked by stimulation of the vagus nerve were recorded to separate direct effects on HR from those acting on vagal pathways.
We interpret the increase in systolic BP (ΔBP) following drug administration as a measure of postjunctional NPY activity (Potter et al., 1989), and was calculated using the equation: ΔBP mmHg=BPc−BPt, where BPc is the control systolic BP and BPt is systolic BP during the treatment period.
Dose–response curves were generated for each of the parameters measured after increasing the doses of NPY or Gal, given as an intravenous, bolus dose. The parameters measured were: maximum per cent inhibition of the increase in PI evoked by stimulation of the vagus nerve (ΔPI%); time to half recovery of the inhibitory response (T50, min); maximum increase in BP (ΔBP, mmHg); duration of the change in BP (BP duration, min).
Data analysis
Data were pooled for both males and females following initial studies that showed no difference in responses. Data are presented as mean±standard error of the mean (s.e.m.). All data were normally distributed. Between group comparisons were performed by one-way ANOVA. If a significant difference was indicated, a Student's t-test was used to determine which dose was significantly different from the control response. Where appropriate the P-values were corrected using the Dunn–Sidak method. P-values of <0.05 were considered significant.
Experimental protocol
Drugs were administered in aliquots of 50 μl as an intravenous, bolus dose. Following drug application, the venous cannula was rinsed with 50 μl of warmed heparinised saline. Doses of both GAL and NPY ranged from 2.5 to 40 μg kg−1 (GAL: 0.8 to 13 nmol kg−1 and NPY: 0.5 to 10 nmol kg−1). Doses of NPY higher than 10 nmol kg−1 were considered not within the physiological range. An extended time period between GAL injections was allowed to prevent tachyphylaxis to GAL (Ulman et al., 1992).
Previously, we have reported that NPY inhibits cardiac vagal activity in the rat by activating the Y2 receptor (Smith-White et al., 2001); therefore, a selective antagonist for the Y2 receptor, (S)-N2-[[1-[2-[4-[(R,S)-5,11-dihydro-6(6h)-oxodibenz[b,e]azepin-11-yl]-1-piperazinyl]-2-oxo-ethyl]cyclopentyl]acetyl]-N-[2-[1,2-dihydro-3,5(4H)-dioxo-1,2-diphenyl-3-H-1, 2,4-triazol-4-y]ethyl]-argininamid (BIIE0246; Doods et al., 1999), was administered to control and GAL-1R-KO mice to block the inhibitory effect of NPY on ΔPI following stimulation of the sympathetic nerve and thus show only the effect of endogenously released GAL on ΔPI. The dose of BIIE0246 used here was higher than used in a previous study in the rat (Smith-White et al., 2001), in order to completely block and not just reduce NPY-evoked inhibitory effects on ΔPI. The effect of BIIE0246 was tested by an absence of Y2-mediated cardiac vagal inhibitory activity following administration of the selective NPY Y2 agonist, N acetyl [Leu28,31] NPY 24-36. Since the dose of BIIE0246 was high, recovery of the inhibitory activity was not within the time frame of the experiment.
Drugs
BIIE0246 was a gift from Dr Henri Doods (Boehringer Ingelheim Pharma, Biberach, Germany). Drugs commercially available were: NPY, rat/human (Calbiochem-Novabiochem AG Läufelfingen, Switzerland); GAL, rat (Peninsula Laboratories, Inc., Belmont, CA, U.S.A.); propranolol (Sigma-Aldrich Pty. Ltd, Castle Hill, NSW, Australia). All drugs were dissolved in 0.09% saline. Bolus doses of drug were delivered on a mol kg−1 basis. Propranolol (1 mg kg−1) was administered to mice after initial identification of the sympathetic nerve to eliminate inhibitory activity evoked by NAd and to prevent unwanted reflex effects due to increasing HR on an already fast beating heart. Administration of propranolol did not significantly decrease resting HR in the mouse.
All drugs were easily dissolved in saline except for the Y2 antagonist, BIIE0246. This synthetic antagonist was first treated with methanol followed by saline. The vehicle had no effect on the measured parameters in the mouse.
Results
Resting PI and MAP in control, GAL-KO and GAL-R-KO mice
The resting PI in GAL-KO mice with intact vagus nerves was not significantly different compared to control mice: (control: 176±5 ms, 341 beats min−1, n=10; GAL-KO: 171±10 ms, 351 beats min−1, n=16). MAP in control mice was not different from that in GAL-KO mice (control: 72±6 mmHg; GAL-KO: 67±6 mmHg). This is shown in the upper panel of Figure 1(b).
After the vagus nerves were cut, PI in the control group decreased to 151±8 ms (397 beats min−1), and to 141±11 ms (425 beats min−1) in GAL-KO mice. There was a significant difference in PI before and after the nerves were cut in both groups of mice (P<0.05), with no significant difference between groups. This can be seen in the upper panel of Figure 1(a).
The resting PI in GAL-1R-KO mice with intact vagus nerves was not significantly different compared to control mice; control: 155±5 ms (387 beats min−1), n=10; GAL-1R-KO: 158±5 ms (380 beats min−1), n=10. MAP of 72±4 mmHg in control mice was not different from 70±3 mmHg for GAL-1R-KO mice. This is shown in the bottom panel of Figure 1(b).
After the vagus nerves were cut, PI in the control group decreased to 121±5 ms (496 beats min−1), and to 130±6 ms (462 beats min−1) in GAL-1R-KO mice. While there was a significant difference in PI before and after the nerves were cut in both groups of mice (P<0.01), there was no significant difference between the groups before or after cutting of the vagus nerves. This is shown in the bottom panel of Figure 1(a).
Effect of GAL on cardiac vagal activity in control, GAL-KO and GAL-1R-KO mice
In control mice, an intravenous bolus dose of GAL (0.8–13 nmol kg−1; n=4–6) attenuated the increase in PI evoked by vagal stimulation in a dose-dependent manner. ΔPI in control mice ranged from 33±7 to 78±9.5%. The T50 ranged from 2±0.5 to 7±1 min. GAL increased BP, but this effect was not dose dependent. ΔBP ranged from 11±2 to 15±3 mmHg with a duration of 3±1–4.5±1 min. Figure 2 shows a polygraph trace from one mouse for a 5 nmol kg−1 dose of GAL. Figure 3 shows the dose–response data for the group.
Figure 2.

Polygraph trace shows the effect of an intravenous injection of GAL in a control mouse. GAL attenuated the ability of the vagus nerve to slow the heart and transiently increased BP by 10 mmHg following injection. In this particular mouse, there was a small decrease in MAP, 3 min after injection. GAL had no effect on resting PI.
Figure 3.

Control (▵), GAL-KO mice (▴). Results of a dose–response curve generated for GAL in control and GAL-KO mice. Graph (a) measures the per cent change in the increase in PI (ΔPI%) evoked by stimulation of the vagus nerve, (b) is a measure of the time to half recovery of the inhibitory effect (T50), (c) measures change in BP (ΔBP, mmHg), and (d) measures the duration of the BP effect (BP duration, min). There is an increase in ΔPI% with each dose of GAL in control mice (n=4–6). In GAL-KO mice, the inhibitory effect of GAL was not significantly different from that in control mice (n=5–7). In both control and GAL-KO mice, the T50 at doses above 6 nmol kg−1 was diminished. In both groups of mice, GAL increased BP slightly with no significant difference in either magnitude or duration.
In GAL-KO mice, an intravenous bolus dose of GAL (0.8–13 nmol kg−1; n=5–7) dose dependently attenuated the increase in PI evoked by vagal stimulation. The inhibition of ΔPI in control mice ranged from 37±7 to 72±6%. The T50 ranged from 2.5±1 to 4±1 min. GAL increased BP, as in controls, and this effect was not dose dependent. ΔBP ranged from 8±2 to 9±1.5 mmHg with a duration of 4±1.5–5±2 min. The pressor response and inhibitory effect on cardiac vagal activity evoked by intravenous injection of GAL in control mice was not significantly different from the responses in GAL-KO mice, as shown in Figure 3.
In GAL-1R-KO mice, intravenous injection of a bolus dose of GAL (0.8–13 nmol kg−1; n=3–4) did not attenuate the increase in PI evoked by vagal stimulation at any dose within the range. GAL increased BP, as in controls, and this effect was not dose dependent. ΔBP ranged from 8±2 to 9.5±1 mmHg with a duration of 4.5±1.5–5±1.5 min. The ΔBP following injection of GAL in control mice was not significantly different from the ΔBP in GAL-1R-KO mice (not shown).
Effect of NPY on BP and cardiac vagal activity in control and GAL-KO mice
In control mice, an intravenous bolus dose of NPY (0.5–10 nmol kg−1; n=5–7) attenuated the ΔPI evoked by vagal stimulation in a dose-dependent manner. ΔPI in control mice ranged from 13±10% to 67±7%. The T50 ranged from 0.5 to 5±1 min. The ΔBP following an intravenous, bolus dose of NPY was dose dependent. ΔBP ranged from 25±6 to 60±3 mmHg with duration of 7±1–12±1 min.
In GAL-KO mice, the attenuation of ΔPI evoked by vagal stimulation following an intravenous, bolus dose of NPY was dose dependent. ΔPI in GAL-KO mice ranged from 11±4% to 72±2%. The T50 ranged from 1±0.5 to 6±0.5 min. The ΔBP following an intravenous, bolus dose of NPY was dose dependent. ΔBP ranged from 22±3 to 60±10 mmHg, with a duration of 6±0.5–10±2 min. The pressor response and inhibitory effect on cardiac vagal activity evoked by intravenous injection of NPY in control mice was not significantly different from the responses in GAL-KO mice, as shown in Figure 4.
Figure 4.

Control (□), GAL-KO mice (▪). Results of a dose–response curve generated for NPY in control and GAL-KO mice. Graph (a) measures per cent change in the increase in PI (ΔPI%) evoked by stimulation of the vagus nerve, (b) is a measure of the time to half recovery of the inhibitory effect (T50), (c) measures change in BP (ΔBP, mmHg), and (d) measures the duration of the BP effect (BP duration, min). There is an increase in ΔPI% with each dose of NPY in control mice (n=5–7). In GAL-KO mice, the inhibitory effect of GAL was not significantly different from that in control mice (n=7). In both control and GAL-KO mice, the T50 increased with increasing doses of NPY, with no significant difference in the response. In both groups of mice, NPY increased BP in a dose-dependent manner with no significant difference in either magnitude or duration.
Cardiac sympathetic stimulation in the mouse
Stimulation of the sympathetic nerve in three mice decreased PI by approximately 75 ms (i.e. increased HR by ∼260 beats min−1; from ∼340 to 600). BP was also increased during stimulation of the nerve. Stimulation of the nerve at 6 Hz, 1 ms, 10 V (S6) for a period of 2 min attenuated the ΔPI evoked by stimulation of the vagus nerve. A polygraph trace from one mouse is shown in Figure 5, panel (a). The inhibitory effect on vagal action was short lasting and abolished in the presence of propranolol, 1 mg kg−1 (not shown). Following a higher stimulation frequency, 10 Hz, 1 ms, 25 V (S10) for a period of 2 min, ΔPI evoked by vagal stimulation was inhibited for a prolonged period, ΔPI=92±2%, T50=7±1 min, n=3, as shown in Figure 5, panel (b). In the presence of propranolol (1 mg kg−1), ΔPI was inhibited by 80±3% with a T50 of 6±1 min, n=4, an effect that was significantly different from the control response for ΔPI but not T50, as shown in Figure 6, panel (a).
Figure 5.

Polygraph trace (a) from the chart recorder for one control mouse showing the ΔPI following a period of sympathetic nerve stimulation at 6 Hz for 2 min (S6). Following sympathetic stimulation, there was attenuation of the ΔPI evoked by vagal stimulation that was of short duration. BP increased and PI decreased during stimulation of the sympathetic nerve. Polygraph trace (b) from the chart recorder in the same mouse showing an inhibitory effect on ΔPI following a period of sympathetic nerve stimulation (black line) at an increased frequency of 10 Hz for 2 min (S10). ΔPI inhibition and ΔBP are greater at the higher frequency of stimulation. The stimulus to the vagus nerve was discontinued during the period of sympathetic stimulation.
Figure 6.

Polygraph trace (a) from a chart recorder showing sympathetic stimulation (black line) in the presence of propranolol (10 Hz for 2 min, S10). A prolonged attenuation of the ΔPI evoked by stimulation of the vagus nerve was still evident following stimulation of the sympathetic nerve (top panel). Trace (b) shows the effect on ΔPI of cardiac sympathetic nerve, 30 min after a bolus dose of Y2 antagonist BIIE0246. The attenuation of ΔPI was reduced in the presence of the antagonist. There was no significant increase in BP during sympathetic stimulation in the presence of propranolol. The vagus nerve stimulation was ceased during sympathetic stimulation.
At 30 min following a bolus dose of Y2 antagonist BIIE0246 (1 μmol kg−1), the inhibitory effect of sympathetic stimulation on vagal action was attenuated, ΔPI, 57±3% with a T50 of 2.5±0.5 min, n=3. The evoked inhibition was significantly different from both the control response and the response seen in the presence of propranolol, although only the T50 for the control showed a significant difference. A polygraph trace from one mouse is shown in Figure 6, panel (b). Group data are shown in Figure 8.
Figure 8.

Panel (a) is a summary of results from four control mice showing the effect on maximum per cent inhibition of ΔPI and time to half recovery following stimulation of the cardiac sympathetic nerve (open bar), in the presence of a bolus dose of propranolol (hatched bar) and in the presence of propranolol and the NPY Y2 antagonist, BIIE0246 (cross-hatched bar). In the presence of both antagonists, there is some remaining attenuation of the increase in PI following sympathetic stimulation. Panel (b) is a summary of results in three GAL-1R-KO mice showing the effect on maximum per cent inhibition of ΔPI and time to half recovery following stimulation of the cardiac sympathetic nerve (open bar), in the presence of propranolol (hatched bar) and in the presence of propranolol and the NPY Y2 antagonist, BIIE0246 (cross-hatched bar). After a bolus dose of propranolol, the attenuation was significantly reduced and then abolished after subsequent administration of BIIE0246. Significant differences are shown; **P<0.01; ***P<0.001.
The effect of stimulation of the sympathetic nerves on cardiac vagal activity, in GAL-1R-KO mice, alone and in the presence of the Y2 antagonist BIIE0246
Following a period of stimulation of the sympathetic nerve (S10) in the GAL-1R-KO mouse, cardiac vagal activity was inhibited by 93±7% with a T50 of 2.5±0.25 min, n=3. In the presence of propranolol (1 mg kg−1), cardiac vagal activity was inhibited by 56±2% with a T50 of 1.5±0.5 min, n=5. This is shown in Figure 7, top panel.
Figure 7.

Panel (a) shows a polygraph trace from a chart recorder for one GAL-1R-KO mouse showing the effect on ΔPI of a period of cardiac sympathetic nerve stimulation at 10 Hz, 25 V for 2 min (S10). Following stimulation of the sympathetic nerve and in the presence of propranolol, cardiac vagal activity was attenuated. Note that there is no increase in PI following stimulation of the sympathetic nerve in the presence of propranolol. Panel (b) shows a polygraph trace for one GAL-1R-KO mouse showing the effect on ΔPI of a period of cardiac sympathetic nerve stimulation at 10 Hz, 25 V for 2 min (S10) in the presence of propranolol. At 30 min after a bolus dose of the Y2 antagonist BIIE0246, the inhibition of ΔPI following sympathetic stimulation was abolished.
At 30 min following a bolus dose of the Y2 antagonist BIIE0246 (1 μmol kg−1), the inhibitory effect of sympathetic stimulation on vagal action was abolished, P<0.001, n=3. A polygraph trace from one mouse is shown in Figure 7, bottom panel. Group data and a summary of effects of sympathetic stimulation in the mouse are shown in Figure 8.
Discussion
The aim of this study was to determine the role of GAL in autonomic control of cardiovascular function. In anaesthetised mice, GAL administered intravenously attenuated the ability of the vagus nerve to slow the heart in a dose-dependent manner. The inhibitory effect of GAL was not modified by the absence of GAL in GAL-KO mice, but was absent in GAL-1R-KO mice.
High doses of GAL were found to exhibit tachyphylaxis in control and GAL-KO groups, an effect more evident in the recorded duration of the response. GAL also increased BP in both control and GAL-KO mice; however, in half the mice studied, the increase was followed by a secondary decrease in BP. The biphasic response might explain why GAL does not increase BP in a dose-dependent manner. The effect on BP is in contrast with the potent effects evoked by NPY in the mouse (Smith-White et al., 2002) and other species (Revington et al., 1987; Moriarty et al., 1992; Ulman et al., 1992). In GAL-KO mice the cardiac vagal inhibitory and BP effects of exogenous NPY were unchanged. These results suggest that in the absence of GAL, neither GAL- nor NPY receptor-mediated effects are altered.
Electrical stimulation of the cardiac sympathetic nerve is shown here in the mouse to increase HR, as shown previously in the other species (Liddell & Sherrington, 1929; Yasunaga & Nosaka, 1979). Following a period of sympathetic stimulation in the mouse, there is a prolonged attenuation of subsequent cardiac vagal activity. Observations in other species suggest this effect to be mediated by multiple transmitters released by the sympathetic nerve during stimulation (Potter, 1985; Revington et al., 1990; Courtice et al., 1993; Smith-White et al., 1999). While NAd has been shown to inhibitcardiac vagal activity, its effect is very short lasting (Potter, 1988). The inhibitory effect of NAd on vagal transmission was reported to be mediated by activation of a prejunctional α1-adrenoceptor, but the activity diminished quickly after sympathetic activity ceased (Wetzel et al., 1985). We show an inhibitory effect on vagal activity, followed by stimulation of the sympathetic nerve that is prolonged. The inhibitory effect was of long duration and was reduced in the presence of propranolol and further reduced by the NPY Y2 antagonist BIIE0246. In control mice, propranolol reduced the inhibitory effect that followed sympathetic stimulation by approximately 10%, with a further reduction of 20% following administration of BIIE0246. The remaining inhibition is attributed to the effect of GAL in these mice. This suggests that NPY, under the conditions of the experiment, plays a minor role in modulation of cholinergic transmission in this species.
Stimulation of the sympathetic nerve in GAL-1R-KO mice evoked an inhibitory effect on cardiac vagal activity that was similar in magnitude to that observed in control mice, but was reduced by 50% in the presence of propranolol and abolished following administration of Y2 antagonist BIIE0246. The amplification of inhibitory effect of adrenoceptor activation suggests that the GAL-1R receptor may affect the release of NAd from the sympathetic nerve; however, it was beyond the scope of this study to determine if the effect was at the ganglion or nerve terminal. The time to half recovery of the inhibitory effect attributed to NAd and NPY following administration of selective antagonists was not significantly altered in control mice, although the overall decrease in duration suggests that GAL had a significant influence on prolonging the inhibitory effect on vagal action. We noted that GAL-1R-KO mice responded differently to anaesthesia compared to control and GAL-KO mice, requiring more frequent supplementation of anaesthetic during the experimental period; therefore, it is possible that the decrease in time to half recovery observed in these mice may be attributed to wakefulness.
In anaesthetised GAL-KO and GAL-1R-KO mice, resting PI (or HR) and BP were not different from control mice. The decrease in PI (or increase in HR) seen after cutting the vagus nerves was similar in both knockout and control groups of mice, suggesting that autonomic nervous activity to the heart is not altered by the absence of GAL in the nerves or the absence of the GAL-1R receptor. This suggests that GAL acts peripherally to evoke its inhibitory action on vagal transmission.
The findings of this study support the view that cardiac parasympathetic neurons in the mouse possess both GAL and NPY receptors that, when activated, reduce cholinergic transmission. We propose that following stimulation of the sympathetic nerve, both GAL and NPY act to attenuate subsequent cardiac vagal activity, with the actions of GAL predominating. To date, the mouse is the first species to show that both GAL and NPY inhibit cardiac vagal activity, as in all species studied previously it is either GAL or NPY that mediates this important conservation of function. The preferential release of GAL by the sympathetic nerve in this species may serve to extend the effects of sympathetic stimulation without the potent increases in pressor activity associated with NPY.
Acknowledgments
We thank Dr Henri Doods for the gift of BIIE0246, Professor David Wynick for supplying the GAL-KO mice, Y.J. Hort for technical assistance and the National Health and Medical Research Council of Australia for supporting this work.
Abbreviations
- ACh
acetylcholine
- BIIE0246
((S)-N2-[[1-[2-[4-[(R,S)-5,11-dihydro-6(6h)-oxodibenz[b,e]azepin-11-yl]-1-piperazinyl]-2-oxo-ethyl]cyclopentyl]acetyl]-N-[2-[1,2-dihydro-3,5(4H)-dioxo-1,2-diphenyl-3-H-1,2,4-triazol-4-y]ethyl]-argininamid)
- dmnX
dorsal motor nucleus of the vagus
- GAL
galanin
- GAL-KO
galanin knockout
- GAL-1R-KO
galanin 1R receptor knockout
- NAd
noradrenaline
- NPY
neuropeptide Y
- nTS
nucleus of the tractus solitarius
- PI
pulse interval
- ΔPI
change in pulse interval
References
- CAREY D.G., IISMAA T.P., HO K.Y., RAJKOVIC I.A., KELLY J., KRAEGEN E.W., FERGUSON J., INGLIS A.S., SHINE J., CHISHOLM D.J. Potent effects of human galanin in man: growth hormone secretion and vagal blockade. J. Clin. Endocrin. Metabol. 1993;77:90–93. doi: 10.1210/jcem.77.1.7686918. [DOI] [PubMed] [Google Scholar]
- COURTICE G.P., POTTER E.K., MCCLOSKEY D.I. Inhibition of cardiac vagal action by galanin but not neuropeptide Y in the brush-tailed possum Trichosurus vulpecula. J. Physiol. 1993;461:379–386. doi: 10.1113/jphysiol.1993.sp019518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRAWLEY J.N. Minireview. Galanin–acetylcholine interactions: relevance to memory and Alzheimer's disease. Life Sci. 1996;58:2185–2199. doi: 10.1016/0024-3205(96)00093-8. [DOI] [PubMed] [Google Scholar]
- DEGLI UBERTI E.C., BONDANELLI M., MARGUTTI A., AMBROSIO M.R., VALENTINI A., CAMPO M., FRANCESCHETTI P., ZATELLI M.C., PANSINI R., TRASFORINI G. Acute administration of human galanin in normal subjects reduces the potentiating effect of pyridostigmine-induced cholinergic enhancement on release of norepinephrine and pancreatic polypeptide. Neuroendocrinology. 1996;64:398–404. doi: 10.1159/000127143. [DOI] [PubMed] [Google Scholar]
- DOODS H., GAIDA W., WIELAND H.A., DOLLINGER H., SCHNORRENBERG G., ESSER F., ENGEL W., EBERLEIN W., RUDOLF K. BIIE0246: a selective and high affinity neuropeptide YY2 receptor antagonist. Eur. J. Pharm. 1999;384:R3–R5. doi: 10.1016/s0014-2999(99)00650-0. [DOI] [PubMed] [Google Scholar]
- FISONE G., BARTFAI T., NILSSON S., HÖKFELT T. Galanin inhibits the potassium-evoked release of acetylcholine and the muscarinic receptor-mediated stimulation of phosphoinositide turnover in slices of monkey hippocampus. Brain Res. 1991;568:279–284. doi: 10.1016/0006-8993(91)91409-t. [DOI] [PubMed] [Google Scholar]
- HARFSTRAND A., FUXE K., MELANDER T., HÖKFELT T., AGNATI L.F. Evidence for a cardiovascular role of central galanin neurons: focus on interactions with alpha 2-adrenergic and neuropeptide Y mechanisms. J. Cardiovasc. Pharmacol. 1987;10:S199–S204. [PubMed] [Google Scholar]
- HOWARD A.D., TAN C., SHIAO L.L., PALYHA O.C., MCKEE K.K., WEINBERG D.H., FEIGHNER S.D., CASCIERI M.A., SMITH R.G., VAN DER PLOEG L.H., SULLIVAN K.A. Molecular cloning and characterization of a new receptor for galanin. FEBS Lett. 1997;405:285–290. doi: 10.1016/s0014-5793(97)00196-8. [DOI] [PubMed] [Google Scholar]
- IISMAA T.P., SHINE J. Galanin and galanin receptors. Res. Prob. Cell. Diff. 1999;26:257–291. doi: 10.1007/978-3-540-49421-8_12. [DOI] [PubMed] [Google Scholar]
- JACOBY A.S., HOLMES F.E., HORT Y.J., SHINE J., IISMAA T.P. Phenotypic analysis of Galr1 knockout mice reveals a role for GALR1 galanin receptor modulating seizure activity but not nerve regeneration. Lett. Pept. Sci. 2002a;8:139–146. [Google Scholar]
- JACOBY A.S., HORT Y.J., CONSTANTINESCU G., SHINE J., IISMAA T.P. A critical role for GALR1 galanin receptor in galanin regulation of neuroendocrine function and seizure activity. Mol. Brain Res. 2002b;107:195–200. doi: 10.1016/s0169-328x(02)00451-5. [DOI] [PubMed] [Google Scholar]
- KOLAKOWSKI L.F., JR, O'NEILL G.P., HOWARD A.D., BROUSSARD S.R., SULLIVAN K.A., FEIGHNER S.D., SAWZDARGO M., NGUYEN T., KARGMAN S., SHIAO L.L., HRENIUK D.L., TAN C.P., EVANS J., ABRAMOVITZ M., CHATEAUNEUF A., COULOMBE N., NG G., JOHNSON M.P., THARIAN A., KHOSHBOUEI H., GEORGE S.R., SMITH R.G., O'DOWD B.F. Molecular characterization and expression of cloned human galanin receptors GALR2 and GALR3. J. Neurochem. 1998;71:2239–2251. doi: 10.1046/j.1471-4159.1998.71062239.x. [DOI] [PubMed] [Google Scholar]
- KUMMER W. Galanin- and neuropeptide Y-like immunoreactivities coexist in paravertebral sympathetic neurones of the cat. Neurosci. Lett. 1987;78:127–131. doi: 10.1016/0304-3940(87)90620-3. [DOI] [PubMed] [Google Scholar]
- LEGAKIS I.N., MANTZOURIDIS T., SARAMANTIS A., PHENEKOS C., TZIORAS C., MOUNTOKALAKIS T. Human galanin secretion is increased upon normal exercise test in middle-age individuals. Endocr. Res. 2000;26:357–364. doi: 10.3109/07435800009066173. [DOI] [PubMed] [Google Scholar]
- LIDDELL E.G.T., SHERRINGTON C. Mammalian Physiology, A course of practical exercises 1929London: Clarendon Press; 38–47.ed. Liddell, E.G.T. & Sherrington, C [Google Scholar]
- LINDH B., LUNDBERG J.M., HÖKFELT T. NPY-, galanin-, VIP/PHI-, CGRP- and substance P-immunoreactive neuronal subpopulations in cat autonomic and sensory ganglia and their projections. Cell Tissue Res. 1989;256:259–273. doi: 10.1007/BF00218883. [DOI] [PubMed] [Google Scholar]
- MELANDER T., HOKFELT T., ROKAEUS A., FAHRENKRUG J., TATEMOTO K., MUTT V. Distribution of galanin-like immunoreactivity in the gastro-intestinal tract of several mammalian species. Cell Tissue Res. 1985;239:253–270. doi: 10.1007/BF00218003. [DOI] [PubMed] [Google Scholar]
- MORIARTY M., GIBBINS I.L., POTTER E.K., MCCLOSKEY D.I. Comparison of the inhibitory roles of neuropeptide Y and galanin on cardiac vagal action in the dog. Neurosci. Lett. 1992;139:275–279. doi: 10.1016/0304-3940(92)90570-w. [DOI] [PubMed] [Google Scholar]
- MORRIS J.L., GIBBINS I.L., HOLMGREN S. Galanin is more common than NPY in vascular sympathetic neurons of the brush-tailed possum. Regul. Pept. 1992;37:101–109. doi: 10.1016/0167-0115(92)90659-i. [DOI] [PubMed] [Google Scholar]
- PARKER E.M., IZZARELLI D.G., NOWAK H.P., MAHLE C.D., IBEN L.G., WANG J., GOLDSTEIN M.E. Cloning and characterization of the rat GALR1 galanin receptor from Rin14B insulinoma cells. Brain Res. Mol. Brain Res. 1995;34:179–189. doi: 10.1016/0169-328x(95)00159-p. [DOI] [PubMed] [Google Scholar]
- PARKER P., CELLER B.G., POTTER E.K., MCCLOSKEY D.I. Vagal stimulation and cardiac slowing. J. Auton. Nerv. Syst. 1984;11:226–231. doi: 10.1016/0165-1838(84)90080-8. [DOI] [PubMed] [Google Scholar]
- POTTER E.K. Prolonged non-adrenergic inhibition of cardiac vagal action following sympathetic stimulation: neuromodulation by neuropeptide Y. Neurosci. Lett. 1985;54:117–121. doi: 10.1016/s0304-3940(85)80065-3. [DOI] [PubMed] [Google Scholar]
- POTTER E.K. Neuropeptide Y as an autonomic neurotransmitter. Pharmacol. Ther. 1988;37:251–273. doi: 10.1016/0163-7258(88)90028-9. [DOI] [PubMed] [Google Scholar]
- POTTER E.K., MITCHELL L., MCCLOSKEY M.J.D., TSENG A., GOODMAN A.E., SHINE J., MCCLOSKEY D.I. Pre- and postjunctional actions of neuropeptide Y and related peptides. Regul. Pept. 1989;25:167–177. doi: 10.1016/0167-0115(89)90258-9. [DOI] [PubMed] [Google Scholar]
- REVINGTON M.L., MCCLOSKEY D.I., POTTER E.K. Effects of neuropeptide Y on the pressor responses to phenylephrine and to activation of the sympathetic nervous system in anaesthetized rats. Clin. Exp. Pharmacol. Physiol. 1987;14:703–710. doi: 10.1111/j.1440-1681.1987.tb01895.x. [DOI] [PubMed] [Google Scholar]
- REVINGTON M., POTTER E.K., MCCLOSKEY D.I. Prolonged inhibition of cardiac vagal action following sympathetic stimulation and galanin in anaesthetized cats. J. Physiol. 1990;431:495–503. doi: 10.1113/jphysiol.1990.sp018342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH-WHITE M.A., HARDY T.A., BROCK J.A., POTTER E.K. Effects of a selective neuropeptide Y Y(2) receptor antagonist, BIIE0246, on Y(2) receptors at peripheral neuroeffector junctions. Br. J. Pharmacol. 2001;132:861–868. doi: 10.1038/sj.bjp.0703879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH-WHITE M.A., HERZOG H., POTTER E.K. Role of neuropeptide Y Y(2) receptors in modulation of cardiac parasympathetic neurotransmission. Regul. Pept. 2002;103:105–111. doi: 10.1016/s0167-0115(01)00368-8. [DOI] [PubMed] [Google Scholar]
- SMITH-WHITE M.A., WALLACE D., POTTER E.K. Sympathetic–parasympathetic interactions at the heart in the anaesthetised rat. J. Auton. Nerv. Syst. 1999;75:171–175. doi: 10.1016/s0165-1838(98)00169-6. [DOI] [PubMed] [Google Scholar]
- SULLIVAN K.A., SHIAO L.L., CASCIERI M.A. Pharmacological characterization and tissue distribution of the human and rat GALR1 receptors. Biochem. Biophys. Res. Commun. 1997;233:823–828. doi: 10.1006/bbrc.1997.6542. [DOI] [PubMed] [Google Scholar]
- TATEMOTO K., ROKAEUS A., JORNVALL H., MCDONALD T.J., MUTT V. Galanin – a novel biologically active peptid from porcine intestine. FEBS Lett. 1983;164:124–128. doi: 10.1016/0014-5793(83)80033-7. [DOI] [PubMed] [Google Scholar]
- ULMAN L.G., POTTER E.K., MCCLOSKEY D.I. Effects of sympathetic activity and galanin on cardiac vagal action in anaesthetized cats. J. Physiol. 1992;448:225–235. doi: 10.1113/jphysiol.1992.sp019038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG S., HE C., MAGUIRE M.T., CLEMMONS A.L., BURRIER R.E., GUZZI M.F., STRADER C.D., PARKER E.M., BAYNE M.L. Genomic organization and functional characterization of the mouse GalR1 galanin receptor. FEBS Lett. 1997;411:225–230. doi: 10.1016/s0014-5793(97)00695-9. [DOI] [PubMed] [Google Scholar]
- WETZEL G.T., GOLDSTEIN D., BROWN J.H. Acetylcholine release from rat atria can be regulated through an alpha 1-adrenergic receptor. Circ. Res. 1985;56:763–766. doi: 10.1161/01.res.56.5.763. [DOI] [PubMed] [Google Scholar]
- WYNICK D., SMALL C.J., BACON A., HOLMES F.E., NORMAN M., ORMANDY C.J., KILIC E., KERR N.C., GHATEI M., TALAMANTES F., BLOOM S.R., PACHNIS V. Galanin regulates prolactin release and lactotroph proliferation. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12671–12676. doi: 10.1073/pnas.95.21.12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU Y., JOHANSSON O., ROKAEUS A. Distribution and chromatographic analysis of galanin immunoreactivity in the heart. Peptides. 1995;16:73–79. doi: 10.1016/0196-9781(94)00147-x. [DOI] [PubMed] [Google Scholar]
- YASUNAGA K., NOSAKA S. Cardiac sympathetic nerves in rats: anatomical and functional features. Jpn. J. Physiol. 1979;29:691–705. doi: 10.2170/jjphysiol.29.691. [DOI] [PubMed] [Google Scholar]