

Abstract
Ragaglitazar [(−) DRF 2725; NNC 61-0029] is a coligand of PPARα and PPARγ.
In ob/ob mice, ragaglitazar showed significant reduction in plasma glucose, triglyceride and insulin (ED50 values <0.03, 6.1 and <0.1 mg kg−1). These effects are three-fold better than rosiglitazone and KRP-297. In Zucker fa/fa rats, ragaglitazar showed dose-dependent reduction in triglyceride and insulin, hepatic triglyceride secretion and triglyceride clearance kinetics (maximum of 74, 53, 32 and 50% at 3 mg kg−1), which are better than rosiglitazone and KRP-297.
In a high-fat-fed hyperlipidaemic rat model, the compound showed an ED50 of 3.95, 3.78 mg kg−1 for triglyceride and cholesterol lowering, and 0.29 mg kg−1 for HDL-C increase. It also showed improvement in clearance of plasma triglyceride and hepatic triglyceride secretion rate. All these effects are 3–10-fold better than fenofibrate and KRP-297.
Ragaglitazar treatment showed significant reduction in plasma Apo B and Apo CIII levels, and increase in liver CPT1 and CAT activity and ACO mRNA. Significant increase of both liver and fat LPL activity and fat aP2 mRNA was also observed.
In a high-fat-fed hamster model, ragaglitazar at 1 mg kg−1 showed 83 and 61% reduction in triglyceride and total cholesterol, and also 17% reduction in fat feed-induced body weight increase. In these hyperlipidaemic animal models, PPARγ ligands failed to show any significant efficacy. Taken together, ragaglitazar shows better insulin-sensitizing and lipid-lowering potential, as compared to the standard compounds.
Keywords: Antidiabetic, hypolipidaemic, insulin sensitizer, peroxisome proliferator-activated receptor (PPAR)
Introduction
Type II diabetes is characterized by insulin resistance, hyperglycaemia, and in most cases hyperlipidaemia (Defronzo et al., 1992). If untreated, it leads to several secondary complications such as hypertension, atherosclerosis, coronary artery disease, neuropathy and nephropathy (Porte & Schwartz, 1996). Thiazolidinediones (TZDs) are the first chemical class of insulin sensitizers launched for the treatment of type II diabetes. In addition to lowering blood glucose and insulin levels and improving insulin sensitivity, TZDs have a marginal plasma lipid-lowering effect (Aronoff et al., 2000). Several lines of evidence indicate that peroxisome proliferator-activated receptor gamma (PPARγ) might be the molecular target for these molecules (Schoonjans et al., 1996b; Wilson et al., 1996). PPARγ belongs to the nuclear receptor family of PPARs. PPARs regulate the expression of genes that control lipid and glucose metabolism. Three subtypes of PPAR have been identified: α, γ and δ, each with a specific role and tissue distribution (Desvergene & Wahli, 1999). PPARα is predominantly expressed in the liver, kidney, heart and skeletal muscle, where it controls fatty acid catabolism (Desvergene & Wahli, 1999). Fibrates, discovered decades ago, are effective at lowering serum triglycerides and raising HDL cholesterol levels in humans (Staels et al., 1998). Recent reports have indicated PPARα isoform as the primary target for fibrates (Staels & Auwrex, 1997). PPARγ on the other hand is highly expressed in the adipose tissue and intestine, triggers cellular differentiation, promotes lipid storage and modulates the action of insulin (Desvergene & Wahli, 1999). Although there are some reports of its possible role in embryonic development, the specific function of PPARδ is yet to be determined.
As coligands of both these receptors could offer a better therapeutic option for the treatment of dyslipidaemia and insulin resistance, we initiated a program to discover novel, nonthiazolidinedione dual PPARα and PPARγ agonists. This effort led to the discovery of ragaglitazar ((−) DRF 2725, NNC 61-0029), a phenoxazine analogue of phenyl propanoic acid having dual (PPARα and PPARγ) agonist property (Lohray et al., 2001). Recently, several other groups have also reported the efficacy of such coligands (Murami et al., 1998, Shinkai, 2001; Etegen et al., 2002). Here we report the detailed pharmacological profile of ragaglitazar in different animal models of diabetes and dyslipidaemia. We have compared the efficacy of ragaglitazar with PPARγ activator rosiglitazone, PPARα activator fenofibrate and dual activator KRP-297. We have also compared the insulin-sensitizing effect with metformin.
In insulin-resistant, dyslipidaemic and hyperglycemic ob/ob mice, ragaglitazar showed dose-dependent improvement in plasma glucose, lipid and insulin levels. The treated animals showed significant improvement in oral glucose tolerance. In insulin-resistant Zucker fa/fa rats, ragaglitazar showed dose-dependent improvement in plasma lipid and insulin levels. They also showed significant reduction in hepatic triglyceride secretion, and improved clearance of exogenous lipid. In both these models, ragaglitazar showed better efficacy than rosiglitazone, KRP-297 and metformin. We have developed several dyslipidaemic but normoglycemic animal models such as high-fat-fed Sprague–Dawley (SD) rats and hamsters. In all these models, ragaglitazar showed better improvement in lipid profile than fenofibrate and KRP-297. Rosiglitazone failed to show any significant effect in these models. In the fat-fed rat model, ragaglitazar treatment also showed significant reduction in triton-induced hepatic lipid secretion, and improved clearance of exogenous lipid. Treatment with ragaglitazar also produced increase in fat and liver LPL, liver CPT1 and CAT activity, along with induction of liver acyl CoA oxidase (ACO) and fat aP2 mRNA, which might be responsible for its lipid-lowering activity.
Methods
Animals
C57 BL/6J-ob/ob mice were obtained from Jackson Laboratory, Bar Harbour, ME, U.S.A., at 6 weeks of age. Zucker fa/fa rats were procured from IFFA-CREDO, L'ARBRESLE CEDEX, France. Golden Syrian hamsters were obtained from the National Institute of Nutrition (NIN), Hyderabad, India. SD rats and Swiss albino mice (SAM) were bred at Dr Reddy's Research Foundation (DRF) animal house. All animals were maintained at a controlled temperature (25±1°C) under 12 h light (0600–1800) and 12 h dark (1800–0600) cycle. All animals were given standard laboratory chow (NIN) and water ad libitum. Male SD rats weighing 180–200 gm were made hyperlipidaemic by feeding a high-fat diet containing 2% cholesterol and 1% sodium cholate mixed with standard laboratory chow. All animal experiments were approved by the DRF animal experimental ethics committee, and were in accordance with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Social Justice and Environment, Government of India. Rules of CPCSEA are laid down as per ILAR (Institute of Laboratory Animal Resources, U.S.A.) guidelines.
Chemicals
Ragaglitazar was synthesized by the Medicinal Chemistry Department, DRF, Hyderabad, India. Rosiglitazone, KRP-297 and metformin HCl were synthesized by the published procedure, and were found to be 99% pure. Fenofibrate and Triton WR 1339 (Tyloxapol) were purchased from Sigma Chemicals (St Louis, U.S.A.). Intralipid (20%) was purchased from Pharmacia AB (Stockholm, Sweden). Carboxy methyl cellulose (CMC) was purchased from Loba Chemical Pvt. Ltd (Mumbai, India).
PPAR transactivation
The response element (UASGAL4 × 5) was cloned upstream of the Pgl2-sv 40-Luc reporter (Promega, Madison, WI, U.S.A.), which contains the Simian virus early promoter for luciferase assay. GAL4 fusions were made by fusing human PPARγ1 or PPARα ligand-binding domain (amino acids: 174–475) to the C-terminal end of the yeast GAL4 DNA-binding domain (amino acids: 1–147) of the pM1 vector. pAdVantage (Promega, Madison, WI, U.S.A.) vector was used to enhance luciferase expression.
HEK 293T cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% foetal bovine serum (DMEM-FBS) at 37°C in 5% CO2. At 1 day prior to transfection, cells were plated to 50–60% confluence in DMEM containing 10% delipidated FBS (DMEM-DFBS). Cells were transfected by Superfect as per the manufacturer's protocol. At 3 h after transfection, the reagent was removed and cells were maintained in DMEM-DFBS. At 42 h after transfection, the cells were placed in phenol red-free DMEM-DFBS, and treated for 18 h with the test compounds or vehicle alone. The cells were lysed and assayed for luciferase activity. Luciferase activity was determined by using Luclite kit (Packard, CT, U.S.A.) in a Packard Top –count, and expressed as fold activation relative to untreated cells.
Drug treatment and blood sampling
ob/ob Mice were used at 10 weeks of age. Ragaglitazar and rosiglitazone were administered by oral gavage for 9 days, at the doses mentioned. Animals in the control group received vehicle only (0.25% CMC, 10 ml kg−1). For glucose tolerance test, animals were fasted for 5 h (starting at 0600), and then challenged with 3 g kg−1 glucose orally. The blood samples were collected at 0, 15, 30, 60 and 120 min after glucose load, for plasma glucose estimation. Male Zucker fa/fa rats were used at 18 weeks of age. Ragaglitazar and rosiglitazone were administered orally at the doses mentioned for 9 days. For lipid clearance test, 20% intralipid was administered through the intravenous route (5 ml kg−1), and plasma triglyceride levels were measured at 0, 1, 10, 30, 60 and 120 min after injection. Triton-induced hepatic triglyceride output was measured by intravenous injection of Triton WR 1339 at 250 mg kg−1 (5 ml kg−1 in saline). Plasma triglyceride levels were measured at 0, 2, 4, 6, 24 and 48 h after injection.
Hyperlipidaemic SD rats were treated orally with the compounds for 6 days. Plasma lipids, apolipoprotein (Apo) B and Apo CIII were measured at the end of the study. Triton-induced hepatic triglyceride output was measured by intravenous injection of Triton WR 1339 at 250 mg kg−1 (5 ml kg−1 in saline). Plasma triglyceride levels were measured at 0, 2, 4, 6, 24 and 48 h after injection. Plasma lipid clearance test was performed in the control and treated animals after 5 ml kg−1 intravenous injection of 20% intralipid. Plasma triglyceride levels were measured at 0, 1, 10, 30, 60 and 120 min after injection.
Male Syrian hamsters were fed high-fat diet for 15 days, with and without ragaglitazar (1 mg kg−1, oral). Plasma cholesterol and triglyceride levels were measured at the end of the study. Body weight was measured at the beginning and at the end of the study.
Blood samples were collected in fed state from the animals under mild ether anaesthesia from retro-orbital sinus, 1 h after drug administration (∼1100 hours). No significant effect of ether anaesthesia was observed on the plasma parameters measured.
Estimation of adipocyte fatty acid-binding protein (aP2) and ACO mRNA in the adipose and liver
High-fat-fed rats were treated with ragaglitazar (3 mg kg−1), fenofibrate and rosiglitazone (both at 30 mg kg−1) for 6 days. Liver and epididymal adipose tissues were excised under aseptic condition, and total RNA was extracted by TRIzol reagent (Life Technologies, U.S.A.), following the manufacturer's instruction. First-strand cDNA was generated from 1 μg of RNA in a 20 μl volume, by using the random primer in the first-strand cDNA synthesis kit (Boehringer-Mannheim, Germany). The reverse transcription reaction mixture (2.5 μl) was amplified in 100 μl volume, with primers specific for ACO, adipocyte fatty acid-binding protein (aP2) and β-actin as control. All sequences of the primers, the expected fragment length and Tm values of the primers are as shown:
Linearity of the PCR was tested by amplification of 200 ng of total RNA per reaction from 15 to 40 cycles. The linearity range was found to be between 15 and 35 cycles. In no case did the amount of RNA used for PCR exceed 200 ng per reaction. The samples were amplified for 30 cycles for ACO, 33 cycles for aP2 and 30 cycles for β-actin, by using the following parameters: 94°C-3 min-1cycle, followed by 94°C-30 s, 55°C-30 s and 72°C-1 min.
PCR products (10 μl) were electrophoresed on 1.5% agarose gel. Band intensity was quantitated (under UV light) by the gel documentation system using 1D intermediate software (UVP, U.K.). Levels of mRNA were expressed as the ratio of band intensity for the target gene relative to that of β-actin.
Measurement of CAT and CPT1 and LPL activity
Fat-fed rats were treated with ragaglitazar (10 mg kg−1) and rosiglitazone (30 mg kg−1) for 6 days, after which the rats were killed and liver samples collected in liquid nitrogen and stored at −80°C until further use. All procedures were performed at 0–4°C unless otherwise mentioned. A 20% homogenate was prepared in 10 mM Tris-HCl buffer (pH 7.5) containing 0.35 M sucrose, and centrifuged at 3300 r.p.m. for 5 min. The supernatant was collected and subjected to another centrifugation at 10,000 r.p.m. for 15 min. The pellet containing mitochondria was used for the assay of CPT1 and CAT activity. CPT1 and CAT activity were measured by established procedure (Bieber et al., 1972; Bieber & Markwell, 1981). LPL activity was measured in epididymal adipose, and the liver tissue of fat-fed rats Ho treated with ragaglitazar and rosiglitazone as mentioned above. LPL activity in the tissue homogenate was measured as described (Iverius & Lindqvist, 1986).
Analytical methods
Plasma glucose, triglyceride, cholesterol, HDL and LDL cholesterol and free fatty acid (FFA) were measured spectrophotometrically using commercially available kits (Pointe Scientific, U.S.A. and Roche Diagnostics, Germany). Plasma insulin was measured using RIA kit from Linco Research Inc., U.S.A. Plasma Apo B was measured by the immunoturbidometric method, using a kit from Pointe Scientific, U.S.A. Apo CIII was measured by the immunoturbidometric method, using a kit from Daiichi Pure Chemicals Co. Ltd, Tokyo, Japan.
Data analysis and statistics
The percent reduction was calculated according to the formula
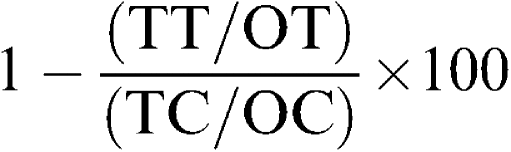 |
where TT is the test day treated, OT the zero day treated, TC the test day control and OC the zero day control. Comparisons among experimental groups were analysed by one-way analysis of variance (ANOVA), followed by Bonferroni/Dunnett's test to evaluate the statistical difference between two groups. P<0.05 was considered significant.
Results
Ragaglitazar is a dual activator of PPARγ and PPARα
Comparative dose–response study of ragaglitazar was performed with rosiglitazone for PPARγ, and with WY 14,643 for PPARα. Ragaglitazar showed significant activation of both the isoforms. Both ragaglitazar and rosiglitazone showed similar activation of PPARγ (Figure 1a). Though ragaglitazar showed less potency (EC50=324 nM) as compared to rosiglitazone (EC50=196 nM), both showed similar maximal activation. Ragaglitazar showed more potent (EC50=270 nM) PPARα activation than WY 14,643 (EC50=8.1 μM) (Figure 1b). Fenofibrate at 300 μM concentration showed only 1.5-fold activation of PPARα. While rosiglitazone did not show any significant PPARα activation, WY 14,643 and fenofobrate did not show any significant PPARγ activation. Ragaglitazar did not show any PPARδ activation even at 50 μM concentration.
Figure 1.

Activation of (a) PPARγ and (b) PPARα by ragaglitazar. HEK 293T cells were transfected with Gal4-PPARγ1 or PPARα-LBD, pGL2(Gal 4X5)-SV 40-Luc reporter and pAdvantage constructs. The values are an average of three experiments conducted in triplicate.
Antidiabetic effect in ob/ob mice
Ragaglitazar showed dose-related reduction in plasma glucose, triglyceride, FFA and insulin levels (Table 1 ), after 9 days of treatment. Drug treatment also showed a dose-dependent improvement in oral glucose tolerance (maximum reduction of 60% in AUCglucose curve, Figure 2). When compared with rosiglitazone, ragaglitazar showed much better activity (Table 2 ). KRP-297, another dual activator, showed ED50 values of 3, 10, 8 and 10 mg kg−1 for PG, TG, insulin and FFA. Metformin on the other hand showed only 42, 30, 40 and 10% reduction in PG, TG, insulin and FFA levels at 250 mg kg−1. No significant difference in food consumption was observed between the treated and control group.
Table 1.
Effect of ragaglitazar in ob/ob mice
| Group | Plasma glucose (mg ml−1) | Triglyceride (mg ml−1) | Free fatty acid (mmol l−1) | Insulin (ng ml−1) |
|---|---|---|---|---|
| Control | 2.49±0.37 | 0.71±0.04 | 1.703±0.052 | 42.07±12.75 |
| Ragaglitazar (0.3 mg kg−1) | 1.24±0.15* | 0.54±0.02 | Not done | 6.35±2.52* |
| Ragaglitazar (1 mg kg−1) | 1.18±0.08* | 0.40±0.07* | 1.33±0.08 | 4.15±1.00* |
| Ragaglitazar (3 mg kg−1) | 1.15±0.02* | 0.35±0.07* | 0.798±0.04* | 3.28±1.65* |
| Ragaglitazar (10 mg kg−1) | 1.10±0.06* | 0.38±0.02* | 0.671±0.04* | 1.84±0.37* |
All values are measured at the end of treatment (9th day), and expressed as mean±s.e. (n=5)
P<0.05 as compared to control (ANOVA).
Figure 2.

Effect of ragaglitazar on glucose tolerance test in ob/ob mice. Animals were treated with the compound at 0.3, 1, 3 and 10 mg kg−1 dose for 9 days, and then subjected to oral glucose load (3 gm kg−1) after a 5 h fast. The values are expressed as mean±s.e.m. (n=5); *P<0.05 as compared to control (ANOVA).
Table 2.
Comparative effect of rosiglitazone and ragaglitazar in ob/ob mice
| Ragaglitazar | Rosiglitazone | |||
|---|---|---|---|---|
| Plasma parameters | ED50 (mg kg−1) | Emax (mg kg−1) | ED50 (mg kg−1) | Emax (mg kg−1) |
| Plasma glucose | <0.3 | 60% at 1 | 3.63 | 53% at 3 |
| Triglyceride | 6.1 | 50% at 3 | 10.0 | 53% at 10 |
| Insulin | <0.1 | 90% at 1 | 5 | 72% at 10 |
| Free fatty acid | 4.2 | 61% at 10 | >10 | 48% at 10 |
ED50 was calculated according to the regression analysis of the dose–response curve.
Lipid- and insulin-lowering effect in Zucker fa/fa rats
In Zucker fa/fa rats, ragaglitazar treatment led to dose-dependent reduction in plasma triglyceride, FFA and insulin levels (Table 3 ). A maximum reduction of 74% in triglyceride, 53% in FFA and 53% in insulin was observed at 3 mg kg−1 dose. KRP-297 at a similar dose showed 60, 50 and 48% reduction in TG, FFA and insulin levels. Metformin at 100 mg kg−1 showed 15, 40 and 30% reduction in TG, FFA and insulin levels. No treatment-related effect on food consumption was observed. Ragaglitazar at 1 mg kg−1 dose showed similar efficacy to 3 mg kg−1 dose of rosiglitazone (Table 4 ). The treated animals (3 mg kg−1 p.o.) showed significant (P<0.05) reduction (3.95 vs 5.74 mg ml−1 h−1 of control) in triton-induced hepatic triglyceride secretion (Figure 3a). Lean rats showed a secretion rate of 2.5 mg ml−1 h−1. There was also significant improvement in plasma triglyceride kinetics (50% reduction in AUCtriglyceride curve) when challenged with an exogenous lipid emulsion (20% intralipid) (Figure 3b).
Table 3.
Effect of ragaglitazar on Zucker fa/fa rats
| Group | Triglyceride (mg ml−1) | Free fatty acid (mmol l−1) | Insulin (ng ml−1) |
|---|---|---|---|
| Control | 4.34±0.45 | 0.77±0.03 | 13.28±1.3 |
| Ragaglitazar (0.1 mg kg−1) | 3.56±0.43 | 0.83±0.01 | 10.33±2.06 |
| Ragaglitazar (0.3 mg kg−1) | 2.52±0.22* | 0.46±0.02* | 11.43±2.16* |
| Ragaglitazar (1 mg kg−1) | 1.53±0.10* | 0.37±0.02* | 6.51±1.4* |
| Ragaglitazar (3 mg kg−1) | 1.12±0.06* | 0.36±0.02* | 6.19±0.70* |
All values are measured at the end of treatment (9th day), and expressed as mean±s.e. (n=5)
P<0.05 as compared to control (ANOVA).
Table 4.
Comparative effect of ragaglitazar and rosiglitazone in Zucker fa/fa rats
| Group | Triglyceride (mg ml−1) | Free fatty acid (mmol l−1) | Insulin (μU ml−1) |
|---|---|---|---|
| Control | 3.60±0.50 | 0.51±0.03 | 441.00±189.00 |
| Ragaglitazar (1 mg kg−1) | 1.84±0.27* | 0.25±0.02* | 64.50±11.97* |
| Rosiglitazone (3 mg kg−1) | 1.67±0.17* | 0.216±0.04* | 68.44±11.97* |
All values are measured at the end of treatment (9th day), and expressed as mean±s.e. (n=5)
P<0.05 as compared to control (ANOVA).
Figure 3.

Effect of ragaglitazar on (a) hepatic triglyceride secretion and (b) plasma triglyceride clearance in Zucker fa/fa rats. Animals were treated with the compound for 9 days, and injected with triton WR 1339 or 20% intralipid, as described in Methods. The values are expressed as mean±s.e. (n=5); *P<0.05 as compared to control (ANOVA).
Improvement of hyperlipidaemia in high-fat-fed rats
SD rats, when fed with a high-fat-containing diet, showed significant increase in plasma total and LDL-cholesterol, and also plasma triglyceride levels, with a concomitant decrease in HDL-cholesterol levels. No change in plasma glucose level was observed. When treated with ragaglitazar, these animals showed a dose-dependent improvement in plasma lipid levels (Figure 4a–d). Normal feed-fed animals showed plasma TG, TC, HDL-C and LDL-C levels as 0.37±0.13, 1.13±0.04, 0.64±0.02 and 0.42±0.04 mg ml−1. Ragaglitazar did not have any effect on food consumption of the treated rats. The compound showed significantly better activity than fenofibrate (Table 5 ). KRP-297 at 100 mg kg−1 dose showed 52 and 49% reduction in TC and TG, and 98% increase in HDL-C. The treated animals (10 mg kg−1 day−1) showed 66% reduction in plasma Apo B level (0.18±0.07 vs 0.52±0.009 mg ml−1 of controls). There was a significant (P<0.05) reduction in plasma Apo CIII levels in ragaglitazar (10 mg kg−1 day−1)-treated rats (0.03±0.002 vs 0.03±0.002 mg ml−1 of control, P<0.05).
Figure 4.

Effect of ragaglitazar on plasma (a) total cholesterol, (b) triglyceride, (c) HDL-C and (d) LDL-C in high-fat-fed rats. Animals were kept on high-fat diet, and compound treatment was done for 6 days at 0.1, 0.3, 1, 10 and 30 mg kg−1. The values are expressed as mean±s.e. (n=5); *P<0.05 as compared to control (ANOVA).
Table 5.
Comparative effect of ragaglitazar and fenofibrate in high-fat-fed rats
| Ragaglitazar | Fenofibrate | |||
|---|---|---|---|---|
| Parameters | ED50 (mg kg−1) | Emax (mg kg−1) | ED50 (mg kg−1) | Emax (mg kg−1) |
| Triglyceride | 3.95 | 61% at 10 | 12 | 48% at 60 |
| Total cholesterol | 3.78 | 73% at 30 | 45 | 65% at 60 |
| HDL-C | 0.29 | 151% at 30 | 30 | 114% at 60 |
| LDL-C | 2.12 | 85% at 30 | 6 | 73% at 60 |
| VLDL-C | 3.78 | 61% at 10 | 10 | 58% at 60 |
ED50 is calculated according to the regression analysis of the dose–response curve.
Fat-fed rats, treated with ragaglitazar (3 mg kg−1 p.o.) showed a significant (P<0.05) reduction (0.82 vs 2.6 mg ml−1 h−1 of controls) in triton-induced hepatic triglyceride secretion (Figure 5a), and an improvement (50%) in lipid clearance (Figure 5b) when challenged intravenously with a lipid emulsion (20% intralipid). Normal diet-fed rats showed a triglyceride secretion rate of 1.7 mg ml−1 h−1. Rosiglitazone or pioglitazone, on the other hand, did not show any significant activity in this model of hyperlipidaemia, even at 30 mg kg−1 dose. aP2 mRNA in the white adipose tissue and ACO mRNA in the liver were studied from ragaglitazar- (3 mg kg−1), fenofibrate- (30 mg kg−1) and rosiglitazone- (30 mg kg−1) treated fat-fed rats. Ragaglitazar and rosiglitazone, but not fenofibrate, showed significant induction (6-, 5- and 0.1-fold) of aP2 mRNA (Figure 6a), whereas both ragaglitazar and fenofibrate, but not rosiglitazone, showed a significant induction (2.5-, 1.8- and 0.2-fold) of ACO mRNA in the liver (Figure 6b). Animals treated with ragaglitazar (10 mg kg−1) showed 78 and 167% increase of LPL activity in both white adipose (14,07,589±15,400 c.p.m. g−1 tissue vs 7,90,508± 11,600 c.p.m. g−1 tissue of control, P<0.05) and the liver (48,007±532 c.p.m. g−1 tissue vs 17,991±424 c.p.m. g−1, P<0.05). Rosiglitazone at 30 mg kg−1 showed only 42% increase in fat LPL, but no effect on liver LPL activity. Ragaglitazar treatment also showed 120 and 819% increases in the liver CPT1 (16.54±1.84 U mg−1 protein vs 7.54±0.85 U mg−1 protein of control, P<0.05) and CAT activity (40.24±2.18 U mg−1 protein vs 4.38±0.66 U mg−1 protein of control, P<0.05). Rosiglitazone (30 mg kg−1) failed to show any effect on these enzymes.
Figure 5.

Effect of ragaglitazar on (a) hepatic triglyceride secretion and (b) plasma triglyceride clearence in high-fat-fed rats. Animals were treated with the compound at 3 mg kg−1 for 6 days, and injected with triton or 20% intralipid, as described in Methods. The values are expressed as mean±s.e. (n=5); *P<0.05 as compared to control (ANOVA).
Figure 6.

Induction of aP2 and ACO mRNA. High-fat-fed rats were treated with ragaglitazar (3 mg kg−1), rosiglitazone and fenofibrate (both at 30 mg kg−1) for 6 days. Extraction of RNA and RT–PCR procedure as described in Methods. (a) Represents aP2 mRNA (top panel), and the corresponding β-actin (bottom panel). (b) Represents ACO mRNA (top panel) and β-actin (bottom panel). Lane 1: control; 2: ragaglitazar; 3: fenofibrate and 4: rosiglitazone.
Lipid- and body-weight-reducing action in fat-fed hamster
Hamsters showed significant increase in plasma lipid levels, and also in body weight, after 15 days of high-fat diet. When these animals were treated simultaneously with ragaglitazar at 1 mg kg−1 oral dose, the treated animals showed 61% reduction in plasma cholesterol (1.14±0.11 vs 2.96±0.18 mg ml−1 of control, P<0.05) and 83% reduction in triglyceride levels (1.06±0.22 vs 6.28±0.2 mg ml−1 of control, P<0.05), as compared to fat-fed controls. KRP-297 at a similar dose did not show any significant effect on any of the plasma parameters. There was also a significant reduction (17%) in the body weight increase as compared to untreated fat-fed animals (101.75±0.63–109.00±3.08 g for ragaglitazar as compared to 92.25±0.48–119.25±3.87 g of control, P<0.05).
Discussion
Insulin resistance in type II diabetes is not only associated with hyperglycaemia, but also with hyperlipidaemia and atherosclerosis (Reaven, 1988). A drug that simultaneously ameliorates insulin resistance and hyperlipidaemia facilitates better management of type II diabetes. PPARs are ligand-activated transcription factors, which offer a promising therapeutic approach for the treatment of metabolic syndrome. Ragaglitazar, an alkoxypropanoic acid derivative, showed dual activation of α and γ isoforms of PPAR. We studied the antidiabetic and hypolipidaemic potential of ragaglitazar in insulin-resistant ob/ob mice, Zucker fa/fa rats and high-fat-diet-induced hyperlipidaemic models.
In an in vitro transactivation assay, ragaglitazar showed significant activation of both PPARα and PPARγ. Among the TZDs in the market, rosiglitazone (Patel et al., 1997; Balfour & Plosker, 1999) shows most potent PPARγ activation. Hence, we have used it for comparison. Ragaglitazar treatment showed similar activation of PPARγ as compared to rosiglitazone. Since fibrates are low-affinity ligands of PPARα, we have used WY 14,643, a more potent PPARα-specific ligand (Wilson et al., 2000) for comparison. Ragaglitazar showed better transactivation potential than WY 14,643. In our study, rosiglitazone failed to show any significant PPARα activation even at 100 μM concentration. WY 14,643 did not show any significant PPARγ activation at 100 μM. The specificity of both the standard compounds, as noticed in our study, corroborates with the previous reports (Lehmann et al., 1995; 1996; Forman et al., 1997).
Regulation of blood glucose has been the cornerstone of both type I and type II diabetes treatment for decades. The diabetes control and complication trials (DCCT) have convincingly demonstrated the importance of tight glucose regulation in type I diabetes. A Japanese trial (Okubo et al., 1995) with an intensive insulin regimen in type II patients showed remarkably similar results with respect to the reduction of microvascular complications, as found in DCCT trial with type I diabetes patients. The United Kingdom Prospective Diabetes Study (UKPDS) has demonstrated a 16% reduction (P=0.052) in the risk of combined fatal and nonfatal myocardial infarction, and a sudden death in intensively treated type II diabetes patients (UKPDS, 1998). As the primary goal of our compound was to treat diabetic patients, we have profiled ragaglitazar in different animal models of type II diabetes.
Table 6.
| Gene | Sequence of sense primer | Sequence of antisense primer | Expected fragment size | Tm°C (5′/3′) Sense/antisense |
| ACO | 5′..GCCCTCAGCTATGGTATTAC..3′ | 5′..AGGAACTGCTCTCACAATGC..3′ | 634 bp | 60/57 |
| β-actin | 5′..TTGTAACCAACTGGGACGATATGG..3′ | 5′..GATCTTGATCTTCATGGTGCTAGG..3′ | 764 bp | 57/54 |
| aP2 | 5′..GACCTGGAAACTCGTCTCCA..3′ | 5′..CATGACACATTCCACCACCA..3′ | 340 bp | 62/60 |
The insulin-sensitizing property of ragaglitazar was studied in two different insulin resistant models – hyperglycemic, hyperinsulinemic and impaired glucose-tolerant C57 BL/6J-ob/ob mice (Genuth et al., 1971), and hyperinsulinemic, hyperlipidaemic but normoglycemic Zucker fa/fa rats (Assimacopoulas-Jannet & Jeanrenaud, 1976). In both these models, ragaglitazar showed significant dose-dependent reduction in plasma triglyceride, FFA and insulin levels after 9 days of treatment. In ob/ob mice, ragaglitazar also showed dose-dependent reduction in plasma glucose, and marked improvement in glucose tolerance. Zucker rats show increased hepatic triglyceride secretion and reduced efficacy in clearing plasma triglyceride, as compared to lean rats. Treatment with ragaglitazar improved both the hepatic triglyceride secretion and plasma triglyceride clearence kinetics in this model. Among the TZDs in the market, rosiglitazone is claimed to be the most potent and efficacious (Patel et al., 1997); we have compared the efficacy of ragaglitazar with that of rosiglitazone. In both the models, ragaglitazar showed better efficacy than rosiglitazone in all the parameters studied. Among the dual PPAR activators, KRP-297 (Murami et al., 1998) and JTT-501 (Shinkai, 2001) had entered Phase II clinical trial. KRP-297, which is now being developed by Merck, has successfully completed Phase II clinical trial, and entered Phase III. Our preliminary studies have also suggested better efficacy for KRP-297 (data not shown); so we have chosen this dual activator to compare with ragaglitazar. In ob/ob mice, ragaglitazar showed better efficacy than KRP-297, and in Zucker fa/fa rats raga showed comparable-to-better efficacy.
Our studies in insulin-resistant animal models clearly indicate that ragaglitazar is more efficacious than rosiglitazone, even though the in vitro assay showed similar PPARγ activation for both the compounds. There was no significant difference in the pharmacokinetic profiles of ragaglitazar and rosiglitazone in normal animals (AUC, t1/2 and Cmax values for ragaglitazar: 49.5±3.7 μg h ml−1; 3.0±0.15 h and 10.2±0.68 μg ml−1 (25 μM). Corresponding values for rosiglitazone were 45.6±1.9 μg h ml−1; 2.5±0.2 h and 11.5±0.75 μg ml−1, respectively). Both the compounds were dosed orally at 3 mg kg−1, which suggests the noninvolvement of PK-related issues in different pharmacodynamic profiles. Interestingly, there are several recent observations suggesting an insulin-sensitizing effect of PPARα agonists (Kobayashi et al., 1988; Ogawa et al., 2000). Although, the mechanism of this insulin-sensitization action of PPARα ligands is not very clear yet, the hypothesis is that it might be secondary to the effect on lipid metabolism. PPARα agonists increase the hepatic oxidation of fatty acids (FA), and reduce the synthesis and secretion of triglycerides (Desvergene & Wahli, 1999). This in turn will increase diversion of FA from peripheral tissues (e.g., skeletal muscle and fat tissue) to the liver, and thereby decrease both FA synthesis and delivery of triglyceride to peripheral tissues. By their actions on FA, PPARα agonists probably increase the insulin-stimulated glucose disposal in the skeletal muscle (Randle et al., 1963; Boden, 1994), and thereby ameliorate insulin resistance. Besides, through their plasma triglyceride-lowering effect, PPARα agonists can lower skeletal muscle triglyceride content, which is significantly related to insulin resistance (Goodpaster & Kelly, 1998). We believe that due to its PPARα activation, in addition to its PPARγ agonism, ragaglitazar shows better insulin-sensitizing property, which is reflected in different animal models.
Insulin resistance is not only associated with hyperglycaemia, but also with lipoprotein abnormalities, such as hypertriglyceridaemia, high levels of VLDL and small dense LDL (Taskinen, 1995), which are the risk factors for coronary heart disease. These metabolic abnormalities, together with type II diabetes, may cluster in the same individual, constituting the lethal metabolic syndrome or insulin resistance-associated disorder (IRAD) (Reaven, 1988). Results from the Helsinki heart study and recently concluded Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial (VA-HIT) study demonstrate that fibrates significantly reduce the incidence of cardiovascular disease in patients with type II diabetes, by raising HDL levels and lowering TG levels without lowering the LDL levels (Huttunen et al., 1991; Rubins et al., 1999). As we have mentioned previously, the main objective behind developing a dual activator was to have a compound with both insulin-sensitizing and lipid-lowering activity. Studies in different genetic models described so far have confirmed the lipid-lowering activity of ragaglitazar. We wanted to study the lipid-lowering potential of the compound in further detail. For this, ragaglitazar was profiled in two different animal models of hyperlipidaemia, where PPARγ ligands failed to show any significant effect, whereas PPARα ligands showed excellent lipid-lowering property. We have compared the activity of ragaglitazar with the fibrate group of drugs, and with dual activator KRP-297 in these animal models. In a preliminary study, we found that fenofibrate was more potent than clofibrate, bezafibrate and gemfibrozil (data not shown). Therefore, in subsequent studies, the hypolipidaemic potential of ragaglitazar was compared with that of fenofibrate.
High-fat-fed rat models had been used previously to study the efficacy of fibrates (Petit et al., 1988). Male SD rats, when fed with high-fat diet, develop hypercholesterolemia and hypertriglyceridaemia, but are nondiabetic. Normally, rodent plasma cholesterol contains a very high proportion of HDL cholesterol and very low LDL cholesterol. This makes therapeutic interpretation of cholesterol lowering in normal rodents difficult. In our hyperlipidaemic rat model, plasma cholesterol is predominantly LDL cholesterol, which reflects the clinical situation more closely. Ragaglitazar treatment showed significant reduction in plasma total and LDL cholesterol, and increase in HDL cholesterol levels. The efficacy of ragaglitazar has been found to be several folds better than that of fenofibrate and KRP-297. PPARγ ligands rosiglitazone and pioglitazone failed to show any significant effect even at 30 mg kg−1 dose. Our PPAR activation study was performed with human constructs. In order to confirm that both α and γ isoforms are involved in the action of ragaglitazar, levels of aP2 and ACO mRNA, two well-accepted markers of PPARγ and PPARα were measured in the white adipose and liver of fat-fed rats treated with ragaglitazar, rosiglitazone and fenofibrate. As expected, PPARγ-specific ligand rosiglitazone failed to show any induction on liver ACO, whereas PPARα-specific ligand fenofibrate failed to show any effect on fat aP2 mRNA. Ragaglitazar on the other hand showed significant activation of both aP2 and ACO mRNA, confirming its dual PPAR activation in vivo. The fat-fed hamster represents a potentially important model of nondiabetic hyperlipidaemia. Although real-time PCR is the optimum method for quantitative PCR, we believe that, with proper internal control, regular PCR, as done here, gives conclusive proof of quantitative mRNA expression. Lipoprotein metabolism in hamster more closely reflects that of humans than those of rats and mice (Sullivan et al., 1993). This model has been used to study the lipid-lowering and body-weight-reducing property of PPARα agonists (Glaxo Wellcome Group, 1997; Minnich et al., 2001). We have studied the efficacy of ragaglitazar on plasma lipids and body weight in this model. When fed with high-fat diet, these animals develop hypercholesterolemia and hypertriglyceridaemia, and show significant increase in body weight. Concomitant treatment with ragaglitazar inhibits both the increase in plasma lipids as well as the body weight. Treatment with TZDs, probably due to adipogenesis, is associated with significant increase in body weight in clinics. The effect of ragaglitazar (which also has PPARα activity) on the body weight increase in hamsters, if translated in humans, will give the compound an added advantage, specially in treating obese patients.
The mechanism responsible for hypertriglyceridaemia associated with insulin resistance may be complex, but a commonly proposed feature for most of the mechanisms involves overproduction of VLDL triglyceride and VLDL ApoB by the liver, and decreased triglyceride uptake in peripheral tissues (Gliemann et al., 1972; Grundy et al., 1979; Howard, 1987; Reaven and Chen, 1998). Ragaglitazar treatment in both Zucker fa/fa and high-fat-fed rats showed reduction in hepatic triglyceride secretion, and improved clearance of plasma triglyceride. In high-fat-fed rats, ragaglitazar treatment also significantly reduced plasma ApoB levels, which is indicative of the positive effect of the drug on VLDL ApoB metabolism. It is known that lipoprotein lipase (LPL) plays an important role in the removal of plasma triglyceride, by hydrolysing the triglycerides of VLDL and chylomicron particles (Goldberg, 1996). ApoCIII on the other hand appears to antagonize plasma triglyceride metabolism, as it inhibits triglyceride hydrolysis by LPL and hepatic lipase (Quarfordt et al., 1982; Ginsberg et al., 1986). Hypotriglyceridaemic action of PPARα ligand fibrates is known to involve an increase in LPL activity (Larsen & Illingworth, 1993; Murphy et al., 1993), and a reduction of ApoCIII expression in liver (Staels et al., 1995). It is known that PPARγ also plays an important role in the induction of LPL activity through the PPRE in the LPL promoter (Schoonjans et al., 1996a). As ragaglitazar is a coactivator of both PPARα and PPARγ isoforms, we studied the effect of ragaglitazar on both of these two important proteins of lipid metabolism. Ragaglitazar treatment increases the LPL activity associated with fat tissue, as well as in liver. Ragaglitazar treatment also reduces the plasma ApoCIII levels, which in turn will help the peripheral metabolism of triglyceride. PPARγ ligand rosiglitazone, on the other hand, can induce only fat LPL activity, and had no effect on plasma Apo CIII levels (data not shown). We believe that increased lipolysis through LPL, and subsequent peripheral uptake, might be responsible for the increased clearance of plasma triglyceride after the drug treatment. Administration of PPARα agonists increases the expression of peroxisomal and mitochondrial β-oxidation enzymes, in particular ACO, CAT, CPT1, and several other related enzymes in hepatocellular compartments in rodents (Dryer et al., 1992; Schoonjans et al., 1996c). We have mentioned before the induction of ACO mRNA, a key peroxisomal enzyme by ragaglitazar. We have also investigated the effect of ragaglitazar on CPT1 and CAT activity, two major enzymes in mitochondrial β-oxidation. The increased activity of liver CPT1 and CAT enzymes in the ragaglitazar-treated rats is suggestive of its role in fatty acid uptake and catabolism in mitochondria. As expected, rosiglitazone failed to show any effect on these fatty acid-catabolizing enzymes. Taken together, ragaglitazar treatment normalized increased hepatic triglyceride secretion and ApoB levels, increased the peripheral clearance of triglyceride, and increased the mitochondrial and peroxisomal catabolism of fatty acid, which leads to the improvement of dyslipidaemia in hyperlipidaemic animal models.
Conclusion
To conclude, ragaglitazar, by virtue of its dual PPARα and PPARγ-activating property, acts both on the liver and adipose tissue, and thereby shows better amelioration of not only hyperglycaemia and hyperinsulinemia, but also abnormal lipid metabolism, than marketed PPARα- or PPARγ-selective agonists. Phase II clinical data of ragaglitazar (Saad et al., 2002; Strand et al., 2002) validated our findings in animal studies.
Acknowledgments
We are thankful to Dr Reddy's Laboratories for their support, and to Novo Nordisk, Denmark, for providing the PPAR constructs. DRF publication No. 147.
Abbreviations
- ACO
Acyl CoA oxidase
- Apo
apolipoprotein
- CAT
carnitine acetyltransferase
- CPT
carnitine palmitoyltransferase
- ED50
50% effective dose
- FFA
free fatty acid
- LPL
lipoprotein lipase
- PPAR
peroxisome proliferator-activated receptor
- PPRE
peroxisome proliferators responsive element
- SD
Sprague–Dawley
References
- ARONOFF S., ROSENBALT S., BRAITHWAITE S., EGAN J.W., MATHISEN A.L., SCHNEIDER R.L. Pioglitazone hydrochloride monotherapy improves glycemic control in the treatment of patients with type 2 diabetes. Diabetes Care. 2000;23:1605–1611. doi: 10.2337/diacare.23.11.1605. [DOI] [PubMed] [Google Scholar]
- ASSIMACOPOULAS-JANNET F., JEANRENAUD B. The hormonal and metabolic basis of experimental obesity. Clin. Endocrinol. Metab. 1976;5:337–365. doi: 10.1016/s0300-595x(76)80025-4. [DOI] [PubMed] [Google Scholar]
- BALFOUR J.A.B., PLOSKER G.L. Rosiglitazone. Drugs. 1999;57:921–930. doi: 10.2165/00003495-199957060-00007. [DOI] [PubMed] [Google Scholar]
- BIEBER L.L., ABRAHAM T., HELMRATH T. A rapid spectrophotometric method for carnitine palmitoyltransferase. Anal. Biochem. 1972;50:509–518. doi: 10.1016/0003-2697(72)90061-9. [DOI] [PubMed] [Google Scholar]
- BIEBER L.L., MARKWELL M.A.K. Peroxisomal and microsomal carnitine acetyltransferase. Methods Enzymol. 1981;71:351–358. doi: 10.1016/0076-6879(81)71044-9. [DOI] [PubMed] [Google Scholar]
- BODEN G. Mechanism of fatty acid induced inhibition of glucose uptake. J. Clin. Invest. 1994;93:2438–2446. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEFRONZO R.A., BONDANA R.C., FERRANINI E. Pathogenesis of NIDDM: a balanced overview. Diabetes Care. 1992;15:318–367. doi: 10.2337/diacare.15.3.318. [DOI] [PubMed] [Google Scholar]
- DESVERGENE B., WAHLI W. Peroxisome proliferator activated receptors: nuclear control of metabolism. Endocrinol. Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- DRYER C., KREY G., KELLER H., GIVEL F., HELFTENBEIN G., WAHLI W. Control of peroxisomal β-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- ETEGEN G.J., OLDHAM B.A., JOHNSON W.T., BRODERICK C.L., MONTROSE C.R., BROZINICK J.T., MISENER E.A., BEAN J.S., BENSCH W.R., BROOKS D.A., SHUKER AJ RITO C.J., MCCARTHY JR ARDECKY R.J., TYHONAS J.S., DANA S.L., BILAKOVICS J.M., PATERNITI J.R., OGILVIE K.M., LIU S., KAUFFMAN R.F. A tailored therapy for metabolic syndrome. Diabetes. 2002;51:1083–1087. doi: 10.2337/diabetes.51.4.1083. [DOI] [PubMed] [Google Scholar]
- FORMAN B.M., CHOU J., EVANS R.M. Hypolipidemic drugs, polyunsaturated fatty acids and eicosanoids are ligands for peroxisome proliferator activated receptor α and δ. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GENUTH S.M., PRZYBYLSKI R.J., ROSENBERG D.M. Insulin resistance in genetically obese, hyperglycemic mice. Endocrinology. 1971;88:1230–1238. doi: 10.1210/endo-88-5-1230. [DOI] [PubMed] [Google Scholar]
- GINSBERG H.N., LE N.A., GOLDBERG I.J., GIBSON J.C., RUBINSTEIN A., WANGIVERSON P., NORUM R., BROWN W.V. Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. J. Clin. Invest. 1986;78:1287–1295. doi: 10.1172/JCI112713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLAXO WELLCOME GROUP Use of agonists of peroxisome proliferator activated receptor alpha for treating obesity 1997(WO 9736579)
- GLIEMANN J., OSTERLAND K., VINTEN J., GAMMELTOFT S. A procedure for measurement of distribution spaces in isolated fat cells. Biochim. Biophys. Acta. 1972;286:1–9. doi: 10.1016/0304-4165(72)90082-7. [DOI] [PubMed] [Google Scholar]
- GOLDBERG I.J. Lipoprotein lipase and lipolysis. J. Lipids Res. 1996;37:693–707. [PubMed] [Google Scholar]
- GOODPASTER B.H., KELLY D.E. Role of muscle in triglyceride metabolism. Curr. Opin. Lipidol. 1998;9:231–236. doi: 10.1097/00041433-199806000-00008. [DOI] [PubMed] [Google Scholar]
- GRUNDY S.M., MOK H.Y., ZECH L., STEINBERG D., BERMAN M. Transport of very low density lipoprotein triglycerides in varying degrees of obesity and hypertriglyceridemia. J. Clin. Invest. 1979;63:1274–1283. doi: 10.1172/JCI109422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOWARD B.V. Lipoprotein metabolism in diabetes mellitus. J. Lipids Res. 1987;28:613–628. [PubMed] [Google Scholar]
- HUTTUNEN J., MANNIEN V., MANTARRI M., KOSKINEN P., ROMO M., TENKANEN I., HEINONEN O., FRICK M. The Helisinki heart study: central findings and clinical implications. Ann. Med. 1991;23:155–159. doi: 10.3109/07853899109148041. [DOI] [PubMed] [Google Scholar]
- IVERIUS P.H., LINDQVIST A.M.O. Preparation, characterization and measurement of lipoprotein lipase. Methods Enzymol. 1986;129:691–704. doi: 10.1016/0076-6879(86)29099-0. [DOI] [PubMed] [Google Scholar]
- KOBAYASHI M., SHIGETA Y., HIRATA Y., OMORI Y., SAKAMOTO N., NAMBU S., BABU S. Improvement of glucose tolerance in NIDDM by clofibrate. Randomised double-blind study. Diabetes Care. 1988;11:495–499. doi: 10.2337/diacare.11.6.495. [DOI] [PubMed] [Google Scholar]
- LARSEN M.L., ILLINGWORTH D.R. Triglyceride-lowering agents: fibrates and nicotinic acid. Curr. Opin. Lipidol. 1993;4:34–40. [Google Scholar]
- LEHMANN J.M., MOORE L.B., OLIVER-SMITH T.A., WILKINSON W.O., WILSON T.M., KLIEWER S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator activator receptor gamma. J. Biol. Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- LOHRAY B.B., LOHRAY V.B., BAJJI A.C., KALCHAR S., POONDRA R., PADAKANTI S., CHAKRABARTI R., VIKRAMADITHYAN R.K., MISRA P., SURESH J., MAMIDI N.V.S.R., RAJAGOPALAN R. (−)3-[4-[2-(Phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxypropanoic acid [(−) DRF 2725]: a dual PPAR agonist with potent antihyperglycemic and lipid modulating activity. J. Med. Chem. 2001;44:2675–2678. doi: 10.1021/jm010143b. [DOI] [PubMed] [Google Scholar]
- MINNICH A., TIAN N., BYAN L., BILDER G. A potent PPARα agonist stimulates mitochondrial fatty acid β-oxidation in liver and skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2001;280:E270–E279. doi: 10.1152/ajpendo.2001.280.2.E270. [DOI] [PubMed] [Google Scholar]
- MURAMI K., TOBE K., IDE T., MOCHIZUKI T., OHASHI M., AKANUMA Y., YAZAKI Y., KADOWAKI T. A novel insulin sensitizer acts as a coligand for peroxisome proliferator activated receptor-α (PPARα) and PPARγ. Diabetes. 1998;47:1841–1847. doi: 10.2337/diabetes.47.12.1841. [DOI] [PubMed] [Google Scholar]
- MURPHY M.C., ZAMPELAS A., PUDDICOMBE S.M., FURLONGER N.P., MORGAN L.M., WILLIAMS C.M. Pretranslational regulation of the expression of lipoprotein lipase gene by dietary fatty acids in the rat. Br. J. Nutr. 1993;70:727–736. doi: 10.1079/bjn19930168. [DOI] [PubMed] [Google Scholar]
- OGAWA S., TAKEUCHI K., SUGIMURA K., FUKUDA M., LEE R., ITO S., SATO T. Bezafibrate reduces blood glucose in type 2 diabetes mellitus. Metabolism. 2000;49:331–334. doi: 10.1016/s0026-0495(00)90176-8. [DOI] [PubMed] [Google Scholar]
- OKUBO Y., KISHIKAWA H., ARAKI E. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin dependent diabetes mellitus: a randomized prospective 6 year study. Diabetes Res. Clin. Practice. 1995;28:103–117. doi: 10.1016/0168-8227(95)01064-k. [DOI] [PubMed] [Google Scholar]
- PATEL J., MILLER E., HU J., GRANETT J.BRL 49653 (a thiazolidinedione) improves glycemic control in NIDDM patients Diabetes 199746Suppl 1A150(Abstract 0578) [Google Scholar]
- PETIT D., BONNEFIS M.T., REY C. Effects of ciprofibrate and fenofibrate on liver lipids and lipoprotein synthesis in normo and hyperlipidemic rats. Atherosclerosis. 1988;74:215–225. doi: 10.1016/0021-9150(88)90240-7. [DOI] [PubMed] [Google Scholar]
- PORTE D., JR, SCHWARTZ M.W. Diabetes complications: Why glucose potentially toxic. Science. 1996;27:699–700. doi: 10.1126/science.272.5262.699. [DOI] [PubMed] [Google Scholar]
- QUARFORDT S.H., MICHALOPOULOS G., SCHIRMER B. The effect of human C apolipoproteins on the in vitro hepatic metabolism of triglyceride emulsions in the rat. J. Biol. Chem. 1982;257:14642–14647. [PubMed] [Google Scholar]
- RANDLE P.J., GARLAND P.B., HALES C.N., NEWSHOLME E.A. The glucose fatty acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- REAVEN G.M. Role of insulin resistance in human disease. Diabetes. 1988;37:1495–1507. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- REAVEN G.M., CHEN Y.D. Role of insulin in regulation of lipoprotein metabolism in diabetes. Diabetes Metab. Rev. 1998;4:639–652. doi: 10.1002/dmr.5610040703. [DOI] [PubMed] [Google Scholar]
- RUBINS H.B., ROBINS S.J., COLLINS D. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 1999;341:410–418. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- SAAD M.F., OSEI K., LEWIN A.J., PATEL N., GRECO S., NUNEZ M., HUANG W.C., REINHARDT R.R.Ragaglitazar improves lipid profile and glycemic control in hypertriglyceridemic type 2 diabetes Diabetes 200251Suppl 2A35(143-OR) [DOI] [PubMed] [Google Scholar]
- SCHOONJANS K., PEINADO-ONSURBE J., HEYMAN R., BRIGGS M., CAYET D., DEEB S., STAELS B., AUWREX J. PPARα and PPARγ activators direct a distinct tissue specific transcriptional response via a PPRE in lipoprotein lipase gene. EMBO J. 1996a;15:5336–5343. [PMC free article] [PubMed] [Google Scholar]
- SCHOONJANS K., STAELS B., AUWERX J. The peroxisome proliferator activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochem. Biophys. Acta. 1996b;1302:93–109. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- SCHOONJANS K., STAELS B., AUWERX J. Role of peroxisome proliferator activated receptor in mediating the effects of fibrates and fatty acids on gene expression. J. Lipids Res. 1996c;37:907–925. [PubMed] [Google Scholar]
- SHINKAI H. The chemical structure and pharmacological properties of a novel isoxazolidinedione insulin sensitizer, JTT-501. Nippon Rinsho. 2001;59:2207–2210. [PubMed] [Google Scholar]
- STALES B., AUWREX J. Role of PPAR in the pharmacological regulation of lipoprotein metabolism by fibrates and thiazolidinediones. Curr. Pharm. Design. 1997;3:1–14. [Google Scholar]
- STAELS B., DALLONGEVILLE J., AUWERX J., SCHOONJANS K., LEITERSDORF E., FRUCHART J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98:2088–2093. doi: 10.1161/01.cir.98.19.2088. [DOI] [PubMed] [Google Scholar]
- STAELS B., VU-DAC N., KOSYKH V., SALADIN R., FRUCHART J.C., DOLLONGEVILLE J., AUWREX J. Fibrates downregulate apolipoprotein CIII expression independent of induction of Acyl COA: a potential mechanism for the hypolipidemic action of fibrates. J. Clin. Invest. 1995;95:705–712. doi: 10.1172/JCI117717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRAND J., SANDEMAN D., SKOVSTED B., EDSBERG B., MULLER P.Ragaglitazar (NNC 61-0029, (−)DRF 2725): the efficacy and safety of a novel dual PPARα and PPARγ agonist in patients with type 2 diabetes Diabetes 200251Suppl 2A144583-P [Google Scholar]
- SULLIVAN M.P., CERDA J.J., ROBBINS FL BURGIN C.W., BEATTY R.J. The gerbil, hamster and guinea pig as rodent models for hyperlipidemia. Lab. Anim. Sci. 1993;43:575–578. [PubMed] [Google Scholar]
- TASKINEN M.R. Insulin resistance and lipoprotein metabolism. Curr. Opin. Lipidol. 1995;6:153–160. doi: 10.1097/00041433-199506000-00007. [DOI] [PubMed] [Google Scholar]
- THE UNITED KINGDOM PROSPECTIVE DIABETES STUDY Intensive blood–glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- WILSON T.M., BROWN P.J., STERNBACH D.D., HENKE B.R. The PPARs: from orphan receptors to drug discovery. J. Med. Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- WILSON T.M., COBB J.E., COWAN D.J., WIETHE R.W., CORREA I.D., PRAKASH S.R., BECK KD MOORE L.B., KLIEWER S.A., LEHMANN J.M. The structure–activity relationship between PPARγ agonism and the antihyperglycemic activity of thiazolidinediones. J. Med. Chem. 1996;39:665–668. doi: 10.1021/jm950395a. [DOI] [PubMed] [Google Scholar]