

Abstract
Rivastigmine is an acetylcholinesterase inhibitor used in Alzheimer's disease therapy. In the present study, we investigated the effects of rivastigmine on the transient outward K+ current (IK(A)) and the delayed rectifier K+ current (IK(DR)) in acutely dissociated rat hippocampal pyramidal neurons using the whole-cell patch-clamp technique.
Rivastigmine inhibited the amplitudes of IK(A) and IK(DR) in a reversible and concentration-dependent manner. At a concentration of 100 μM, rivastigmine inhibited IK(A) and IK(DR), recorded when the cells were depolarized from −50 to +40 mV, by 65.9 (P<0.01) and 67.3% (P<0.01), respectively. The IC50 values for IK(A) and IK(DR) were 3.8 and 1.7 μM, respectively.
The decay time constant of IK(A), recorded following a test pulse to +40 mV, was prolonged reversibly by rivastigmine at concentrations of 10 and 100 μM (both P<0.05).
Rivastigmine affected the voltage dependence of IK(A) and IK(DR). At a concentration of 10 μM, it shifted the steady-state inactivation curve of IK(A) towards more negative potentials by −11 mV (P<0.05), but had no effect on the steady-state activation curve or the recovery from inactivation. Regarding the kinetic properties of IK(DR), 10 μM rivastigmine shifted the steady-state activation and inactivation curves towards more negative potentials by −10 (P<0.05) and −27 mV (P<0.01), respectively.
Our findings that rivastigmine inhibits IK(A) and IK(DR) in rat hippocampal pyramidal neurons suggest that this agent has other pharmacological actions besides its antiacetylcholinesterase activity.
Keywords: Rivastigmine, hippocampus, pyramidal neurons, K+ current, patch clamp
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by a slow, progressive decline in cognitive and memory function. A reduction of acetylcholine (ACh) and a central cholinergic deficit have been consistently identified in the development of AD (Francis et al., 1999), which provides the rationale for the treatment of AD by enhancing ACh in the brain. Acetylcholinesterase (AChE) inhibitors such as tacrine, donepezil, rivastigmine and galantamine (Grutzendler & Morris, 2001) have produced promising outcomes in clinical trials. Their actions on other targets in the brain, for example, central K+ channels, have also been extensively studied. Harvey & Rowan (1990) proposed the hypothesis that AChE inhibitors may affect K+ channels. Tacrine, the first approved AChE inhibitor, inhibits the transient outward K+ current (IK(A)) in rat hippocampal neurons (Rogawski, 1987) and the delayed rectifier K+ current (IK(DR)) in the larval muscles of Drosophila (Kraliz & Singh, 1997). Donepezil (Zhong et al., 2002), galantamine (Pan et al., 2003) and huperzine A, a promising AChE inhibitor isolated from a Chinese herb (Li & Hu, 2002a, 2002b), inhibit IK(A) and/or IK(DR) in rat hippocampal neurons. This research work raises the inspiring possibility that manipulations aimed at reducing outward K+ currents in the brain may provide a means of enhancing cognitive abilities in AD.
Rivastigmine is a novel AChE inhibitor, displaying specificity for central AChE over peripheral AChE. In clinical trials, it has proved more efficient than other AChE inhibitors at improving memory (Gottwald & Rozanski, 1999), but to date no data about its actions on central K+ currents have been reported. In the present study, we aimed at investigating the effects of rivastigmine on the two main voltage-activated outward K+ currents, IK(A) and IK(DR), in acutely dissociated rat hippocampal pyramidal neurons.
Methods
Cell preparation
Male Wistar rats aged 21–28 days were purchased from the Experimental Animal Center of the Chinese Academy of Medical Sciences. The experiments were performed in accordance with the current laws governing animal experimentation in the United Kingdom. Single rat hippocampal pyramidal neurons were acutely dissociated by enzymatic digestion and mechanical dispersion according to the methods of Kay & Wong (1986), with slight modifications. Briefly, 400-μm-thick hippocampal slices were cut in ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl 126, KCl 5, NaH2PO4 1.25, MgSO4 2, NaHCO3 26, glucose 10, CaCl2 2 (pH 7.20), bubbled with 95%O2–5%CO2, and incubated for 1 h at room temperature. The slices were then incubated at 32°C in ACSF containing 0.5 g l−1 trypsin for 30 min, and then in ASCF containing 0.5 g l−1 protease E for a further 30 min. Thereafter, the tissue was washed with enzyme-free solution and kept at room temperature. Neurons were isolated by triturating the slices in a series of Pasteur pipettes with decreasing tip diameters. After the final cell suspension had settled on the bottom of the recording chamber, neurons with a bright, smooth appearance and no visible organelles were selected for recording.
Whole-cell patch-clamp technique
Voltage-clamp recordings were performed in the whole-cell patch-clamp configuration (Hamill et al., 1991). Patch pipettes were pulled in two steps from borosilicate glass capillaries 1.5 mm in diameter, and had a tip resistance of 3–5 MΩ when filled with pipette solution containing (in mM): KCl 140, MgCl2 0.5, HEPES 10, EGTA 10 and Na2ATP 2, adjusted to pH 7.2 with KOH. The acutely isolated neurons were perfused by gravity with an extracellular solution of the following composition (in mM): NaCl 150, KCl 5, MgCl2 1.1, CaCl2 2.0, glucose 10, HEPES 10 and TTX 0.001, pH set at 7.3. Rivastigmine was dissolved in the extracellular solution and bath applied for 5 min. Tight seals (>1 GΩ) were obtained during the recordings. Data were recorded with an EPC-7 amplifier (List, Germany), filtered at 3 kHz, and stored in a PC 486 personal computer using a Labmaster TL-1 interface and pCLAMP 5.5.1 software (Axon, U.S.A.). All experiments were performed at room temperature (21–24°C).
Data analysis and statistics
Voltage protocols and data analysis for IK(A) and IK(DR) are described in Results. All data were analyzed using pCLAMP 6.0.1 (Axon Instrument) and SigmaPlot software, and are expressed as means±s.e.m. Significant differences between groups were assessed by paired Student's t-test. The criterion for significance was P<0.05 in all the analyses.
Drugs
Rivastigmine hydrogen tartrate (ENA-713) was generously provided by Novartis Basle, Switzerland. HEPES, EGTA, Na2ATP and TTX were purchased from Sigma Chemical Co. (St Louis, MO, U.S.A.). Other chemicals were obtained from Beijing Chemical Factory (Beijing, China).
Results
Isolation of IK(DR) and IK(A)
Total voltage-activated outward K+ currents were stimulated with a 150 ms depolarizing test pulse from a holding potential of −50 to +60 mV, in 10 mV increments, following a conditioning prepulse to −110 mV. The current displayed two components, the transient outward K+ current (IK(A)) and the delayed rectifier K+ current (IK(DR)) (Figure 1a). IK(DR) was isolated by using the same voltage protocol as for total K+ currents, but with a 50 ms interval inserted after the prepulse. The currents obtained at the end of the depolarizing pulse were referred to as IK(DR) (Figure 1b). IK(A) was obtained by subtracting the isolated IK(DR) traces from the total outward K+ current. The peak of the subtracted currents was referred to as IK(A) (Figure 1c).
Figure 1.
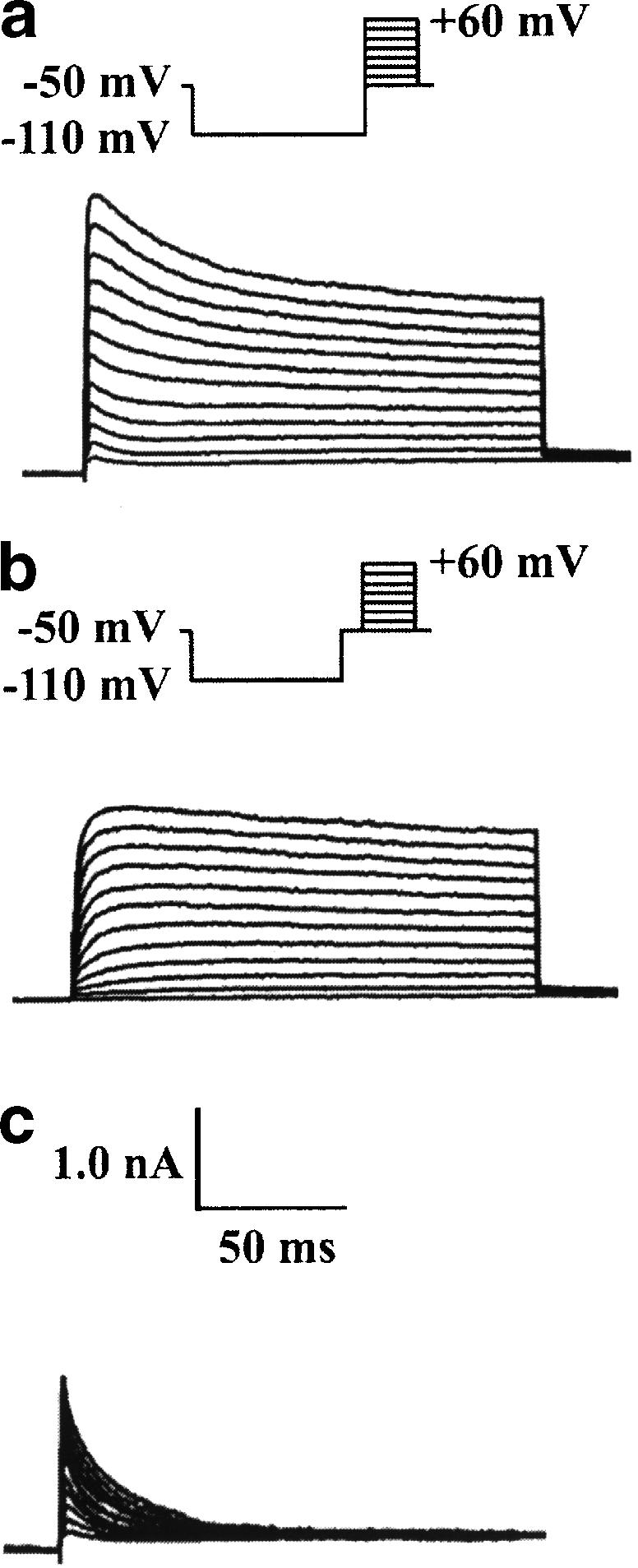
Outward K+ current components in a hippocampal pyramidal neuron. (a) Total outward K+ current stimulated with 150 ms depolarizing pulses from −50 to +60 mV in 10 mV steps following a hyperpolarizing prepulse of 400 ms to −110 mV. (b) IK(DR) stimulated with a similar protocol to that used in (a), except that a 50 ms interval at −50 mV was inserted after the prepulse. Currents recorded at the end of the depolarizing pulse were referred to as IK(DR). (c) Isolated IK(A) obtained by subtracting the current traces in (b) from those in (a). The peak of the subtracted currents was referred to as IK(A).
Effects of rivastigmine on IK(A) and IK(DR)
In order to observe the effects of rivastigmine on IK(A) and IK(DR), the pulse protocols and subtraction procedure shown in Figure 1 were used. The depolarizing test pulse was to +40 mV. In blank controls without rivastigmine (time matched controls), IK(A) and IK(DR) were altered by 4.1±4.3 (n=8) and 2.9±5.1% (n=8) after 15 min of current recording, respectively, indicating that rundown of current was negligible. Inhibition by rivastigmine occurred rapidly over 1–3 min and reached a steady state by ∼5 min. In the presence of 100 μM rivastigmine, IK(A) and IK(DR) were inhibited by 65.9±4.1 (n=6, P<0.01) and 67.3±3.1% (n=6, P<0.01), respectively. On removal of rivastigmine, IK(A) and IK(DR) rapidly returned to near control values within 2 min (Figure 2a). The currents after washout recovered to 80.6±8.1% of control (n=6) for IK(A) and 76.4±5.7% of control (n=6) for IK(DR), showing that the blocking effects of rivastigmine were readily reversible. Figure 2b illustrates the concentration dependence of the action of rivastigmine on IK(A) and IK(DR). Data were fitted with the equation (Wooltorton & Mathie, 1993): I/Icontrol= 100−[max/1+(IC50/C)n], where Icontrol and I are current amplitudes measured in control conditions and in the presence of rivastigmine, max is the maximum inhibition attainable, C is the concentration of rivastigmine in the external solution, IC50 is the half-maximal inhibitory concentration and n is the slope factor (Hill coefficient). For IK(A), the IC50 and Hill coefficient were calculated as 3.8±0.5 μM and 0.88±0.11, respectively. For IK(DR), the values of IC50 and the Hill coefficient were 1.7±0.3 μM and 0.67±0.08, respectively.
Figure 2.

Effects of rivastigmine on IK(A) and IK(DR) recorded following a depolarizing test pulse to +40 from −50 mV. (a) Superimposed current traces obtained at +40 mV in control conditions, 5 min after the application of 100 μM rivastigmine and after washout of the drug. IK(A) was obtained by digital subtraction. The effects of rivastigmine were readily reversible on washout of the drug. The decay time constant of IK(A) was fitted by a monoexponential function. In the presence of 100 μM rivastigmine, the rate of IK(A) inactivation was 24.3 ms compared with 7.1 ms in control. (b) Concentration–response curves for the blocking action of rivastigmine on IK(A) and IK(DR). Data were fitted with the equation: I/Icontrol=100−[max/1+(IC50/C)n]. For IK(A), IC50=3.8 μM, n (the Hill coefficient)=0.88 and max=65.9%. For IK(DR), IC50=1.7 μM, n=0.67 and max=67.3%. The numbers above each data point indicate the numbers of patches used to construct the points.
Rivastigmine also reversibly slowed down the decay of IK(A), in addition to causing a reduction in its peak amplitude. The decay time constant was fitted by a monoexponential equation (Belluzzi et al., 1985). With a test pulse to +40 mV, the decay time constant was prolonged from 6.1±0.7 to 13.7±3.6 ms by the addition of 10 μM rivastigmine (n=6, P<0.05) and from 7.1±1.7 to 24.3±4.0 ms with 100 μM rivastigmine (n=5, P<0.05). The decay time constant recovered to 14.2±3.1 ms after washout of 100 μM rivastigmine (Figure 2a).
Effects of rivastigmine on the voltage dependence of the steady-state activation of IK(A) and IK(DR)
The current–voltage (I–V) curves of both IK(A) and IK(DR) were obviously depressed by 10 μM rivastigmine (Figure 3b). The amplitudes of IK(A) and IK(DR) were converted to conductance (G) using the equation: G=I/(V−VK), where V is membrane potential and VK is the potassium reversal potential (calculated as −86 mV for the potassium concentrations used). The normalized conductance was fitted with the Boltzmann function: G/Gmax=1/{1+exp[−V−V1/2/k]}, where V is the membrane potential, V1/2 is the potential for half-maximal activation and k is the slope factor. The values of V1/2 for IK(A) were −10.7±3.1 mV in control and −9.6±3.6 mV in the presence of 10 μM rivastigmine (n=5), and the values for k were 18.7±0.9 and 19.5±1.3 mV, respectively. The values of V1/2 for IK(DR) were −6.1±3.0 mV in control and −15.7±2.1 mV in the presence of 10 μM rivastigmine (n=5, P<0.05), and the values for k were 16.1±0.5 and 18.6±1.7 mV, respectively (Figure 3c). Thus, rivastigmine caused a hyperpolarizing shift of the steady-state activation curve of IK(DR) of about −10 mV, and did not alter the voltage dependence of IK(A) activation.
Figure 3.

Effects of 10 μM rivastigmine on the current–voltage (I–V) curves and the voltage dependence of activation of IK(A) (left) and IK(DR) (right). (a) The original current traces of IK(A) and IK(DR) before and after the addition of 10 μM rivastigmine. The pulse protocols and subtraction procedure were the same as in Figure 1. (b) The effect of 10 μM rivastigmine on the I–V curves of IK(A) and IK(DR). (c) The effect of 10 μM rivastigmine on the steady-state activation curves of IK(A) and IK(DR). The amplitudes of IK(A) and IK(DR) were converted to conductance and normalized to maximal conductance. Normalized data points were fitted with the Boltzmann equation: G/Gmax=1/{1+exp[−(V−V1/2)/k]}. Each point represents the mean±s.e.m. of five cells. V1/2 values for IK(A) were −10.7 mV in control and −9.6 mV in the presence of 10 μM rivastigmine. V1/2 values for IK(DR) were −6.1 mV in control and −15.7 mV in the presence of 10 μM rivastigmine.
Effect of rivastigmine on the voltage dependence of the steady-state inactivation of IK(A) and IK(DR)
Cells with prominent IK(A) were selected. The steady state inactivation behavior of IK(A) was determined by applying 1 s conditioning prepulses to between −130 and −50 mV in 10 mV increments, followed by a 150 ms depolarizing pulse to +60 mV. To minimize the contamination by IK(DR), IK(A) was measured as the peak current obtained after subtracting the slow component of the current at the end of the test pulse. The steady-state inactivation behavior of IK(DR) was determined by applying 1 s conditioning prepulses to between −120 and +40 mV in 10 mV increments, followed by a delay of 50 ms at −50 mV to inactivate IK(A) and a 150 ms depolarizing pulse to +40 mV. The difference between the current at the end of the test pulse and that at the end of the conditioning pulse was referred to as IK(DR) (Figure 4a).
Figure 4.

Effect of 10 μM rivastigmine on the voltage dependence of IK(A) (left) and IK(DR) (right). (a) The original current traces before and after the addition of 10 μM rivastigmine. Pulse protocol is shown in the inset. (b) The effect of 10 μM rivastigmine on the steady-state inactivation curves of IK(A) and IK(DR). Normalized data points were fitted with the Boltzmann equation: I/Imax=1/{1+exp[(V−V1/2)/k]}. Each point represents the mean±s.e.m. of five cells for IK(A) and IK(DR). In the presence of 10 μM rivastigmine, V1/2 was −115.0 mV compared with −104.4 mV in control for IK(A). For IK(DR), V1/2 was −74.5 mV with rivastigmine compared with −47.6 mV in control.
The steady-state inactivatiovn curve was fitted with the Boltzmann function: I/Imax=1/{1+exp[(V−V1/2)/k]}, where V is the membrane potential, V1/2 is the potential for half-maximal inactivation and k is the slope factor. The values of V1/2 for IK(A) before and after the addition of 10 μM rivastigmine were −104.4±2.7 and −115.0±2.4 mV (n=5, P<0.05), respectively, and the respective k values were 8.3±0.7 and 5.9±0.4 mV (n=5, P<0.05). The values of V1/2 for IK(DR) before and after the addition of 10 μM rivastigmine were −47.6±6.2 and −74.5±4.6 mV (n=5, P<0.01), respectively, and the respective k values were 22.6±1.1 and 16.1±2.2 mV (Figure 4b). Thus, rivastigmine caused hyper-polarizing shifts of the steady-state inactivation of both IK(A) and IK(DR) of −11 and −27 mV, respectively.
Effect of rivastigmine on the recovery from inactivation of IK(A)
Cells with prominent IK(A) were selected and voltage clamped at −30 mV to fully inactivate IK(A). A 150 ms depolarizing test pulse to +40 mV was applied after a conditioning prepulse to −110 mV. The prepulses were increased from 0 to 150 ms in 10 ms steps. IK(A) was measured as the peak current after subtracting the slow current component mediated by IK(DR) at the end of the depolarizing pulse (Figure 5a). The curve for recovery from inactivation of IK(A) was fitted with the monoexponential function: I/Imax=1−exp(−t/τ), where t is the recovery interval of the conditioning prepulse and τ is the time constant for the recovery from inactivation of IK(A). The values of τ were 34.5±2.9 ms in control and 33.7±3.1 ms (n=5) in the presence of 10 μM rivastigmine (Figure 5b). Rivastigmine did not affect the recovery from inactivation of IK(A).
Figure 5.

Effect of 10 μM rivastigmine on the recovery from inactivation of IK(A). (a) The original current traces before and after the addition of 10 μM rivastigmine. (b) The effect of 10 μM rivastigmine on the recovery from inactivation curve of IK(A). Normalized data points were fitted with the monoexponential equation: I/Imax=1−exp(−t/τ). Each point represents mean±s.e.m. of five cells. τ was 34.5 ms in control and 33.7 ms in the presence of 10 μM rivastigmine.
Discussion and conclusions
IK(A) and IK(DR) are the two main neuronal voltage-activated K+ currents, and they play a critical role in maintaining neuronal excitability (Pongs, 1999). In the present study, we found that, in the presence of rivastigmine, a novel AChE inhibitor (Gottwald & Rozanski, 1999), the amplitudes of IK(A) and IK(DR) were reduced. Some important features of the mechanism of channel block can be deduced from our data. The inhibitory effects of rivastigmine were induced quickly and were reversible on washing out the drug, suggesting that rivastigmine probably interacts directly with the channels from the extracellular side of the membrane. The Hill coefficients for IK(A) and IK(DR) were smaller than 1, indicating that rivastigmine binds to the corresponding K+ channels in a negatively cooperative manner.
The kinetic properties of IK(A) and IK(DR) were also significantly affected by rivastigmine. The current decay of IK(A) was slowed down. The observation that the channels conducting IK(A) close more slowly in the presence of rivastigmine suggests that the open-state inactivation is affected. The steady-state inactivation curves of IK(A) and IK(DR) were shifted in a hyperpolarizing direction. This implies that rivastigmine inhibited IK(A) and IK(DR) voltage-dependently, mainly due to earlier inactivation of both currents. Thus, we speculated that rivastigmine produces changes in the intrinsic inactivation-gating properties of both channels. It was interesting to find that the steady-state activation curve of IK(DR) was also shifted towards more negative potentials. The shift was −10 mV, which indicates earlier activation and normally, but not always, results in an increase in current amplitude. However, a decrease in IK(DR) was clearly observed in the present study. One possible reason could be that although the IK(DR) activation curve was slightly shifted by about −10 mV, the inactivation curve was shifted by −27 mV. Therefore, the voltage-dependent inhibition induced by rivastigmine is much stronger than the channel activation resulting from the negative shift of the activation curve. This observation has also been noted for other AChE inhibitors whose inhibitory effects on K+ currents have been reported. Donepezil (Zhong et al., 2002), galantamine (Pan et al., 2003) and huperzine A (Li & Hu, 2002b), all shifted the steady-state activation curve of IK(DR) in a hyperpolarizing direction in acutely dissociated rat hippocampal pyramidal neurons.
In our study, we also found that the recovery of IK(A) from inactivation was unchanged in the presence of rivastigmine, which indicates that rivastigmine does not affect the change between the inactivation and the resting states of the channel conducting IK(A). It is known that ion channels can be activated only in the resting state.
It is generally accepted that the massive neuronal death which occurs in AD is due to apoptosis (Shimohama, 2000). It has recently been suggested that early loss of total intracellular K+ (apparently associated with the enhancement of IK(DR)) is an essential event in the development of some forms of apoptosis. Attenuating outward K+ current with tetraethylammonium (TEA) or elevated extracellular K+ reduced apoptosis (Yu et al., 1997; 1998). One possible pharmacological implication of our findings is that the blockade of voltage-activated K+ currents by rivastigmine may lead to the suppression of apoptosis and a substantial increase in cell survival.
Tacrine was the earliest approved AChE inhibitor, and was the first known such inhibitor with a K+ channel-blocking effect. Its therapeutic concentration in the cerebrospinal fluid of AD patients is 22 nM (5.21 μg l−1) (Jann et al., 2002). Kraliz & Singh (1997) found that, at concentrations as low as 10 μM, tacrine selectively blocked IK(DR) and broadened the action potential in the larval muscle of Drosophila. They proposed that the K+ channel-blocking effect of tacrine increased transmitter release in the brain, and thus contributed to its effectiveness in the treatment of AD. We report, for the first time, that rivastigmine inhibits IK(A) (IC50 3.8 μM) and IK(DR) (IC50 1.7 μM) in acutely dissociated rat hippocampal neurons. It has been reported that the concentration of rivastigmine in the cerebro-spinal fluid of AD patients is about 14 nM (5.42 μg l−1) (Gobburu et al., 2001). Like tacrine, rivastigmine may target some K+ channels as well as AChE at higher concentrations. However, in acute rat hippocampal slices, rivastigmine had no effect on glutamatergic synaptic transmission at concentrations up to 100 nM (Santos et al., 2002), which suggests that in in vitro studies presynaptic enhancement of transmission requires relatively high concentrations of the drug. Thus, the relevance of our in vitro findings to the clinical effects of rivastigmine is uncertain, because it is usually very difficult to speculate about in vivo concentrations based on concentrations found to be effective in vitro. Whether the effects of rivastigmine on K+ currents contribute to its effectiveness in AD therapy remains to be elucidated in further studies.
Acknowledgments
This project was supported by grants from the National 973 Fundamental Project of China (No. G1998051106).
Abbreviations
- ACh
acetylcholine
- AChE
acetylcholinesterase
- ACSF
artificial cerebrospinal fluid
- AD
Alzheimer's disease
- IK(A)
transient outward K+ current
- IK(DR)
delayed rectifier K+ current
- I–V
current–voltage
- TEA
tetraethylammonium
References
- BELLUZZI O., SACCHI O., WANKE E. A fast transient outward current in the rat sympathetic neurone studied under voltage-clamp conditions. J. Physiol. 1985;358:91–108. doi: 10.1113/jphysiol.1985.sp015542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANCIS P.T., PALMER A.M., SNAPE M., WILCOCK G.K. The cholinergic hypothesis of Alzheimer's disease: a review of progress. J. Neurol. Neurosurg. Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOBBURU J.V., TAMMARA V., LESKO L., JHEE S.S., SRAMEK J.J., Cutler N.R., YUAN R. Pharmacokinetic–pharmacodynamic modeling of rivastigmine, a cholinesterase inhibitor, in patients with Alzheimer's disease. J. Clin. Pharmacol. 2001;41:1082–1090. doi: 10.1177/00912700122012689. [DOI] [PubMed] [Google Scholar]
- GOTTWALD M.D., ROZANSKI R.I. Rivastigmine, a brain-region selective acetylcholinesterase inhibitor for treating Alzheimer's disease: review and current status. Expert. Opin. Investig. Drugs. 1999;8:1673–1682. doi: 10.1517/13543784.8.10.1673. [DOI] [PubMed] [Google Scholar]
- GRUTZENDLER J., MORRIS J.C. Cholinesterase inhibitors for Alzheimer's disease. Drugs. 2001;61:41–52. doi: 10.2165/00003495-200161010-00005. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P., HUGUENARD J.R., PRINCE D.A. Patch-clamp studies of voltage-gated currents in identified neurons of the rat cerebral cortex. Cereb. Cortex. 1991;1:48–61. doi: 10.1093/cercor/1.1.48. [DOI] [PubMed] [Google Scholar]
- HARVEY A.L., ROWAN E.G. Effects of tacrine, aminopyridines, and physostigmine on acetylcholinesterase, acetylcholine release, and potassium currents. Adv. Neurol. 1990;51:227–233. [PubMed] [Google Scholar]
- JANN M.W., SHIRLEY K.L., SMALL G.W. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin. Pharmacokinet. 2002;41:719–739. doi: 10.2165/00003088-200241100-00003. [DOI] [PubMed] [Google Scholar]
- KAY A.R., WONG R.K. Isolation of neurons suitable for patch-clamping from adult mammalian central nervous systems. J. Neurosci. Methods. 1986;16:227–238. doi: 10.1016/0165-0270(86)90040-3. [DOI] [PubMed] [Google Scholar]
- KRALIZ D., SINGH S. Selective blockade of the delayed rectifier potassium current by tacrine in Drosophila. J. Neurobiol. 1997;32:1–10. [PubMed] [Google Scholar]
- LI Y., HU G.Y. Huperzine A, a nootropic agent, inhibits fast transient potassium current in rat dissociated hippocampal neurons. Neurosci. Lett. 2002a;324:25–28. doi: 10.1016/s0304-3940(02)00167-2. [DOI] [PubMed] [Google Scholar]
- LI Y., HU G.Y. Huperzine A inhibits the sustained potassium current in rat dissociated hippocampal neurons. Neuro-sci. Lett. 2002b;329:153–156. doi: 10.1016/s0304-3940(02)00620-1. [DOI] [PubMed] [Google Scholar]
- PAN Y.P., XU X.H., WANG X.L. Galantamine blocks delayed rectifier, but not transient outward potassium current in rat dissociated hippocampal pyramidal neurons. Neurosci. Lett. 2003;336:37–40. doi: 10.1016/s0304-3940(02)01235-1. [DOI] [PubMed] [Google Scholar]
- PONGS O. Voltage-gated potassium channels: from hyperexcitability to excitement. FEBS Lett. 1999;452:31–35. doi: 10.1016/s0014-5793(99)00535-9. [DOI] [PubMed] [Google Scholar]
- ROGAWSKI M.A. Tetrahydroaminoacridine blocks voltage-dependent ion channels in hippocampal neurons. Eur. J. Pharmacol. 1987;142:169–172. doi: 10.1016/0014-2999(87)90670-4. [DOI] [PubMed] [Google Scholar]
- SANTOS M.D., ALKONDON M., PEREIRA E.F., ARACAVA Y., EISENBERG H.M., MAELICKE A., ALBUQUERQUE E.X. The nicotinic allosteric potentiating ligand galantamine facilitates synaptic transmission in the mammalian central nervous system. Mol. Pharmacol. 2002;61:1222–1234. doi: 10.1124/mol.61.5.1222. [DOI] [PubMed] [Google Scholar]
- SHIMOHAMA S. Apoptosis in Alzheimer's disease – an update. Apoptosis. 2000;5:9–16. doi: 10.1023/a:1009625323388. [DOI] [PubMed] [Google Scholar]
- WOOLTORTON J.R., MATHIE A. Block of potassium currents in rat isolated sympathetic neurones by tricyclic antidepressants and structurally related compounds. Br. J. Pharmacol. 1993;110:1126–1132. doi: 10.1111/j.1476-5381.1993.tb13931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU S.P., FARHANGRAZI Z.S., Ying H.S., YEH C.H., CHOI D.W. Enhancement of outward potassium current may participate in beta-amyloid peptide-induced cortical neuronal death. Neurobiol. Dis. 1998;5:81–88. doi: 10.1006/nbdi.1998.0186. [DOI] [PubMed] [Google Scholar]
- YU S.P., YEH C.H., SENSI S.L., GWAG B.J., CANZONIERO L.M., FARHANGRAZI Z.S., YING H.S., TIAN M., DUGAN L.L., CHOI D.W. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- ZHONG C.B., ZHANG W., WANG X.L. Effects of donepezil on the delayed rectifier-like potassium current in pyramidal neurons of rat hippocampus and neocortex. Acta Pharm. Sin. 2002;37:415–418. [PubMed] [Google Scholar]