

Abstract
The alternatively spliced, short and long cholecystokinin receptors (CCK2S and CCK2L) were expressed in NIH3T3 cells, and compared using radioligand-binding assays with identical buffer and incubation conditions.
As judged by a saturation analysis, the selective CCK2-receptor antagonist radioligand [3H]-JB93182 did not discriminate between the CCK2S or CCK2L receptors.
A global analysis of competition studies, using a range of structurally diverse, CCK-receptor selective ligands, provided further evidence that these receptor subtypes were pharmacologically indistinguishable. However, when analysed individually a number of small, yet significant differences were observed with some of the compounds.
These data are consistent with previous study that suggested a possible pharmacological difference between these isoforms, at least in terms of the CCK2-receptor antagonist, L-365,260. However, it would appear that the pharmacological profile of these compounds is not consistent with their affinity at the putative G1/G2 receptors previously described by Harper et al.
Keywords: Cholecystokinin, CCK2 receptor, [3H]-JB93182, human, short and long isoforms, G1 and G2 receptors
Introduction
The cholecystokinin (CCK) receptor family is composed of two distinct gene products, the CCK1 and CCK2 receptors, and, as such, is one of the smallest G-protein-coupled receptor (GPCR) families known. Despite the lack of multiple distinct proteins, pharmacological heterogeneity of the CCK1 and CCK2 receptors has been proposed on numerous occasions (Sankaran et al., 1980; Durieux et al., 1986; Harper et al., 1996; Roberts et al., 1996; Bellier et al., 1997; Lena et al., 1999). In the majority of these cases, any functional significance of the heterogeneity has not been established although the possibility remains that multiple receptor subtypes are involved in the physiological modulation of CCK- and gastrin-regulated systems.
Although no additional CCK2-receptor subtypes have been identified, splice variants of the CCK2 receptor have been reported (Song et al., 1993; Miyake, 1995; Hellmich et al., 2000). These CCK2-receptor variants include the amino-terminal truncated receptor subtype (Miyake, 1995), the alternatively spliced short and long CCK2 receptors (CCK2S and CCK2L; Song et al., 1993) and most recently the CCK-Bri4sv, which is a variant of the CCK2 receptor that retains a region of intron four in the coding sequence of the receptor (Hellmich et al., 2000). Radioligand-binding studies have revealed differences in the affinity of the full-length and truncated receptor subtypes for the endogenous ligands CCK-8S and gastrin (Miyake, 1995). The truncated receptor displayed ∼1 log concentration unit lower affinity for CCK-8S than the wild-type receptor and an increased selectivity for CCK and gastrin from ∼10-fold (wild-type) to ∼100-fold at the truncated receptor. Similarly, a comparison between the CCK-Bri4sv and the wild-type CCK2 receptor revealed that the variant protein displayed differences in the binding of the endogenous agonists gastrin-17 (G-17) and glycine-extended gastrin-17 (G-Gly; Hellmich et al., 2000). It was shown that the CCK-Bri4sv receptor displayed biphasic competition curves for G-17 and, furthermore, G-Gly was ∼3-fold more potent at the variant receptor subtype (Hellmich et al., 2000). It has not been established whether these differences in agonist-binding potencies for these receptor subtypes arise from actual alterations in the receptor structure or from the formation of different G-protein-regulated affinity states of the receptors. In contrast, the CCK2S and CCK2L receptors, which have been shown to coexist in human gastric tissue samples (Song et al., 1993) and have also been demonstrated in human tumour cell lines (Biagini et al., 1997), do not differentiate between the natural hormones CCK and gastrin (Ito et al., 1994), whereas, the nonpeptide antagonist L-365,260 displayed ∼3-fold greater affinity at the short receptor isoform (Wank et al., 1994). Interestingly, this antagonist has been previously shown to discriminate between the pharmacologically defined subtypes termed G1 and G2 (Harper et al., 1996; Roberts et al., 1996). Therefore, these splice variants appeared to be potential molecular counterparts of these G1 and G2 subtypes.
The aim of this study was to pharmacologically characterise the CCK2S and the CCK2L receptor splice isoforms using a range of CCK-receptor ligands that have been previously shown to discriminate between the G1- and G2-receptor subtypes (Harper et al., 1999). Therefore, it should be possible, firstly, to determine if the coexpression of these molecularly distinct CCK2 receptors could account for previously reported receptor heterogeneity and, secondly, to establish which ligands (if any) could be used in the characterisation of the CCK2S and the CCK2L receptors in fresh human tissue, where these receptors have previously been shown to be coexpressed (Song et al., 1993; Biagini et al., 1997).
Methods
Cell culture of NIH3T3 mouse fibroblast cells
NIH3T3 cells that had undergone stable transfection with either the short (CCK2S) or long (CCK2L) human receptor isoforms were a generous gift from Professor T. Matsui, Department of Medicine, Kobe University School of Medicine, Japan. The cells were maintained in Dulbecco's Modified Eagles Medium (DMEM), supplemented with 10% newborn calf serum, 2 mM L-glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (all cell culture materials from GibcoBRL, U.K.). The cells were harvested at approximately 70% confluence by 2 min incubation in 0.25 × trypsin–EDTA solution (GibcoBRL, U.K.), followed by centrifugation (600 × g, 5 min) and freezing at −70°C. Cells were subcultured a maximum of 10 times before being reseeded from original stocks.
Membrane preparation
The cell pellets were homogenised in 20 ml of buffer A (pH 7.2 at 21±3°C; composition (mM): NaCl 130, KCl 4.7, MgCl2 5, HEPES 10, EGTA 1 and bacitracin 0.089), using a polytron PT-10 (3 × 1 s). The homogenate was centrifuged at 39,800 × g for 15 min at 4°C and the supernatant discarded. This stage was repeated once more and the final pellet resuspended in 50 mM Tris-HCl (Sigma, U.K.) with 0.089 mM bacitracin (pH 6.9 at 21±3°C) by polytron homogenisation. For competition, saturation and kinetic studies, the pellets were resuspended at a cell concentration of 2.5 × 105 cells per tube for CCK2S and 3.5 × 105 cells per tube for CCK2L.
Incubation conditions for CCK2S and CCK2L receptor assays
Determination of optimal cell number
All dilutions were conducted using 50 mM Tris-HCl (pH 6.9, 21±3°C) with 1 μM dypyridamole (preliminary studies revealed that coincubation of 1 μM dipyridamole resulted in 10±3% increase in the specific binding, data not shown). [3H]-JB93182 (2 nM) was incubated with increasing cell concentrations in a final assay volume of 500 μl for 2.5 h at room temperature (21±3°C). Nonspecific binding was defined with 1 μM YM220, a potent and selective CCK2-receptor antagonist, which is structurally unrelated to JB93182 (pKI∼10 in mouse cortex; Harper et al., 1999). All incubations were conducted in triplicate and assays were terminated by rapid filtration using a Brandell cell harvester through presoaked Whatman GF/B filters (Whatman, U.K.), which were washed (3 × 3 ml) with ice-cold 50 mM Tris-HCl (pH 6.9 at 21±3°C). Filters were transferred into scintillation vials, 5 ml Canberra Packard Ultima liquid scintillation cocktail added and, after overnight incubation, the bound radioactivity was determined by counting (3 min) in a Beckman LS6000C liquid scintillation counter. This termination procedure was used for all binding assays.
Saturation studies
NIH3T3 cell membranes were incubated in a final volume of 0.5 ml, with increasing concentrations of radiolabel (0.01–2 nM; [3H]-JB93182), for 2.5 h at 21±3°C. Total and nonspecific binding was defined using 50 μl buffer and 50 μl YM220 (final concentration 1 μM), respectively.
Kinetic studies
[3H]-JB93182 (50 μl; 20 nM) was incubated with the cell membranes for varying time intervals (0.1–180 min) for determination of the time course of association for the radiolabel (termination of the reaction was conducted as described above). To investigate the radiolabel dissociation kinetics by displacement, an excess of the CCK2-receptor selective antagonist YM220 (10 μl; 50 μM) was added after the complete association of [3H]-JB93182 (2.5 h). Total bound radioactivity, nonspecific and the residual bound, after displacement of [3H]-JB93182 by YM220, was determined at increasing time intervals (1–160 min).
Competition studies
NIH3T3 cell membranes were incubated for 2.5 h at 21±3°C with 50 μl of competing ligand (concentration range 0.1 pM-1 mM) and 50 μl of 20 nM [3H]-JB93182. YM220 (1 μM) was used to define the nonspecific binding, and was replaced with 50 μl buffer for determination of the total binding.
Data analysis
Saturation data were analysed by fitting all the individual data points to the Hill equation with and without the slope (nH) constrained to unity (one-site and free-fit model, respectively). Analysis of the saturation data using a free-fit model provided a test of the goodness of fit to the one-site model.
The individual competition curve data were expressed as the percentage in the decrease of specific [3H]-JB93182 binding (B) within each experiment. These data were then analysed using a four-parameter logistic (Equation (1); GraphPad Prism 3.02; Motulsky, 1999) with the upper (αmax) and lower (αmin) asymptotes weighted to 100 and 0% by including these values two log units above and below the lowest and highest concentrations of the competitor, respectively. The equilibrium dissociation constants (KI) values were calculated from the midpoint locations (IC50) following Cheng & Prusoff (1973; Equation (2)).
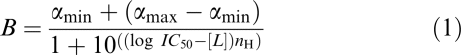 |
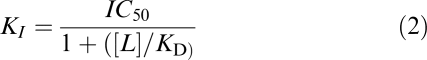 |
Statistical comparison of model parameter estimates
The dissociation constant (pKI) values, estimated from the competition studies, were compared by fitting three straight-line models (y=x, y=x+c and y=mx+c) to evaluate the relationship between these values. The sum-of-squares was reduced by minimising the perpendicular distance between the data points and the best-fit line, using the principal components analysis described by Meester et al. (1998). Preliminary affinity estimate comparisons using this procedure resulted in apparently linear relationships being interpreted as significantly different from the model describing a straight line (y=mx+c). Therefore, y=mx+c was considered to be a measure of baseline variance within the system, to which the other straight-line models were compared using an F-test (see Meester et al. (1998) for details).
Materials
[3H]-JB93182 (specific activity ∼26 Ci mmol−1) was supplied by Amersham, Buckinghamshire, U.K. JB93182 (5[[[(1S)-[[3,5-dicarboxyphenyl)amino]carbonyl]-2-phenylethyl] amino]-carbonyl]-6-[[(1-adamantylmethyl)amino]carbonyl]-indole), SR27 897 (1-[[2-(4-(2-chlorophenyl)thiazol-2-yl)-aminocarbonyl]indolyl] acetic acid), 2-NAP (2-naphthalenesulphonyl 1-aspartyl-(2-phenylethyl)amide), YM220 ((R)-1-[2,3-dihydro-2-oxo-1-pivaloylmethyl-5-(2′-pyridyl)-1H-1,4-benzodiazepin-3-yl]-3-(3-methyl-phenyl)urea), PD-134,308 ([R-(R*,R*)]-4-[[2-[[3-(1H-indol-3-yl)-2-methyl-1-oxo-2-[[(tricyclo[3.3.1.13,7]dec-2-yloxy)carbonyl]amino]propyl]amino-1-phenylethyl]amino]-4-oxobutanoic acid) and L-364,718 were synthesised by James Black Foundation chemists. Bacitracin and Trizma base® were obtained from the Sigma Chemical Co., Poole, Dorset, U.K. MgCl2, NaCl, KCl, MgCl2, HEPES and EGTA were obtained from Merck-BDH, U.K. All compounds were dissolved in dimethyl formamide to give stock concentrations of 1 mM and further dilutions were made in 50 mM Tris-HCl buffer.
Results
Relationship between the cell number of CCK2S- and CCK2L-receptor-expressing NIH3T3 cells and the binding of [3H]-JB93182
For both the CCK2S and the CCK2L receptor assays, total binding, nonspecific binding and specific binding of [3H]-JB93182 increased with increasing cell number (Figure 1a, c). No specific binding was detected in the wild-type NIH3T3 cells at cell concentrations up to 1 × 106 cells per tube (data not shown). The specific binding increased linearly over the cell number range tested for both CCK2S and CCK2L (Figure 1b, d). The membranes prepared from NIH3T3 cells expressing the CCK2S receptor subtype bound significantly more radiolabel than those expressing CCK2L receptor (total binding of [3H]-JB93182 at 1 × 106 cells per tube, value±s.e.m. (d.p.m.): CCK2S=5463±680 (n=3) and CCK2L=2987±351 (n=4); t=3.565, d.f.=16). Additionally, the specific binding was significantly higher in the CCK2S receptor membranes than in the CCK2L receptor membranes (% specific bound [3H]-JB93182 at 1 × 106 cells per tube, value±s.e.m.: CCK2S=82±1% (n=3), CCK2L=52+3% (n=4); t=7.733, d.f.=5). For the CCK2S receptor-expressing cells, a concentration of 2.5 × 105 cells per tube resulted in 68±8% specific binding (n=3), and this concentration was used in all further experiments. For the CCK2L receptor, cell concentrations of 2.5 × 105 and 4 × 105 cells per tube resulted in 37±4 and 50±2% specific binding, respectively (n=4). An intermediate concentration of 3.5 × 105 cells per tube was chosen for all further experiments using the CCK2L receptor. These concentrations of cells provided a good ‘window' of specific binding with the total binding being less than 10% of the total added radioligand (CCK2S: total added bound at 2.5 × 105 cells=3.5±0.8%; CCK2L: total added bound at 4 × 105 cells=2.5±0.5%; n=4). Additionally, at these cell concentrations the total and specific binding of [3H]-JB93182 was within the linear portion of the tissue concentration curve for both CCK2S and CCK2L.
Figure 1.

Total, nonspecific and specific binding of [3H]-JB93182 (2 nM) plotted as a function of increasing CCK2S- and CCK2L-receptor cell number (panels a, c). Increasing concentrations of membranes (400 μl) were incubated, in triplicate, with 2 nM [3H]-JB93182 (50 μl; 1 nM) for 150 min at 21±3°C. Total binding and nonspecific binding were defined with 50 μl of buffer B and 50 μl of 10 μM YM220, respectively. Data represent the mean±s.e.m. of four experiments. The linear relationship between cell number and the specific binding of [3H]-JB93182 is also shown (b, d; hatched lines demonstrate the 95% confidence intervals of the linear regression).
Analysis of [3H]-JB93182 saturation-binding data to CCK2S and CCK2L receptors
The specific binding of [3H]-JB93182, to both the CCK2S and the CCK2L receptors, was saturable (Figure 2). The estimated Hill slope parameters were not significantly different from unity, and there was no significant difference between the pKD values estimated for CCK2S and CCK2L (CCK2S pKD=9.00±0.03, nH=0.91±0.06 and CCK2L pKD=9.10± 0.05, nH=1.08±0.11, respectively). A significantly greater number of sites were labelled in the membranes prepared from the CCK2S receptor-transfected cells than the CCK2L receptor-transfected cells (mean Bmax±s.e.m.; CCK2S=11.74±1.08 fmol 1 × 105 cells−1, CCK2L=3.69±0.40 fmol 1 × 105 cells−1; d.f.=4, t=7.01).
Figure 2.

Saturation analysis of the binding of [3H]-JB93182 at the CCK2S and CCK2L receptors. Cell membranes (400 μl) were incubated in triplicate with increasing concentrations of [3H]-JB93182 (50 μl; 0.01–6 nM) and 50 μl of buffer B or 50 μl of 10 μM YM220 to define total and nonspecific binding, respectively. The incubation was terminated after 150 min at 21±3°C. The inset shows the linear transformation of the data as a Hill plot (hatched line represents 95% confidence interval).
Analysis of the kinetic properties of [3H]-JB93182 binding to the CCK2S and CCK2L receptor isoforms
The specific binding of [3H]-JB93182 reached equilibrium within a 5-min incubation period in both the CCK2S and the CCK2L receptor assays (Figure 3), and the association and dissociation data could be fitted by pseudo-first-order and first-order rate equations, respectively. There were no significant differences between the one-site model parameter values estimated for the CCK2S and CCK2L receptors (Table 1 ). The pKD value estimated at the CCK2S receptor by kinetic analysis was significantly different from that estimated by saturation analysis at this isoform (from saturation study pKD±s.e.m.=9.00±0.03, from kinetic study pKD±s.e.m.=9.31±0.02; t=−8.32, d.f.=4). For the CCK2L, there was no significant difference between the pKD value estimated by saturation studies and that estimated from kinetic studies (pKD±s.e.m. from saturation study=9.10±0.05, pKD±s.e.m. from kinetic study=9.27±0.22).
Figure 3.

Kinetic analysis of the binding of [3H]-JB93182 at the CCK2S (panel a) and CCK2L (panel b) receptors. Cell membranes (400 μl) were incubated in triplicate with [3H]-JB93182 (50 μl; 20 nM) and 50 μl of buffer B or 50 μl of 10 μM YM220 to define total and nonspecific binding, respectively. The incubation was terminated after varying time intervals (1–150 min) to define the time course of association. For dissociation studies, amount of specific bound radioligand was determined after the addition of a high concentration (1 μM) of the CCK2-receptor selective antagonist YM220.
Table 1.
Estimated parameters from nonlinear regression of kinetic data for CCK2S- and CCK2L-receptor isoforms (n=3)
| Parameter estimates (mean value±s.e.m) | |||||
|---|---|---|---|---|---|
| Isoform | k+1 (× 108 M−1 min−1) | k−1 (min−1) | t1/2 Association (min) | t1/2 Dissociation (min) | pKD |
| CCK2S | 4.85±0.19 | 0.237±0.02 | 0.53±0.02 | 2.96±0.22 | 9.31±0.02 |
| CCK2L | 4.94±0.20 | 0.229±0.02 | 0.66±0.12 | 3.10±0.31 | 9.27±0.22 |
Analysis of CCK-receptor antagonist competition data at the CCK2S and the CCK2L-receptor subtypes
A concentration-dependent inhibition of the binding of [3H]-JB93182, to both the CCK2S- and the CCK2L-receptor isoforms, was observed for all the CCK-receptor selective compounds used in this study (Table 2 ). The Hill slope values estimated for CCK-8S, PD-134,308 and L-740,093 were significantly different from unity. Of these compounds, only the data obtained for CCK-8S were better described by a two-site model. A comparison of the affinity estimates (pKI values) generated for the CCK2S and CCK2L receptors was conducted using principal components analysis. This procedure revealed that the two data sets did not deviate significantly from the line of identity (see Figure 4; principal components analysis: F-ratio for unit slope: (y=x+c), F(1,66)=2.14; F-ratio for zero intercept (y=x), F(1,66)=3.18).
Table 2.
Affinity estimates for CCK-receptor antagonists competition studies using [3H]-JB93182 as radiolabel at the CCK2S and CCK2L receptors
| CCK2S receptor | CCK2L receptor | Literature values | |||||
|---|---|---|---|---|---|---|---|
| Compound | pKI | nH | n | pKI | nH | n | pKI |
| CCK2-receptor selective | |||||||
| YM022 | 9.94±0.09 | 1.25±0.12 | 5 | 10.28±0.06 | 0.96±0.04 | 4 | 9.73a |
| YM220 | 9.32±0.09 | 0.85±0.09 | 5 | 9.66±0.06 | 1.01±0.22 | 3 | 9.72b |
| PD-134,308 | 8.86±0.04 | 0.90±0.01* | 4 | 8.80±0.11 | 0.83±0.02* | 3 | 8.35c |
| PD-140,376 | 8.82±0.07 | 1.06±0.06 | 4 | 9.12±0.06 | 0.74±0.07* | 3 | nd |
| R-L-365,260 | 7.98±0.07 | 0.97±0.03 | 3 | 7.64±0.05 | 0.95±0.06 | 4 | 8.48b |
| S-L-365,260 | 6.33±0.06 | 1.58±0.28 | 3 | 6.37±0.04 | 1.48±0.28 | 3 | nd |
| JB93182 | 8.94±0.06 | 1.02±0.08 | 5 | 9.02±0.09 | 1.12±0.04 | 4 | nd |
| L-740,093 | 9.34±0.04 | 0.93±0.02* | 3 | 9.56±0.11 | 0.84±0.08 * | 3 | 9.72d |
| CCK1-receptor selective | |||||||
| SR27897 | 6.68±0.04 | 1.23±0.07 | 4 | 7.04±0.13 | 1.29±0.39 | 4 | nd |
| L-364,718 | 6.54±0.22 | 1.18±0.10 | 4 | 6.79±0.11 | 1.34±0.34 | 3 | 6.82d |
| 2-NAP | 4.90±0.05 | 1.62±0.10 | 3 | 4.70±0.05 | 1.60±0.31 | 3 | nd |
| Agonist | |||||||
| CCK-8S | 9.05±0.04 | 0.76±0.03* | 3 | 9.27±0.02 | 0.89±0.10 | 3 | 9.92d |
Figure 4.

Comparison of affinity estimates (pKI values) obtained at the CCK2S and CCK2L receptors. The line shown superimposed is the line of identity. The pKI values were analysed using principal components analysis, which revealed no global differences between the two groups (see text for details).
Discussion
The aim of this study was to characterise the radioligand-binding properties of the alternatively spliced CCK2S- and CCK2L-receptor isoforms. Although there appears to be no differences between these splice variants in terms of the CCK- and gastrin-binding affinities or potencies for second messenger activation (Ito et al., 1994), it has been suggested that the CCK2-receptor antagonist L-365,260 interacts with an increased affinity at the CCK2S receptor (Wank et al., 1994). Additionally, L-365,260 has been shown to distinguish between two putative CCK2-receptor populations (G1 and G2) in both functional bioassays and in radioligand-binding studies (Harper et al., 1996; Roberts et al., 1996). However, the molecular counterparts for these receptor subtypes have not been identified.
In this study, the binding of the CCK2-receptor selective antagonist [3H]-JB93182 to both the CCK2S and CCK2L receptors was characterised on the basis that this compound has been shown to display opposite selectivity to L-365,260 at the G1 and G2 receptors. Indeed, the published description of [3H]-JB93182 as a radioligand for CCK2 receptors (Harper et al., 1999) concluded that ‘….it [[3H]-JB93182] may be useful for determining whether there is any correlation between the gastrin G1 and G2 sites and the reported variants of the gastrin/CCKB [i.e. CCK2] receptor gene product (e.g. the long and short isoform; Ito et al., 1994)… '. Furthermore, antagonist radioligand-binding data are inherently simpler than agonist binding data as, in principle at least, the former should not promote multiple receptor states. Therefore, unlike traditional agonist radioligands, for example, [125I]-BH-CCK-8S, the affinity estimates obtained in different assays should be independent of between-assay variation in G-protein content.
These experiments revealed that the specific binding of [3H]-JB93182 to the CCK2S-receptor cell membranes was significantly greater than that observed at the CCK2L-receptor cell membranes. Consistent with this, the estimated Bmax value for the CCK2S receptor was significantly larger than that obtained for the CCK2L receptor. These differences in receptor concentration may have resulted from differences in the efficiency of the initial receptor transfection or variations in the stability of the receptor protein within the respective cell system. Interestingly, the predominant identification of the CCK2S receptor within human tissue samples has been reported (Song et al., 1993; Ito et al., 1994; Biagini et al., 1997), which could also be a consequence of the relative stability of this receptor isoform. In this study, the differences in expression levels between the two cell lines should not affect the data generated with the antagonist ligands used here. This is because, according to current receptor theory, the total receptor expression should only have an effect on altering the midpoint location of agonists. In contrast, antagonists should retain the same binding affinity regardless of receptor density.
The pKD values estimated from the saturation studies suggested that there were no significant differences between the affinity of [3H]-JB93182 at the CCK2S or CCK2L receptors. This finding and, concomitantly, the reproducibility of the assay were confirmed by the similarity of the pKD values generated from the kinetic studies (CCK2S: pKD=9.31±0.02; CCK2L: pKD=9.27±0.22). The estimated affinity value at the CCK2S receptor from the kinetic studies was significantly greater than that determined by saturation analysis. However, the statistical significance of this finding probably reflected the very small standard error associated with these parameter estimates (see values listed above). These data are not consistent with the previous discrimination of G1 and G2 by this compound, which was shown to express ∼1 log unit greater affinity at the G1 receptor subtype (G1 pKI=9.9; G2 pKI=8.6; Harper et al., 1999).
The pharmacological properties of the CCK2S and the CCK2L receptors were investigated using a range of CCK-receptor ligands, which have previously been shown to discriminate between the putative G1 and G2 receptors (Harper et al., 1999). In addition, these compounds are structurally diverse and express affinity values ranging over five log concentration units and, as such, should provide good pharmacological tools to investigate any differences between the CCK2S and CCK2L receptors. A global comparison of the pKI values, using principal components analysis, suggested that there was no overall significant difference between the CCK2S and CCK2L receptors. In addition, the pKI values were not consistent with the previously described G1/G2 receptor pharmacology (Harper et al., 1999). However, further inspection of the data revealed that the pKI values estimated for YM022, PD-140,376 and CCK-8S were significantly higher at the CCK2L than the CCK2S receptor (∼0.35 log unit), whereas R-L-365,260 expressed the same degree of selectivity but reverse potency for these receptor isoforms. This ∼2.5-fold increase in potency at the CCK2S receptor is consistent with the data obtained by Wank et al. (1994). Therefore, these studies suggest that, in theory at least, it is possible to distinguish between the alternatively spliced CCK2-receptor isoforms using YM022, PD-140,376, CCK-8S and R-L-365,260. However, in practice, the small degree of selectivity exhibited by these compounds would be insufficient to differentiate between the receptors in animal or human tissue assays, because the variability is generally greater than that observed in recombinant systems.
As previously discussed, the majority of the CCK-receptor ligands in this study behaved simply within the competition-inhibition studies. However, PD-134,308 and L-740,093 had Hill slope parameters of less than unity at both the CCK2S and CCK2L receptors and PD-140,376 and CCK-8S had Hill slopes of less than one at the CCK2L and CCK2S receptors, respectively. Interestingly, all of the compounds with estimated nH values less than one have previously been shown to exhibit agonist activity (Ding et al., 1995; Kopin et al., 1997) and, therefore, the complex competition curves obtained with these compounds may be due to the formation of multiple agonist-affinity states.
In addition to investigating the pharmacology of the alternatively spliced CCK2 receptor, this study has provided estimated affinity values for the compounds PD-140,376, S-L-365,260, JB93182, SR27897 and 2-NAP at the human CCK2 receptor, which have not been previously reported. These values appear consistent with their observed values in animal studies and therefore, provide no evidence for pharmacological discrimination of species differences using these ligands.
Conclusion
Overall, the pharmacology of the CCK2S and CCK2L receptors does not appear to be consistent with that described for the putative G1 and G2 receptors. There was no global difference in the affinity values estimated for a range of structurally diverse CCK-receptor selective ligands; however, small yet significant differences were observed using four of the ligands investigated. The selectivity of these ligands does not appear to be large enough for characterisation in fresh human tissue, but this finding suggests that, in principle at least, it may be possible to design isoform-selective ligands, which express a greater degree of discrimination.
Abbreviations
- CCK
cholecystokinin
- dpm
disintegrations per minute
References
- BEINBORN M., QUINN S.M., KOPIN A.S. Minor modifications of a cholecystokinin-B/gastrin receptor non-peptide antagonist confer a broad spectrum of functional properties. J. Biol. Chem. 1998;273:14146–14151. doi: 10.1074/jbc.273.23.14146. [DOI] [PubMed] [Google Scholar]
- BELLIER B., MCCORT TRANCHEPAIN I., DUCOS B., DANASCIMENTO S., MEUDAL H., NOBLE F., GARBAY C., ROQUES B.P. Synthesis and biological properties of new constrained CCK-B antagonists: discrimination of two affinity states of the CCK-B receptor on transfected CHO cells. J. Med. Chem. 1997;40:3947–3956. doi: 10.1021/jm970439a. [DOI] [PubMed] [Google Scholar]
- BIAGINI P., MONGES G., VUAROQUEAUX V., PARRIAUX D., CANTALOUBE J.F., DE MICCO P. The human gastrin/cholecystokinin receptors: type B and type C expression in colonic tumors and cell lines. Life Sci. 1997;61:1009–1018. doi: 10.1016/s0024-3205(97)00605-x. [DOI] [PubMed] [Google Scholar]
- BLAKER M., REN Y., GORDON M.C., HSU J.E., BEINBORN M., KOPIN A.S. Mutation within the cholecystokinin-B/gastrin receptor ligand ‘pocket' interconvert the functions of nonpeptide agonists and antagonists. Mol. Pharmacol. 1998;54:857–863. doi: 10.1124/mol.54.5.857. [DOI] [PubMed] [Google Scholar]
- CHENG Y., PRUSOFF W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- DENYER J., GRAY J., WONG M., STOLZ M., TATE S. Molecular and pharmacological characterization of the human CCKB receptor. Eur. J. Pharmacol. 1994;268:29–41. doi: 10.1016/0922-4106(94)90117-1. [DOI] [PubMed] [Google Scholar]
- DING X.Q., CHEN D., HAKANSON R. Cholecystokinin-B receptor ligands of the dipeptoid series act as agonists on rat stomach histidine decarboxylase. Gastroenterology. 1995;109:1181–1187. doi: 10.1016/0016-5085(95)90577-4. [DOI] [PubMed] [Google Scholar]
- DURIEUX C., COPPEY M., ZAJAC J.M., ROQUES B.P. Occurrence of two cholecystokinin binding sites in guinea-pig brain cortex. Biochem. Biophys. Res. Commun. 1986;137:1167–1173. doi: 10.1016/0006-291x(86)90348-7. [DOI] [PubMed] [Google Scholar]
- HARPER E.A., GRIFFIN E.P., SHANKLEY N.P., BLACK J.W. Analysis of the behaviour of selected CCKB/gastrin receptor antagonists in radioligand binding assays performed in mouse and rat cerebral cortex. Br. J. Pharmacol. 1999;126:1496–1503. doi: 10.1038/sj.bjp.0702448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARPER E.A., ROBERTS S.P., SHANKLEY N.P., BLACK J.W. Analysis of variation in L-365,260 competition curves in radioligand binding assays. Br. J. Pharmacol. 1996;118:1717–1726. doi: 10.1111/j.1476-5381.1996.tb15597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HELLMICH M.R., RUI X.L., HELLMICH H.L., FLEMING R.Y., EVERS B.M., TOWNSEND C.M., Jr Human colorectal cancers express a constitutively active cholecystokinin-B/gastrin receptor that stimulates cell growth. J. Biol. Chem. 2000;275:32122–32128. doi: 10.1074/jbc.M005754200. [DOI] [PubMed] [Google Scholar]
- ITO M., IWATA N., TANIGUCHI T., MURAYAMA T., CHILHARA K., MATSUI T. Functional characterisation of two cholecystokinin-B/gastrin receptor isoforms: a preferential splice donor site in the human receptor gene. Cell Growth Differ. 1994;5:1127–1135. [PubMed] [Google Scholar]
- KOPIN A.S., MCBRIDE E.W., GORDON M.C., QUINN S.M., BEINBORN M. Inter- and intraspecies polymorphisms in the cholecystokinin-B/gastrin receptor alter drug efficacy. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11043–11048. doi: 10.1073/pnas.94.20.11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LENA I., SIMON H., ROQUES B.P., DAUGE V. Opposing effects of two CCK(B) agonists on the retrieval phase of a two-trial memory task after systemic injection in the rat. Neuropharmacology. 1999;38:543–553. doi: 10.1016/s0028-3908(98)00223-8. [DOI] [PubMed] [Google Scholar]
- MEESTER B.J., SHANKLEY N.P., WELSH N.J., WOOD J., MEIJLER F.L., BLACK J.W. Pharmacological classification of adenosine receptors in the sinoatrial and atrioventricular nodes of the guinea-pig. Br. J. Pharmacol. 1998;124:685–692. doi: 10.1038/sj.bjp.0701891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIYAKE A. A truncated isoform of human CCK-B/gastrin receptor generated by alternative usage of a novel exon. Biochem. Biophys. Res. Commun. 1995;208:230–237. doi: 10.1006/bbrc.1995.1328. [DOI] [PubMed] [Google Scholar]
- MOTULSKY H.J. Analyzing Data with GraphPad Prism. GraphPad Software, Inc., San Diego, CA, U.S.A; 1999. [Google Scholar]
- ROBERTS S.P., HARPER E.A., WATT G.F., GERSKOWITCH V.P., HULL R.A., SHANKLEY N.P., BLACK J.W. Analysis of the variation in the action of L-365,260 at CCKB/gastrin receptors in rat, guinea-pig and mouse isolated gastric tissue assays. Br. J. Pharmacol. 1996;118:1779–1789. doi: 10.1111/j.1476-5381.1996.tb15604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANKARAN H., GOLDFINE I.D., DEVENEY C.W., WONG K.Y., WILLIAMS J.A. Binding of cholecystokinin to high affinity receptors on isolated rat pancreatic acini. J. Biol. Chem. 1980;255:1849–1853. [PubMed] [Google Scholar]
- SONG I., BROWN D.R., WILTSHIRE R.N., GANTZ I., TRENT J.M., YAMADA T. The human gastrin/cholecystokinin type B receptor gene: alternative splice donor site in exon 4 generates two variant mRNAs. Proc. Natl. Acad. Sci. U.S.A. 1993;90:9085–9089. doi: 10.1073/pnas.90.19.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKINAMI Y., YUKI H., NISHIDA A., AKUZAWA S., UCHIDA A., TAKEMOTO Y., OHTA M., SATOH M., SEMPLE G., MIYATA K. YF476 is a new potent and selective gastrin/cholecystokinin-B receptor antagonist in vitro and in vivo. Aliment. Pharmacol. Ther. 1997;11:113–120. doi: 10.1046/j.1365-2036.1997.110281000.x. [DOI] [PubMed] [Google Scholar]
- WANK S., PISEGNA J.R., POIROT S. Functional significance of potential splice variants of the human cholecystokinin (CCK) B receptor. Gastroenterology. 1994;106:A850. [Google Scholar]