

Abstract
This study investigates the effects of androgens, the antiandrogen flutamide and green tea catechins on glucose transport inhibition in human erythrocytes. These effects may relate to the antidiabetogenic effects of green tea.
Testosterone, 4-androstene-3,17-dione, dehydroepiandrosterone (DHEA) and DHEA-3-acetate inhibit glucose exit from human erythrocytes with half-maximal inhibitions (Ki) of 39.2±8.9, 29.6±3.7, 48.1±10.2 and 4.8±0.98 μM, respectively. The antiandrogen flutamide competitively relieves these inhibitions and of phloretin. Dehydrotestosterone has no effect on glucose transport, indicating the differences between androgen interaction with GLUT1 and human androgen receptor (hAR).
Green tea catechins also inhibit glucose exit from erythrocytes. Epicatechin 3-gallate (ECG) has a Ki ECG of 0.14±0.01 μM, and epigallocatechin 3-gallate (EGCG) has a Ki EGCG of 0.97±0.13 μM. Flutamide reverses these effects.
Androgen-screening tests show that the green tea catechins do not act genomically. The high affinities of ECG and EGCG for GLUT1 indicate that this might be their physiological site of action.
There are sequence homologies between GLUT1 and the ligand-binding domain (LBD) of hAR containing the amino-acid triads Arg 126, Thr 30 and Asn 288, and Arg 126, Thr 30 and Asn 29, with similar 3D topology to the polar groups binding 3-keto and 17-β OH steroid groups in hAR LBD. These triads are appropriately sited for competitive inhibition of glucose import at the external opening of the hydrophilic pore traversing GLUT1.
Keywords: Glucose transport, androgens, green tea catechins, flutamide, GLUT1
Introduction
Androgens are known to inhibit glucose transport in human erythrocytes (Lacko et al., 1975; Krupka & Devés, 1980; May & Danzo, 1988). Androgens, for example, testosterone, also produce a number of clinical effects that are consistent with the inhibition of glucose transport in peripheral tissues (Woodard et al., 1981). Dehydroepiandrosterone (DHEA) is an androgen secreted in relatively large amounts by the adrenal, and is used as a nutritional supplement. DHEA and other androgens, for example, DHEA, epiandrosterone and DHEA 3 –sulphate, are known to be uncompetitive antagonists of glucose 6-phosphate dehydrogenase (G6PD) (Gordon et al., 1995). DHEA inhibits growth and induces apoptosis in BV-2 cells in the absence of glucose, but these effects are reversed by the addition of glucose (5–20 mM) to the growth medium (Yang et al., 2000). These inhibitions are independent of DHEA inhibition of G6PD (Biaglow et al., 2000; Yang et al., 2002). It is unclear as to whether DHEA exerts a significant inhibition of glucose transport in vivo.
Low insulin sensitivity is commonly found in congenital adrenal hyperplasia, and in polycystic ovarian disease. Both conditions lead to testosterone hypersecretion, and are often accompanied by hypersecretion of insulin, hyperlipidaemia and hirsuitism (Speiser et al., 1992; Livingstone and Collison, 2002). These clinical signs improve after treatment with the antiandrogen flutamide (Ibanez et al., 2000). Flutamide is a nonsteroidal antiandrogen, known to antagonise testosterone binding to the androgen receptor (Benten et al., 1999; McDonald et al., 2000; Poujol et al., 2000). It is used mainly in the treatment of androgen-sensitive prostatic adenomas, some of which regress after treatment (Alberts & Blute, 2001).
Although the main actions of androgens are thought to be via a specific nuclear receptor that acts on DNA, nongenomic actions of androgens are also recognised. Membrane-bound androgen receptors have been reported in the brain, macrophages and aorta (Benten et al., 1999; Perusquia & Villalon, 1999; Zhu et al., 1999; Matias et al., 2000). They are thought to activate Ca2+-dependent cell signalling pathways (Perusquia & Villalon, 1999). Some of these nongenomic effects are also sensitive to flutamide (Zhu et al., 1999), others not (Benten et al., 1999; Perusquia & Villalon, 1999).
We decided to investigate if the antiandrogen flutamide antagonises androgen-sensitive inhibition of glucose transport in erythrocytes. We have also explored the structure–affinity relationships of a number of androgens on glucose transport. Although the physiological concentrations of circulating androgens are lower than those used here, much higher local concentrations ≈100 × can occur in the ovary and testis (Jarow et al., 2001; Burger, 2002). The reported nongenomic effects of androgens on Ca2+ channels occur in the 10–100 μM range of androgens (Benten et al., 1999; Perusquia & Villalon, 1999).
Several reports indicate that the green tea polyphenols, albeit at very high concentrations, for example, epicatechin gallate, reduce the intestinal absorption of sugars via the Na+-dependent glucose transporter, reduce glycosuria in diabetics (Ki epicatechin gallate=0.38 mM) (Kobayashi et al., 2000), and reduce the activation of enzymes causing gluconeogenesis (Waltner-Law et al., 2002). Green tea polyphenols have also been reported to reduce prostatic enlargement in benign prostatitis, and in testosterone-dependent metastatic prostatic tumours in a mouse model (Gupta et al., 2001). Here we show that the whole green tea extract and the major catechin gallates present in green tea inhibit glucose transport in erythrocytes in vitro at the same site as androgens, at concentrations equivalent to those found in tea drinkers' plasma.
Additionally, as the effects of androgens on glucose transport show high specificity, the possibility that there are sequence homologies between GLUT1 and the androgen receptor has also been explored similarly to the way in which we investigated oestrogen–GLUT1 interactions (Afzal et al., 2002). Here we show that there are good matches in the outside-facing regions of GLUT1 with the ligand-binding domain (LBD) of the androgen receptor. These may provide a structural basis for the observed interactions between androgens and the glucose transporter. These findings suggest that many of the membrane-associated nongenomic effects of androgens may occur at mimetic sites to the androgen receptor ligand-binding domain (hAR LBD), rather than to the receptor itself.
Methods
Solutions
The erythrocyte suspension medium was phosphate-buffered saline (PBS) adjusted to pH 7.4. D-glucose, phloretin, flutamide, cyproterone acetate, testosterone, dihydrotestosterone, 5α-androstan-17β-ol-3-one, 5α-androstan-3β, 17β-diol, epiandrosterone (5α-androstan-3β-ol-17-one), androsterone (5α-androstan-3α-ol-17-one) (androstenedione 4-androstene-3, 17-dione), etiocholano-3α-ol-17-one, dehydroepiandrosterone-3-acetate (DHEA acetate) and dehydroepiandrosterone-3-sulphate (DHEA sulphate), and all the pure catechins were obtained from Sigma Chemicals Ltd, Poole, Dorset. DHEA and 3β, 17β-dihydroxyandrostenediol were purchased from Steraloids, Inc. (Newport, Rhode Island 02840, U.S.A.). Green tea extract contains, in percent g g−1 extract, 51.94% epigallocatechin gallate, 19.45% epicatechin gallate, 4.99% epicatechin, 4.62% epigallocatechin, 85.4% total catechins, and 99.2% tea polyphenols, with less than 0.1% caffeine. The extracts were analysed at 30°C by HPLC, mobile phase, water : methanol : phosphoric acid=27 : 78 : 0.1, using a UV absorption detector at 280 nm. The extracts were obtained as a gift from Mr Tang Ping Yuan, China Herb Company, 210–504, 4th District, Fuxiang Nan, Yuyao, Zhejiang, 315400, China, http://www.china-tea.com, E-mail: Chinaherb@hotmail.com. A recent report shows that the Ki of caffeine-dependent inhibition of 3-O-methyl-glucose uptake into normal human red cells is 1.5 mM (Ho et al., 2001). This means that the very low xanthine content of the green tea extracts used in this study can be excluded as possible inhibitors of glucose transport; hence, the only inhibitors of glucose transport in green tea extract are catechins. The low caffeine content of the tea extract permits us to exclude this as a possible source of inhibition.
Cells
Fresh human erythrocytes were obtained by venepuncture, and then washed three times in isotonic PBS by repeated centrifugation and resuspension. The cells were then suspended in PBS solutions with 100 mM D-glucose added, final haematocrit 10%, and incubated for at least 2 h at 37°C. The cells were then recentrifuged in 100 mM D-glucose saline to obtain a thick cell suspension ca. 95% haematocrit. This cell suspension was kept at 4°C until required. Cells were always used within 72 h of collection. Aliquots of prewarmed cells (7.5 μl) were added to a 1 cm2 fluorescence cuvette containing 3 ml of saline solution, which had been prewarmed to 24°C. The cell suspensions were mixed vigorously, and photometric monitoring was started within 5 s of mixing.
Photometric monitoring: glucose exit
The exit rates of D-glucose from cells were monitored photometrically using a Hitachi 2000-F fluorescence spectrometer with a temperature-controlled and monitored cuvette; Eex=Eem=650 nm. The output was recorded and stored with a MacLab 2e (AD Instruments). Data were collected at a rate of 0.33–5 points s−1, depending on the time course of exit; each run consisted of 200–2000 data points. The photometric response was found to be approximately linear for osmotic perturbations±50 mM NaCl. In the absence of glucose, an osmotic change results in a step change in output, which remains stationary for at least 30 min, indicating that there is no secondary cause of volume change other than sugar movement.
The time courses of D-glucose exit were fitted to monoexponential curves of the form yt=A{1−B exp (Ct)}, using Kaleidagraph 3.6 (Synergy Software), where the voltage yt was recorded at elapsed time (ts); the coefficient A is a scaling factor that fits the curves to the voltage signal yt, and B and C are the monoexponential coefficients. These fits gave correlation coefficients r>0.98, and standard errors of the means of the rate coefficients. Where the net glucose rate was measured in solutions containing glucose at concentrations >0 mM, the rate coefficient C was multiplied by the factor D=(100−[glucose]ext mM)/100 to account for the decreased extent of the net decrease in intracellular glucose rate; that is, yt=A{1−B exp(DCt)}. Strictly, coefficient A is redundant, but it permits the curve-fitting programme to operate within a narrow range of B and C coefficients, and thus to fit the curves without altering the initial coefficient estimates.
In all cases, the cells were exposed to test substances only during the period of glucose exit. Pre-equilibration for 1 h with varying concentrations of testosterone makes no difference to the inhibition of glucose exit. The external androgen concentration is the determinant of the inhibition constant.
Statistics
All the statistical probabilities were estimated from two-tailed Student's t-values for unpaired means. The n values were estimated from the number of degrees of freedom, and all data points were obtained from the means of 3–5 sets of data.
The Ki values for direct inhibitors of glucose exit were obtained by nonlinear regression of the change in the exponential exit rate of glucose exit, C, against the inhibitor concentration [I], using the equation y=VmaxKi/(Ki+[I]), where Ki is the inhibitor concentration giving 50% decrease of the rate of exit obtained in the absence of inhibitor. The regression coefficient is expressed as the mean±s.e.m. Each Ki plot was obtained from the means of glucose exit rates against at least 3–4 inhibitor concentrations, that is, typically 16–20 glucose exit rates were determined per estimate of each Ki. Each Ki estimate was repeated 3–4 times.
Monitoring the affinity of glucose at the external site (infinite cis Km) and the maximal rate of glucose exit (zero-trans Vm)
With nominally zero glucose concentration in the external solution, exit is defined as the zero-trans net exit condition, and monitors the maximal rate of glucose net exit Vm.
To measure the affinity of glucose for the external side of the transporter, the rates of glucose exit were obtained with varying concentrations of glucose in the external solution. The glucose concentration in the external solution that was required to reduce the rate of net glucose exit by 50% is the infinite cis Km. This mode of exit where the inside concentration is fixed (infinite cis), but the rate of exit varied, was first introduced by Sen & Widdas (1962). The Km is obtained by least-squares fit of the equation v=KmVm/(Km+Gex), where Vm is the maximal rate of glucose exit in the uninhibited state, Km is the concentration of glucose in the external solution Gex required to reduce the exit rate to 50% of the uninhibited rate and v=CD (see above). Androgens were also tested to determine whether they alter the affinity of glucose for the external site, for example, (Ki ic). The Ki (ic glucose/test) is obtained by observing the increase in the apparent Km of glucose binding to the external site as a function of testosterone concentration. This was obtained by plotting the apparent Km (ic glucose) versus [testosterone]. The Ki (ic glucose/test) is obtained from the intercept/slope±s.e.m. of the linear regression line.
The Ki values for indirect inhibition, for example, the effect of flutamide on testosterone, were obtained by linear regression of the apparent Ki values against the inhibitor concentration [I].
As Kapp=Ki1(1+[I]/Ki2), Ki2 is the concentration of modulator, for example, flutamide, required to raise Ki1 of the primary inhibitor (e.g. testosterone (test)) two-fold. This was obtained from (intercept/slope)±s.e.m. of the linear regression line of Kapp versus [I].
Screening for androgen and antiandrogenic activity
Androgenic and antiandrogenic activities were investigated using an androgen-inducible yeast screen (Saccharomyces cerevisiae) expressing the human androgen receptor, and containing expression plasmids carrying androgen-responsive sequences controlling the reporter gene lac-Z. This yeast screen was originally developed in the Genetics Department of Glaxo Wellcome plc (Stevenage, Herts, U.K.), and was a gift from Professor J. Sumpter, Brunel University, U.K. Androgenic activity was determined from the metabolism of chlorophenol red-D-galactopyranoside, by monitoring the absorbance at 540 nm, using 5-α-dihydrotestosterone (DHT) as a standard. Antiandrogenic activity was determined by the ability of test compounds to block the stimulation of 1.25 × 10−9 M DHT (Sohoni & Sumpter, 1998). A standard antiandrogen response was obtained by observing the decrease in a half-maximal response to DHT with flutamide. At concentrations above 0.1 mM, catechins were cytotoxic to the yeast expression system; and so no further action of these agents could be demonstrated above these concentrations.
Searching for sequence homologies between the LBD of the androgen receptor and GLUT1
Homologies were sought between sequences close to the LBD of the human androgen receptor (hAR-LBD) primary accession number P10275, and in GLUT1 using the Swissprot database GLUT-1 (SLC2A1) human primary accession number P11166 as follows:
The program FASTA (Pearson & Lipman, 1988) was used to identify and evaluate the partial matches between GLUT1 and sequences in the hAR LBD that were adjacent to the ligand-binding cleft (Weatherman et al., 1999; Matias et al., 2000; Poujol et al., 2000; Singh et al., 2000; Sack et al., 2001; Marhefka et al., 2001). The searches were restricted to regions matching the outside-facing regions of GLUT1, as predicted by the hydropathy plots (Mueckler et al., 1985). This constraint was observed as the kinetic interactions between phloretin, testosterone, androstenedione and flutamide show that these ligands bind exclusively to the glucose import site on the outside of GLUT1. The matches were applied to the 3-D template structure of GLUT-1 (Zuniga et al., 2001). The atomic coordinates and structure factors (code 1JA5) are in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ, U.S.A. (http://www.rcsb.org/), and can be viewed with Swiss-Pdb viewer, http://www.expasy.ch/spdbv.
Results
Effects of androgens on glucose transport
Several androgens inhibit zero-trans net glucose exit (see Methods) (Table 1 ), as determined by the decreased rates of 100 mM glucose exit from human erythrocytes at 21°C with increasing concentrations of androgens. The Ki for testosterone is 39.2±8.9 μM, androstenedione 29.6±3.7 μM, androsterone 44.0±2.2 μM and DHEA acetate 4.8±0.98 μM. Flutamide also has a weak inhibitor effect on glucose exit flux, Ki=73.4±11.7 μM (Table 1). Although low concentrations of flutamide in the range 0–5 μM have negligible effects on glucose exit, these lower concentrations competitively antagonise the inhibitor effects of androstenedione, androsterone, testosterone and DHEA acetate on glucose exit (Table 1). This is evident from the increases in the apparent Ki's of these androgens in the presence of increasing concentrations of flutamide, and the increased rates of glucose exit seen in the presence of both flutamide and androgens compared with the rates seen with androgen alone (Figure 1a, b). For example, with flutamide=0 μM, the Ki (testosterone) is 39.2±8.9 μM; with flutamide=0.25 μM, the Ki (testosterone) is 52.1±11.3 μM; with flutamide=0.5 μM, the Ki (testosterone) is 121.0±29.6 μM and with flutamide=1 μM, the Ki (testosterone)= 141.3±34.5 μM. Similarly, the Ki (androstenedione) increases from 29.6±3.7 μM in the absence of flutamide to 119.6±27.7 μM, with 0.5 μM flutamide present. The Ki's of flutamide against testosterone Ki (test/Flut)=0.35±0.17 μM and against androstenedione Ki (and/Flut)=0.14±0.05 μM are similar (n.s.) (Table 1).
Table 1.
Structure and affinities of androgens to the human erythrocyte glucose transporter
|
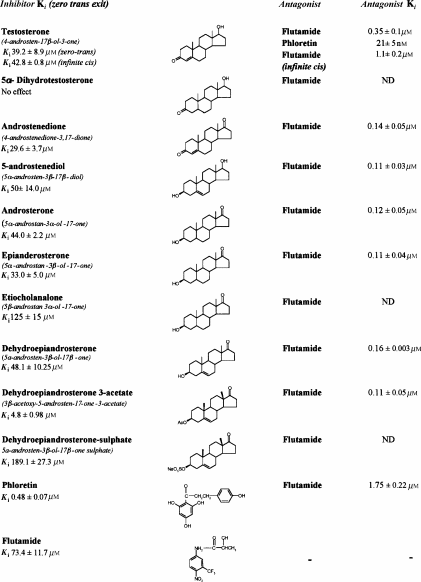
Figure 1.

(a) Effect of flutamide on testosterone-induced inhibition of glucose exit from erythrocytes. Inhibitions of glucose exit by testosterone from human erythrocytes loaded with 100 mM glucose into glucose-free isotonic PBS at 21°C and the effects of flutamide on this inhibition. The rates are estimated by monoexponential fitting, as described in Methods. The lines drawn through the points estimate the Ki as follows: y=VmKi/(I+Ki), where y is the rate of glucose exit (s−1), Vm is the maximal rate of glucose exit with zero inhibitor present and Ki is the concentration of inhibitor that inhibits the rate by 50%. The best-fit lines shown in the figure obtained using the fitting procedures in Kaleidagraph 3.5 (Synergy Systems). The lines show the effects of varying concentrations of testosterone in the absence and presence of flutamide at different concentrations. The Ki (testosterone) increases as the concentration of flutamide is increased; with flutamide=0 μM, the Ki (testosterone) is 39.2±8.9–52.1±11.3 μM; with flutamide=0.25 μM, the Ki (testosterone) increases to 52.1±11.3 μM; with flutamide=0.5 μM the Ki (testosterone) is 121.0±29.6 μM; and with flutamide=1 μM, the Ki (testosterone) is 141.3±34.5 μM. Each data point collected is the average of 3–5 separate fluxes and was repeated at least 4 times i.e. 12–16 fluxes per point. The data shown are from the averaged fluxes of all experiments collected. (b) Comparison of the effects of flutamide on androstenedione and testosterone induced inhibition of glucose exit from erythrocytes. The replots of the Ki's of androstenedione and testosterone with increasing concentrations of flutamide obtained in (b) (androstenedione exit data are not shown) are fitted to a linear regression line and the Ki of flutamide (i.e. the concentration of flutamide that increases the Ki of androstenedione and testosterone 2 ×) is estimated from the intercept/slope.
Infinite cis exit
Glucose exit experiments were carried out where the glucose concentration in the external solution [Gex] was varied according to the technique first employed by Sen & Widdas (1962). The concentration of glucose in this external solution that reduces the rate of glucose exit to half the uninhibited rate measures the affinity of glucose for the external surface ‘Sen–Widdas Km'; see Methods (Km ic exit=1.2±0.3 mM).
Androgens reduce the affinity of glucose, as is apparent from the androgen-dependent increases in the Km of glucose at the external surface. This is consistent with the inhibitor acting on glucose exit at the external surface of the transporter, for example, testosterone (Ki ic/test=42.8±0.8 μM). This Ki is indistinguishable from the Ki zero-trans/test for inhibition of Vm of net glucose exit at an initial intracellular [glucose]=100 mM. This competitive inhibitor effect of testosterone on the infinite-cis Km is also relieved by flutamide (Ki ic test/Flut=1.1±0.2 μM) (Table 1). Competitive inhibition of glucose binding to the external side of the glucose transport system by testosterone is corroborative evidence that it binds externally.
Comparison of the effects of flutamide on phloretin-, genistein- and oestradiol-induced inhibitions of glucose exit
Additional evidence that flutamide acts at the external site of the glucose transporter is provided by the experiment showing that it antagonises phloretin action (Table 1). The apparent Ki of phloretin inhibition of zero-trans glucose exit is shifted from 0.48±0.07 μM with zero flutamide to 1.10±0.18 μM with 2 μM flutamide present (P<0.001). The Ki (phloretin/Flut) is 1.75±0.22 μM (Table 1). Although this Ki (phloretin/Flut) is 5–10 × higher than the Ki (test/Flut) for flutamide-dependent reversal of testosterone, or androstenedione inhibition of zero-trans net glucose exit, it is similar to the Ki of flutamide antagonism of testosterone action on infinite-cis glucose exit (see Table 1). This demonstrates that the drug acts at the external face of the transporter, possibly at a site adjacent to the phloretin-binding site (LeFevre & Marshall, 1959) (see below). Further evidence for this view is provided by the finding that low concentrations of phloretin competitively inhibit testosterone action on glucose exit (Table 1). Phloretin, like flutamide, increases the Ki of testosterone inhibition of zero-trans glucose exit (Ki test/phloretin= 76.3±10.9 nM) although, unlike flutamide, it also reduces the rate of glucose exit (Figure 2) (Basketter & Widdas, 1978). These findings, together with those described above, indicate that phloretin and testosterone bind to contiguous sites.
Figure 2.

Effects of varying concentrations of phloretin on the apparent Ki (testosterone) on glucose exit. (a) The Ki (testosterone) increases as the concentration of phloretin is increased: with 0 μM phloretin, the apparent Ki (testosterone) is 35±1.6 μM; with 100 nM phloretin, Ki (testosterone) is 85.7±12 μM; with 250 nM phloretin, Ki (testosterone) is 132±25 μM; and with 500 μM phloretin, Ki (testosterone) is 288±52. Each point is the average of 3–5 separate fluxes and each point at each concentration is the average of three experiments.
Effects of flutamide on oestradiol and genistein-induced inhibition of glucose exit
Flutamide is without effect on either oestradiol, or genistein-induced inhibition of glucose exit (data not shown). These compounds have been shown to act at the inside face of the glucose transporter (Afzal et al., 2002). This signifies that flutamide's inhibitor actions on glucose transport are specific to steroids acting on the outside face of the glucose transporter (see below).
Effects of green tea catechins and flutamide on glucose transport in erythrocytes
Application of a mixed green tea extract to the erythrocyte suspension inhibits zero-trans exit and reduces the affinity of glucose Ki (ic green tea) (Figure 3; Table 2 ). These effects of green tea extract are reversed by flutamide (Ki (green tea/Flut)= 0.65±0.2 μM). The Ki of green tea is obtained on the basis that the average molecular weight of green tea catechins is ≈500 Da.
Figure 3.

Effects of varying concentrations of green tea extract (estimated average mol wt=500) on zero-trans glucose exit at 21°C in the presence or absence of Flutamide (1.0 μM). The Ki of green tea extract on zero-trans net glucose exit from human red cells loaded with 100 mM is obtained as described in Methods. Ki (green tea) is 1.31±0.11 μM and with flutamide (1 μM) present, Ki (green tea) is 3.49±0.76 μM. Each point is the average of 3–5 separate fluxes, and each point at each concentration is the average of four experiments.
Table 2.
Effect of catechins and flutamide on glucose transport
|
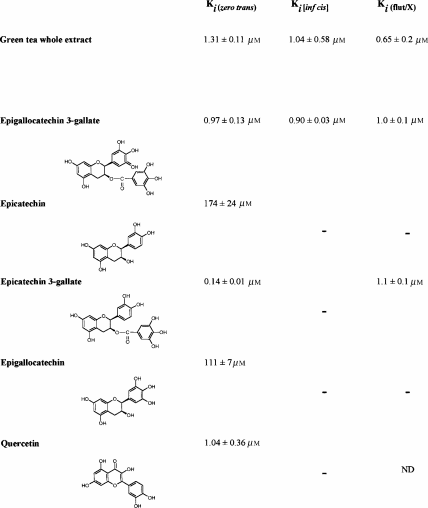
The effects of several pure green tea catechins were tested on zero-trans glucose exit. The Ki's of these substances on glucose exit are shown in Table 2. The catechin with the highest affinity is epicatechin 3-gallate (ECG) Ki (ECG)= 0.14±0.01 μM; the major constituent of green tea, epigallocatechin 3-gallate (EGCG), has a Ki (EGCG)=0.97±0.13 μM. Both the inhibitions of ECG and EGCG are competitively reversed by flutamide (Table 2). These effects are identical to those with the whole green tea extract, and indicate that the major constituents of green tea have the most potent effects on glucose transport. Like testosterone, EGCG competitively inhibits glucose binding on the external face of the carrier, as is evident from its effect on the Ki (ic/EGCG)=0.90±0.03 μM (Table 2). Ungallated catechins, epicatechin and epigallocatechin have only weak effects on glucose transport.
Another flavonoid, quercetin, strongly inhibits glucose exit (Table 2), but flutamide is without any antagonist effect on this inhibition.
Lack of androgenic and antiandrogenic effects of green tea catechins
Screening for androgenic and antiandrogenic effects of green tea catechins shows that in comparison with a standard androgen, dihydrotestosterone (DHT) and a standard antiandrogen, flutamide, the green tea catechins are virtually inactive (Figure 4). The yeast screen responses to DHT and flutamide are similar to those obtained by Sohoni & Sumpter (1998). DHT induces a positive dose-responsive increase in galactosidase expression in the yeast, whereas the antiandrogen flutamide inhibits the dose response generated by the IC50 of DHT=15 nM. It should be noted that all the antiandrogen responses were carried out using a background of 15 nM DHT, so that an inhibition can be observed. In all cases where antiandrogen activity is being observed, there is a background androgenic response of the assay. None of the catechins, or the flavone quercetin (not shown), which is also a strong inhibitor of glucose transport (Ki=1.04±0.04 μM) (Table 2), have any androgenic or antiandrogenic activity, as determined by the yeast assay. Although the assay cannot be used at catechin concentrations >0.1 mM, it shows that there is no significant androgenic or antiandrogenic activity in the concentration range 0.1–5 μM, where there is significant inhibition of glucose transport.
Figure 4.

Demonstration of the absence of any antiandrogen effect of green tea catechins. The androgen-screening test shows that DHT activates and flutamide inhibits chlorophenol red-D-galactopyranoside metabolism. All the results are normalised to the maximal response obtained with DHT. No significant androgenic or antiandrogenic effects were obtained with any of the catechins or flavones. A number of other compounds were tested, among which were epicatechin, epigallocatechin, quercetin and cyproterone. This experiment was repeated three times. Each point is the average of three estimates. The results of a single experiment are demonstrated. The other experiments gave similar results.
Cyproterone, a nonsteroidal antiandrogen (Singh et al., 2000), was found to have a weak androgenic effect with the screening test (data not shown). It has no significant effect on testosterone inhibition of glucose transport in red cells.
Sequence homologies between GLUT1 and AR
Searches for homologies between the ligand-binding sequences of hAR (Weatherman et al., 1999; Matias et al., 2000; Poujol et al., 2000; Sack et al., 2001) and the equivalent sequences in GLUT1 were made using a similar strategy to that previously described for oestradiol interactions with GLUT1 (Afzal et al., 2002). Good matches were obtained between the GLUT1 sequences in transmembrane helical regions TMs 1, 5, 6, 7 and 8, and in the extracellular linker segments joining TMs 3, 4, 9 and 10 (Figure 5).
Figure 5.

Schematic representation showing the predicted transmembrane domains of GLUT1 with homologous sequences colour coded to those of the hAR ligand-binding domain. For correspondences between eight colour-coded sequences and positions in the 3D structure of hAR LBD and the putative model of GLUT1 (Mueckler et al., 1985), see (b–d). The colours in the GLUT1 sequence are coded to show the equivalent positions in the LBD of hAR. (b–d) 3-D structures of the ligand-binding domain of the hAR with dihydrotestosterone in the binding cleft. (b) The homologous sequences to GLUT 1 in the ligand-binding domain are shown as colour-coded chains surrounding the ligand-binding cleft of hAR. Colours correspond to sequences shown in (a). The view shows these homologous sequences in the LBD of hAT with H-bonds linked to 3-oxy from Arg 752 and Thr 807, and Asn 705 to the 17-OH groups. The distances between these three anchoring amino acids, as estimated by Swiss Prot Viewers, are also shown; see Table 3. (c) The colour-coded homologous sequences in GLUT1 to hAR are shown in close view of the outside surface of GLUT1, as modelled by Zuniga et al. (2001). (d) A distance view of homologous sequences in GLUT1, showing their position relative to the outside of the transporter.
The essential H-bonding linkages between the steroid and the LBD in hAR are at Arg 752, which H-bonds to the A ring 3-oxy group, Asn 705 and Thr 877 residues which H-bond to the D ring 17-OH of dihydrotestosterone (Sack et al., 2001). These three H-bonding amino acids anchor the two polar ends of the steroid ligand in the ligand-binding cleft. The separations in hAR were measured using Swiss-Pdb Viewer (Table 3 ). Asn 29 and Thr 30 in TM1, Ser 285 and Asn 288 in TM7 and Asn 317 and Thr 318 in TM8 fall within a sphere of radius 18 Å centred on the guanidinium group of GLUT1 Arg 126. These could all make suitable second anchoring points for the antipodal D-ring 17-β OH group of testosterone, the OH residues of the gallate group of EGCG, or the phenolic OH groups of phloretin (Tables 2,3).
Table 3.
Σχ2 of the deviations from the distances between H-bonding anchoring amino acids in AR-LBD and their equivalents in the putative androgen-binding domains in GLUT1
| Arg | Distance Arg/thr or ser (Å) | Thr/ser | Distance Arg/Asn (Å) | Asn | Distance Thr/Asn (Å) | Σχ2 |
|---|---|---|---|---|---|---|
| 126 | 16 | 30 | 11.5 | 288 | 4.8 | 0.03 |
| 126 | 16 | 30 | 11.5 | 29 | 5.3 | 0.06 |
| 126 | 16.4 | 285 | 15 | 288 | 8.3 | 0.74 |
| 126 | 16.4 | 285 | 17.5 | 317 | 8.3 | 0.82 |
| 126 | 22.7 | 318 | 15 | 288 | 12.4 | 3.4 |
| 126 | 22.7 | 318 | 17.5 | 317 | 12.8 | 3.9 |
The importance of Arg 126 and Thr 30 in the function of GLUT1 is indicated by the fact that these groups are conserved in all species currently in the Swissprot database, namely in GLUT1 from mouse, rat, cattle, sheep, pig, rabbit chicken and human. A mutation in human GLUT1, where Arg 126 is substituted for Leu, R126L, generates GLUT-1 deficiency syndrome (Wang et al., 2000). Children with this mutation have maximal transport rates of 3-O-methyl-glucose entry into erythrocytes that are only 15–20% of the wild type.
Discussion
Evidence that androgens and flutamide bind to the external surface of GLUT1
Previous studies (Krupka & Devés, 1980; May & Danzo, 1988) on androgen interaction with the erythrocyte transport system concluded that testosterone and androstenedione bind to an internal glucose export site. The basis for these conclusions is that testosterone and androstenedione are better inhibitors of the low-affinity sugar D-xylose exit, than of the higher affinity D-glucose exit. In contrast, the action of a sugar transport inhibitor binding exclusively to the external site, for example, phloretin, would be equally effective against both exiting sugars, independent of their affinity for the transporter (LeFevre & Marshall, 1959; Basketter & Widdas 1978; Krupka & Devés, 1980).
May & Danzo (1988) showed that incorporation of labelled androstenedione into the glucose transporter protein was inhibited by cytochalasin B. This finding was taken to corroborate the view that androgens bind at the inside of GLUT1.
The opposite view can be deduced from the results here and previously found (Lacko et al., 1975). Testosterone reduces the Vm of net glucose exit and the affinity of glucose at the external site, consistent with competitive inhibition at the outside site (Table 1). An inhibitor binding to the inside site would alter the Vm of exit without affecting glucose affinity at the external solution (Basketter & Widdas, 1978; Krupka & Devés, 1980). Secondly, flutamide does not alter the affinity of oestradiol or genistein (Table 1), which bind to the inside glucose export site (King et al., 1991; Vera et al., 2001; Afzal et al., 2002). However, flutamide competitively inhibits testosterone, androstenedione, DHEA 3-acetate, DHEA and phloretin-dependent inhibition of glucose exit. Additionally, phloretin competitively reduces the affinity of testosterone inhibition of glucose transport (Figure 2). Since glucose in the external solution and phloretin bind at an outside-facing site (LeFevre & Marshall, 1959), we conclude that androgens and flutamide also bind to an external site and not to an internal site.
The findings of May & Danzo (1988) may be explicable in terms of allosteric protection of the external sites by cytochalasin B rather than androstenedione binding to an internal site (Hamill et al., 1999; Cloherty et al., 2001). Testosterone was found to have less definitive effect in competition for D-xylose and D-glucose exit than cytochalasin B, which competitively inhibited the low-affinity xylose exit more than glucose at the inside (Krupka & Devés, 1980).
Comparison of androgen binding to the erythrocyte and androgen receptors
The mammalian androgen receptor has a higher affinity for DHT than testosterone (Wilson & French, 1976). Androgen binding to human erythrocytes does not conform to the specificity of the hAR. However, two other types of AR have been described in fish (Sperry & Thomas, 1999) and recently isolated (Cavaco et al., 2001). Fish AR1 has a higher affinity for testosterone than DHT, whereas fish AR2 has a similar specificity to mammalian AR. It should be noted that kelpbass AR2 also has a high affinity for xenobiotics, such as the hydroxylated polychlorodiphenols, which have similar chemical structures to catechins (Sperry & Thomas, 1999). Other similarities between the human erythrocyte ‘AR' (herAR) and fish AR1 are the higher affinity of 3-keto-4-ene androgens, for example, androstenedione, androsterone, than either DHT or other 3-keto-androstans (Table 1).
Comparison of the affinities of the steroids herAR tested indicate the following:
Delocalised electrons at the 3-keto group, resulting from a 4,5-ene in the A ring, for example, testosterone, or an acetoxy group at the 3β position, for example, DHEA-3 acetate, increase steroid affinity by >10-fold in comparison with a lone 3-keto group of DHT (P<0.001) (Table 1).
Substitution of strong electronegative 3-O-sulphate in place of a 3-O-acetate decreases the affinity of DHEA-3-sulphate by more than 30-fold over the high-affinity ligand DHEA-3 acetate (P<0.001).
Saturation of the 4,5 position (e.g. 4-androsten-3,17-dione, to 5-β-androstan 3α-ol 17-one, reduces steroid affinity by ≈3–4-fold (P<0.0025)(Table 1).
Alteration from 5α to 5β decreases steroid affinity by 3–4-fold c.f. 5α-androstan and 5β-androstan 3-ol-17-one (P<0.0025) (see androsterone and etiocholanalone, Table 1).
In summary, the following steroid ligand-binding properties in herAR and hAR are similar; 5α in preference to a 5β conformation, unsaturation at either the 4 or 5-position and either a 17β-one, or 17-ol group in the D ring (Ojasoo et al., 1995).
Androgen interactions with the G6PD have similar relative affinities to those shown in Table 1. The Ki's of DHEA, epiandrosterone and DHEA sulphate acting as uncompetitive antagonists of glucose-6-phosphate interaction with G6PD are 8.9±0.3, 3.0±0.1 and 511±70 μM, respectively (Gordon et al., 1995). These findings indicate that DHEA is a more potent inhibitor of G6PD than of glucose transport, and that the negative charge of DHEA 3-sulphate similarly reduces binding affinity by ≈30-fold to both the glucose transporter and to G6PD.
Modelling flutamide action on glucose transport in erythrocytes
A 3-D structural model of GLUT1 shows that the 12α helical transmembrane domains (TMs) are arranged around a hydrophilic core, through which glucose permeates (Zuniga et al., 2001). A similar 3-D structure for GLUT3 has also been described (Dwyer, 2001). Scanning cysteine mutagenesis studies indicate that there is an open cleft in the extracellular surface of GLUT, which permits the hydrophilic alkylating reagents like parachloromercuribenzoic acid sulphonate (PCMBS) to penetrate at least 50% of the distance across the pore (Mueckler & Makepeace, 1997,2002).
X-ray crystallography shows that in the hAR Arg 752 H-bonds to the A ring 3-keto group and Asn 705 and Thr 877 residues H-bond to the D ring 17-OH of dihydrotestosterone (Alberts & Blute, 2001) (Figure 5b). The best match between the topology of the double-anchor amino-acid triad Arg 752, Asn 705 and Thr 877 in the binding cleft of the LBD of hAR and GLUT1 is with Arg 126, Asn 288 and Thr 30 (P<0.05) (Table 3, Figure 5c). These putative steroid-binding sites for androgens are on the rim of the hydrophilic cleft in the external face of GLUT1. In this position, testosterone would obstruct glucose entry into the hydrophilic cleft, and thus behave as a competitive inhibitor of glucose entry.
The 3-D model of GLUT1 (Zuniga et al., 2001) indicates that the external androgen-binding site and oestrogens at the inside surface (Afzal et al., 2002) are separated by 35–45 Å (4–5 glucose diameters) (Figure 5d). This large separation distance between the alternate binding sites indicates that the transporter is likely to be a two-site rather than a one-site model (Naftalin et al., 2002).
Green tea catechins and flavones on glucose transport
Comparison of the effects of the various catechins tested on glucose transport from erythrocytes indicates that gallation of epigallocatechin to EGCG, and of epicatechin to epicatechin-3-gallate increases the affinities of the catechins for the glucose transporter by two to four orders of magnitude (Table 2). Comparison of the structure of EGCG with testosterone shows that the distance between the oxygens in the 3-keto and 17-OH positions is similar to distances between the 5-OH in the catechin A ring and the hydroxyls of the gallate group (1.0–1.1 nm). The similar separation distances between the antipodal OH groups of EGCG and androgens may permit them to compete for similar sites on GLUT1 (Table 3).
Since neither the green tea catechins nor the flavone quercetin (not shown) act as androgens or antiandrogens at a genomic level (Figure 4), it is evident that the reported antiandrogenic actions of green tea are unrelated to their genomic effects (Gupta et al., 2001).
In human subjects, a single oral dose (1.5 mmol) of green tea catechins is readily absorbed into plasma, reaching a concentration in the range 1–2 μM in plasma within 2–5 h (Van Amelsvoort et al., 2001). The results here show that the gallated catechins EGCG (60% g g−1 catechin in whole tea extracts) and ECG (23% g g−1 catechin in whole tea catechins) act in similar ways to androgens on human erythrocyte glucose transport, albeit with much higher affinities for the external site than most androgens. The evidence for this comes from the findings that (a) flutamide competitively antagonises the inhibitory effect of green tea on glucose exit, as well as of ECG and EGCG (Table 2); (b) EGCG inhibits glucose binding to the external site, as deduced from its effect on the infinite-cis Km of glucose binding to the external site (Table 2). Ungallated catechins, epicatechin EC (5.5% g g−1 catechin in whole tea extracts) and epigallocatechin EGC (6% g g−1 catechin in whole tea extracts) only have weak effects on glucose transport, as previously shown by Park (1999).
Physiological response to catechins
There are several possible sites where catechins may act to cause their antidiabetogenic effects. However, most catechin effects at these putative sites are observed only at 1–3 orders of magnitude higher concentration than realistic levels found in the blood of habitual tea drinkers, ≈1 μM. The leptin-sensitive appetite control centre can be eliminated, as green tea catechins produce a similar weight loss in both lean and obese Zucker rats. This latter strain does not express leptin receptor (Kao et al., 2000). High doses of green tea catechins, enough to raise plasma levels to 1 mM EGCG, reduce the elevation of serum glucose levels in normal rats given 2 g glucose kg−1 body weight by gavage (Sabu et al., 2002). In alloxan-treated rats, catechins (20–50 μM) reduce plasma glucose concentration (Sabu et al., 2002). These findings suggest that the observed antidiabetic effects of green tea catechins are due to inhibition of intestinal glucose transport and/or inhibition of renal glucose absorption. However, they are obtained with much higher concentrations of catechins, than are physiological. At these high concentrations, antioxidant effects also feature catechin action, but not within the physiological concentration range.
Another view is that relatively high concentrations of EGCG (>10 μM) prevent hyperglycaemia by inhibiting gluconeogenesis in heptocytes, due to inhibition of the synthesis of phosphoenolpyruvate kinase (Waltner-Law et al., 2002). However, this catechin concentration is also higher than that normally observed in human plasma (Van Amelsvoort et al., 2001) and so may not be of much relevance to the observed hypoglycaemic effects of green tea in man (Kao et al., 2000). It is possible that the inhibition of gluconeogenesis seen with high concentrations of EGCG could in part be due to inhibition of glucose exit from hepatocytes (Waltner-Law et al., 2002). However, there is no information on the catechin-dependent inhibition of glucose transport in these cells, which have a low-affinity GLUT isoform GLUT2.
The main physiological effect of catechins in rats is appetite suppression. Following feeding rats with green tea catechins, reduced food intake accounts for most, although not all, of the reduction in testosterone levels and a concomitant reduction in testis and prostatic weights (Kao et al., 2000; Kobayashi et al., 2000). The green tea catechin-induced prostatic atrophy and retarded development of murine prostatic carcinogenesis (Gupta et al., 2001) may relate to catechin inhibition of glucose transport in Leydig cells (Khanum et al., 1997).
Since the expected concentration of these compounds in human circulation after a moderate oral intake exceeds the concentration of gallated catechins that cause half-maximal inhibition of glucose transport (Van Amelsvoort et al., 2001), it seems likely that glucose transport is a target for these drugs in vivo.
The blocking of glucose uptake into Leydig cells inhibits androstenedione conversion to testosterone due to depletion of ATP (Khanum et al., 1997). Other similar flavones, e.g. quercetin and myricetin in addition to catechins, have been shown to directly inhibit cellular glucose transport Ki for glucose uptake ≈10 μM (Park, 1999), as is confirmed here (Table 2).
Possible conflict between antiandrogen action of flutamide and catechins
The main site of anti-androgen action is on the LBD of the androgen receptor (Weatherman et al., 1999; Matias et al., 2000; Alberts & Blute, 2001; Sack et al., 2001).
Since, in erythrocytes at least, flutamide antagonises the inhibition of glucose transport by green tea catechins, it is possible that there could be a therapeutic conflict between the action of tea catechins and flutamide when simultaneously applied in the treatment of prostatic hyperplasia. Flutamide is given clinically at a dose of 0.5–3 m mol day−1 (Kolvenbag et al., 1998). This gives an average plasma concentration of hydroxyflutamide, ≈1 μM. Hydroxyflutamide is a more active metabolite of flutamide (Niopas and Daftsios, 2001). If tea catechins block glucose uptake into Leydig cells, thereby inhibiting testosterone production (Khanum et al., 1997), and flutamide in the μM concentration range antagonises this effect of catechins, then Flutamide may exert a physiological antagonism by preventing the catechin-dependent inhibition of glucose transport into Leydig cells and hence of testosterone synthesis.
Possible mechanisms of insulin resistance in hyperandrogenism
The clinical improvement of polycystic ovarian syndrome (PCOS) patients treated with Flutamide could arise from direct actions of the drug on GLUTs. However, the current view is that reduced insulin sensitivity, that is, hyperglycaemia with hyperinsulinaemia, in PCOS results from a reduction in insulin receptor substrate proteins rather than on GLUTs (Collison et al., 2000). The plasma concentrations of the most prevalent steroid DHEA and its derivatives in PCOS are 5–10 μM. The other androgens have concentrations at least 2–3 orders lower than their Ki's for the inhibition of glucose transport (Livingstone & Collison, 2002). It therefore seems unlikely that raised androgen concentrations in PCOS are aetiological factors of the disease via inhibition of GLUTs.
Conclusions
Several androgens inhibit glucose exit from human erythrocytes and compete with glucose binding at an external site of the transporter.
The antiandrogen flutamide competitively antagonises the androgen inhibitions of glucose transport. Flutamide also antagonises the inhibition of glucose exit by phloretin, which is known to bind to the external site of the glucose transporter.
Additionally, the major constituents of green tea, that is, EGCG and EGC are strong competitors of glucose binding to the external site of GLUT1. Flutamide also antagonises these effects of EGCG and ECG.
Several sequence homologies exist between GLUT1 and the LBD of hAR. In GLUT1, these homologies contain two amino acid triads at the external surface of the transporter, which have suitable topologies to form H-bonding anchoring groups to the antipodal 3-OH and 17-OH in androgens.
Abbreviations
- DHEA
dehydroepiandrosterone
- DHT
5-α-dihydrotestosterone
- ECG
epicatechin 3-gallate
- EGCG
epigallocatechin 3-gallate
- GLUT1
glucose transporter protein 1
- G6PD
glucose 6-phosphate dehydrogenase
- hAR
human androgen receptor
- Km(ic)
Km of glucose binding to external site infinite cis
- Km (ic glucose/test)
Ki of the testosterone-dependent reduction of glucose affinity at external site
- LBD
ligand-binding domain
- PBS
phosphate-buffered saline
- TMs
helical transmembrane domains
References
- AFZAL I., CUNNINGHAM P., NAFTALIN R.J. Interactions of ATP, oestradiol, genistein and the anti-oestrogens, faslodex (ICI 182780) and tamoxifen, with the human erythrocyte glucose transporter, GLUT1. Biochem. J. 2002;365:707–719. doi: 10.1042/BJ20011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALBERTS S.R., BLUTE M.L. Chemoprevention for prostatic carcinoma: the role of flutamide in patients with prostatic intraepithelial neoplasia. Urology. 2001;57:188–190. doi: 10.1016/s0090-4295(00)00971-7. [DOI] [PubMed] [Google Scholar]
- BASKETTER D.A., WIDDAS W.F. Asymmetry of the hexose transfer system in human erythrocytes: comparison of the effects of cytochalasin B, phloretin and maltose as competitive inhibitors. J. Physiol. 1978;278:389–401. doi: 10.1113/jphysiol.1978.sp012311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENTEN W.P., LIEBERHERR M., STAMM O., WREHLKE C., GUO Z.Y., WUNDERLICH F. Testosterone signalling through internalizable surface receptors in androgen receptor-free macrophages. Mol. Biol. Cell. 1999;10:3113–3123. doi: 10.1091/mbc.10.10.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIAGLOW J.E., AYENE I.S., KOCH C.J., DONAHUE J., STAMATO T.D., TUTTLE S.W. G6PD deficient cells and the bioreduction of disulfides: effects of DHEA, GSH depletion and phenylamine oxide. Biochem. Biophys. Res. Commun. 2000;273:846–852. doi: 10.1006/bbrc.2000.3024. [DOI] [PubMed] [Google Scholar]
- BURGER H.G. Androgen production in women. Fertil. Steril. 2002;77:S3–S5. doi: 10.1016/s0015-0282(02)02985-0. [DOI] [PubMed] [Google Scholar]
- CAVACO J.E.B., BOGERD J., GOOS H., SCHULZ R.W. Testosterone inhibits 11-ketotestosterone-induced spermatogenesis in African catfish (Clarias gariepinus) Biol. Reprod. 2001;65:1807–1812. doi: 10.1095/biolreprod65.6.1807. [DOI] [PubMed] [Google Scholar]
- CLOHERTY E.K., LEVINE K.B., Carruthers A. The red blood cell glucose transporter presents multiple, nucleotide-sensitive sugar exit sites. Biochemistry. 2001;40:15549–15561. doi: 10.1021/bi015586w. [DOI] [PubMed] [Google Scholar]
- COLLISON M., CAMPBELL I.W., SALT I.P., DOMINICZAK A.F., CONNELL J.M., LYALL H., GOULD G.W. Sex hormones induce insulin resistance in 3T3-L1 adipocytes by reducing cellular content of IRS proteins. Diabetologia. 2000;43:1374–1380. doi: 10.1007/s001250051541. [DOI] [PubMed] [Google Scholar]
- DWYER D.S. Model of the 3-D structure of the GLUT3 glucose transporter and molecular dynamics simulation of glucose transport. Proteins Struct. Funct. Genet. 2001;42:531–541. [PubMed] [Google Scholar]
- GORDON G., MACKOW M.C., LEVY H.R. On the mechanism of interaction of steroids with human glucose-6-phosphate dehydrogenase. Arch. Biochem. Biophys. 1995;318:25–29. doi: 10.1006/abbi.1995.1199. [DOI] [PubMed] [Google Scholar]
- GUPTA S., HASTAK K., AHMAD N., LEWIN J.S., MUKHTAR H. Inhibition of prostate carcinogenesis in TRAMP mice by oral infusion of green tea polyphenols. Proc. Natl. Acad. Sci. U.S.A. 2001;98:10350–10355. doi: 10.1073/pnas.171326098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILL S., CLOHERTY E.K., CARRUTHERS A. The human erythrocyte sugar transporter presents two sugar import sites. Biochemistry. 1999;38:16974–16983. doi: 10.1021/bi9918792. [DOI] [PubMed] [Google Scholar]
- HO Y.Y., YANG H., KLEPPER J., FISCHBARG J., WANG D., DE VIVO D.C. Glucose transporter type 1 deficiency syndrome (Glut1DS): methylxanthines potentiate glut1 haploinsufficiency in vitro. Pediatr. Res. 2001;50:254–260. doi: 10.1203/00006450-200108000-00015. [DOI] [PubMed] [Google Scholar]
- IBANEZ L., POTAU N., MARCOS M.V., DE ZEGHER F. Treatment of hirsutism, hyperandrogenism, oligomenorrhea, dyslipidemia, and hyperinsulinism in nonobese, adolescent girls: effect of flutamide. J. Clin. Endocrinol. Metab. 2000;85:3251–3255. doi: 10.1210/jcem.85.9.6814. [DOI] [PubMed] [Google Scholar]
- JAROW J.P., BREMNER W., CHEN H., ZIRKIN B. The effect of testosterone male contraception upon intratesticular testosterone concentration in the human testis. Fertil. Steril. 2001;76:128. [Google Scholar]
- KAO Y.H., HIIPAKKA R.A., LIAO S. Modulation of endocrine systems and food intake by green tea Epigallocatechin Gallate. Endocrinology. 2000;141:980–987. doi: 10.1210/endo.141.3.7368. [DOI] [PubMed] [Google Scholar]
- KHANUM A., BUCZKO E., DUFAU M.L. Essential role of adenosine triphosphate in activation of 17β-hydroxysteroid dehydrogenase in the rat Leydig cell. Endocrinology. 1997;138:1612–1620. doi: 10.1210/endo.138.4.5062. [DOI] [PubMed] [Google Scholar]
- KING A.P.J., TAI P.-K.K., CARTER-SU C. Cytochalasin B interferes with conformational changes of the human erythrocyte glucose transporter induced by internal and external sugar binding. Biochemistry. 1991;30:11546–11553. doi: 10.1021/bi00113a009. [DOI] [PubMed] [Google Scholar]
- KOBAYASHI Y., SUZUKI M., SATSU H., ARAI S., HARA Y., SUZUKI K., MIYAMOTO Y., SHIMIZU M. Green tea polyphenols inhibit the sodium-dependent glucose transporter of intestinal epithelial cells by a competitive mechanism. J. Agric. Food Chem. 2000;48:5618–5623. doi: 10.1021/jf0006832. [DOI] [PubMed] [Google Scholar]
- KOLVENBAG G.J., FURR B.J., BLACKLEDGE G.R. Receptor affinity and potency of non-steroidal antiandrogens: translation of preclinical findings into clinical activity. Prostate Cancer Prostatic Dis. 1998;1:307–314. doi: 10.1038/sj.pcan.4500262. [DOI] [PubMed] [Google Scholar]
- KRUPKA R.M., DEVÉS R. Asymmetric binding of steroids to internal and external sites of the glucose carrier of erythrocytes. Biochim. Biophys. Acta. 1980;598:134–144. doi: 10.1016/0005-2736(80)90271-0. [DOI] [PubMed] [Google Scholar]
- LACKO L., WITTKE B., GECK P. Interaction of steroids with the transport system of glucose in human erythrocytes. J. Cell. Physiol. 1975;86:673–680. doi: 10.1002/jcp.1040860512. [DOI] [PubMed] [Google Scholar]
- LEFEVRE P.G., MARSHALL J.K. The attachment of phloretin and analogues to human erythrocytes in connection with inhibition of sugar transport. J. Biol. Chem. 1959;234:3022–3026. [PubMed] [Google Scholar]
- LIVINGSTONE C., COLLISON M. Sex steroid and insulin resistance. Clin. Sci. 2002;102:151–166. doi: 10.1042/cs1020151. [DOI] [PubMed] [Google Scholar]
- MARHEFKA C.A., MOORE B.M., BISHOP T.C., KIRKOVSKY L., MUKHERJEE A., DALTON J.T., MILLER D.D. Homology modelling using multiple molecular dynamics simulations and docking studies of the human androgen receptor ligand binding domain bound to testosterone and nonsteroidal ligands. J. Med. Chem. 2001;44:1729–1740. doi: 10.1021/jm0005353. [DOI] [PubMed] [Google Scholar]
- MATIAS P.M., DONNER P., COELHO R., THOMAZ M., PEIXOTO C., MACEDO S., OTTO N., JOSCHKO S., SCHOLZ P., WEGG A., BASLER S., SCHAFER M., EGNER U., CARRONDO M.A. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. implications for pathogenic gene mutations. J. Biol. Chem. 2000;275:26164–26171. doi: 10.1074/jbc.M004571200. [DOI] [PubMed] [Google Scholar]
- MAY J.M., DANZO B.J. Photolabeling of the human erythrocyte glucose carrier with androgenic steroids. Biochem. Biophys. Acta. 1988;943:199–210. doi: 10.1016/0005-2736(88)90552-4. [DOI] [PubMed] [Google Scholar]
- MCDONALD S., BRIVE L., AGUS D.B., SCHER H.I., ELY K.R. Ligand responsiveness in human prostate cancer: structural analysis of mutant androgen receptors from LNCaP and CWR22 tumors. Cancer Res. 2000;60:2317–2322. [PubMed] [Google Scholar]
- MUECKLER M., CARUSO C., BALDWIN S.A., PANICO M., BLENCH I., MORRIS H.R., ALLARD W.J., LEINHARD G.E., LODISH H.F. Sequence and structure of a human glucose transporter. Science. 1985;229:941–945. doi: 10.1126/science.3839598. [DOI] [PubMed] [Google Scholar]
- MUECKLER M., MAKEPEACE C. Identification of an amino acid residue that lies between the exofacial vestibule and exofacial substrate-binding site of the Glut1 sugar permeation pathway. J. Biol. Chem. 1997;272:30141–30146. doi: 10.1074/jbc.272.48.30141. [DOI] [PubMed] [Google Scholar]
- MUECKLER M., MAKEPEACE C. Analysis of trans-membrane segment 10 of the Glut1 glucose transporter by cysteine-scanning mutagenesis and substituted cysteine accessibility. J. Biol. Chem. 2002;277:3498–3503. doi: 10.1074/jbc.M109157200. [DOI] [PubMed] [Google Scholar]
- NAFTALIN R.J., AFZAL I., BROWNING J.A., WILKINS R.J., ELLORY J.C. Effects of high pressure on glucose transport in the human erythrocyte. J. Membr. Biol. 2002;186:113–129. doi: 10.1007/s00232-001-0140-z. [DOI] [PubMed] [Google Scholar]
- NIOPAS I., DAFTSIOS A.C. Determination of 2-hydroxyflutamide in human plasma by high-performance liquid chromatography and its application to pharmacokinetic studies. J. Chromatogr. B. Biomed. Sci. Appl. 2001;759:179–183. doi: 10.1016/s0378-4347(01)00204-3. [DOI] [PubMed] [Google Scholar]
- OJASOO T., RAYNAUD J.P., DORÉ J.C. Correspondence factor analysis of steroid libraries. Steroids. 1995;60:458–469. doi: 10.1016/0039-128x(95)00005-b. [DOI] [PubMed] [Google Scholar]
- PARK J.B. Flavonoids are potential inhibitors of glucose uptake in U937 cells. Biochem. Biophys. Res. Commun. 1999;260:568–574. doi: 10.1006/bbrc.1999.0890. [DOI] [PubMed] [Google Scholar]
- PEARSON W.R., LIPMAN D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. U.S.A. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERUSQUIA M., VILLALON C.M. Possible role of Ca2+ channels in the vasodilating effect of 5beta-dihydrotestosterone in rat aorta. Eur. J. Pharmacol. 1999;371:169–178. doi: 10.1016/s0014-2999(99)00161-2. [DOI] [PubMed] [Google Scholar]
- POUJOL N., WURTZ J.M., TAHIRI B., LUMBROSO S., NICOLAS J.C., MORAS D., SULTAN C. Specific recognition of androgens by their nuclear receptor. A structure–function study. J. Biol. Chem. 2000;275:24022–24031. doi: 10.1074/jbc.M001999200. [DOI] [PubMed] [Google Scholar]
- SABU M.C., SMITHA K., KUTTAN R. Anti-diabetic activity of green tea polyphenols and their role in reducing oxidative stress in experimental diabetes. J. Ethnopharmacol. 2002;83:109–116. doi: 10.1016/s0378-8741(02)00217-9. [DOI] [PubMed] [Google Scholar]
- SACK J.S., KISH K.F., WANG C., ATTAR R.M., KIEFER S.E., AN Y., WU GY SCHEFFLER J.E., SALVATI M.E., KRYSTEK S.R., WEINMANN R., EINSPAHR H.M. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc. Natl. Acad. Sci. U.S.A. 2001;98:4904–4909. doi: 10.1073/pnas.081565498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEN A.K., WIDDAS W.F. Determination of the temperature and pH dependence of glucose transfer across the human erythrocyte membrane measured by glucose exit. J. Physiol. 1962;160:392–403. doi: 10.1113/jphysiol.1962.sp006854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINGH S.M., GAUTHIER S., LABRIE F. Androgen receptor antagonists (antiandrogens): structure–activity relationships. Curr. Med. Chem. 2000;7:211–247. doi: 10.2174/0929867003375371. [DOI] [PubMed] [Google Scholar]
- SOHONI P., SUMPTER J.P. Several environmental oestrogens are also anti-androgens. J. Endocrinol. 1998;158:327–339. doi: 10.1677/joe.0.1580327. [DOI] [PubMed] [Google Scholar]
- SPEISER P.W., SERRAT J., NEW M.I., GERTNER J.M. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hyroxylase deficiency. J. Clin. Endocrinol. Metab. 1992;75:1421–1424. doi: 10.1210/jcem.75.6.1464643. [DOI] [PubMed] [Google Scholar]
- SPERRY T.S., THOMAS P. Identification of two nuclear androgen receptors in Kelp Bass (Paralabrax clathratus) and their binding affinities for xenobiotics: comparison with Atlantic Croaker (Micropogonias undulatus) androgen receptors. Biol. Reprod. 1999;61:1152–1161. doi: 10.1095/biolreprod61.4.1152. [DOI] [PubMed] [Google Scholar]
- VAN AMELSVOORT J.M.M., VAN HET HOF K.H., MATHOT J.N.J.J.T., MULDE P.J., WIERSMA R.A., TIJBURG L.B.M. Plasma concentrations of individual tea catechins after a single oral dose in humans. Xenobiotica. 2001;31:891–901. doi: 10.1080/00498250110079149. [DOI] [PubMed] [Google Scholar]
- VERA J.C., REYES A.M., VELASQUEZ F.V., RIVAS C.I., ZHANG R.H., Strobel P., SLEBE J.C., NUNEZ-ALARCON J., GOLDE D.W. Direct inhibition of the hexose transporter GLUT1 by tyrosine kinase inhibitors. Biochemistry. 2001;40:777–790. doi: 10.1021/bi001660j. [DOI] [PubMed] [Google Scholar]
- WALTNER-LAW M.E., WANG X.L., LAW B.K., HALL R.K., NAWANO M., GRANNER D.K. Epigallocatechin gallate, a constituent of green tea, represses hepatic glucose production. J. Biol. Chem. 2002;277:34933–34940. doi: 10.1074/jbc.M204672200. [DOI] [PubMed] [Google Scholar]
- WANG D., KRANZ-EBLE P., DE VIVO D.C. Mutational analysis of GLUT1(SLC2A1) in Glut-1 deficiency syndrome. Hum. Mutat. 2000;16:224–231. doi: 10.1002/1098-1004(200009)16:3<224::AID-HUMU5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- WEATHERMAN R.V., FLETTERICK R.J., SCANLAN T.S. Nuclear-receptor ligands and ligand-binding domains. Annu. Rev. Biochem. 1999;68:559–581. doi: 10.1146/annurev.biochem.68.1.559. [DOI] [PubMed] [Google Scholar]
- WILSON E.M., FRENCH F.S. Binding properties of androgen receptors. J. Biol. Chem. 1976;251:5620–5629. [PubMed] [Google Scholar]
- WOODARD T.L., BURGHEN G.A., KITABCHI A.E.B., WILIMAS J.A. Glucose-intolerance and insulin resistance in aplastic-anemia treated with oxymetholone. J. Clin. Endocrin. Metab. 1981;53:905–908. doi: 10.1210/jcem-53-5-905. [DOI] [PubMed] [Google Scholar]
- YANG N.C., JENG K.C.G., HO W.M., CHOU S.J., HU M.L. DHEA inhibits cell growth and induces apoptosis in BV-2 cells and the effects are inversely associated with glucose concentration in the medium. J. Steroid Biochem. Mol. Biol. 2000;75:159–166. doi: 10.1016/s0960-0760(00)00180-1. [DOI] [PubMed] [Google Scholar]
- YANG N.C., JENG K.C.G., HO W.M., HU M.L. ATP depletion is an important factor in DHEA-induced growth inhibition and apoptosis in BV-2 cells. Life Sci. 2002;70:1979–1988. doi: 10.1016/s0024-3205(01)01542-9. [DOI] [PubMed] [Google Scholar]
- ZHU X., LI H., LIU J.P., FUNDER J.W. Androgen stimulates mitogen-activated protein kinase in human breast cancer cells. Mol. Cell. Endocrinol. 1999;152:199–206. doi: 10.1016/s0303-7207(99)00031-3. [DOI] [PubMed] [Google Scholar]
- ZUNIGA F.A., SHI S., HALLER J.F., RUBASHKIN A., FLYNN D.R., ISEROVICH P., FISCHBARG J. A three dimensional model of the human facilitative glucose transporter GLUT1. J. Biol. Chem. 2001;276:444970–444975. doi: 10.1074/jbc.M107350200. [DOI] [PubMed] [Google Scholar]